Gentle Isolation of Nuclei from the Brain Tissue of Adult African Turquoise Killifish, a Naturally Short-Lived Model for Aging Research
In This Article
Summary
Here we present a protocol to isolate nuclei from the brains of the short-lived vertebrate model Nothobranchius furzeri for downstream applications such as single-nucleus RNA sequencing or single-nucleus assay for transposase-accessible chromatin with sequencing (ATAC-seq).
Abstract
Studying brain aging at single-cell resolution in vertebrate systems remains challenging due to cost, time, and technical constraints. Here, we demonstrate a protocol to generate single-nucleus RNA sequencing (snRNA-seq) libraries from the brains of the naturally short-lived vertebrate African turquoise killifish Nothobranchius furzeri. The African turquoise killifish has a lifespan of 4-6 months and can be housed in a cost-effective manner, thus reducing cost and time barriers to study vertebrate brain aging. However, tailored protocols are needed to isolate nuclei of sufficient quality for downstream single-cell experiments from the brain of young and aged fish. Here, we demonstrate an empirically optimized protocol for the isolation of high-quality nuclei from the brain of adult African turquoise killifish, a critical step in the generation of high-quality single nuclei omic libraries. Furthermore, we show that the steps to reduce contaminating background RNA are important to clearly distinguish cell types. In summary, this protocol demonstrates the feasibility of studying brain aging in non-traditional vertebrate model organisms.
Introduction
Understanding the mechanisms of vertebrate brain aging is critical to addressing age-related neurodegenerative diseases such as Alzheimer's and dementia1. The African turquoise killifish (Nothobranchus furzeri) is the shortest-lived vertebrate that can be bred in captivity, and due to its short lifespan and age-associated cognitive impairment, it is an excellent brain aging model2,3,4,5. Recently, the advent of single-cell "omics" technologies, such as single nuclei RNA-seq (snRNA-seq) and single nuclei assay for transposase-accessible chromatin with sequencing (snATAC-seq), have allowed researchers to interrogate the aging brain at an unprecedented resolution6,7,8. These methods rely on nuclei isolation, since the recovery of brain cells such as neurons is often too challenging to isolate6,7,8,9,10. However, most published nuclei isolation protocols are optimized for mammalian model organisms11,12,13,14,15. Thus, as there is currently an unmet need for isolating brain nuclei in the killifish as an up-and-coming new model organism in the field of aging research2, the goal of this protocol is to establish a method for isolating high-quality nuclei from frozen brain killifish tissue.
Here, a streamlined and robust workflow is established that uses commonly available materials to isolate high-quality nuclei from killifish brains. This protocol was modified from a 10x Genomics protocol for mouse brains to accommodate the lower myelin content brains of African turquoise killifish, the fragility of frozen tissue, and the need to reduce ambient debris content for sequencing-related applications16. Indeed, previously optimized protocols for mammalian brain tissue17,18 lead to poor nuclei quality (i.e., overlysis) and/or high debris content when used on frozen killifish brains, making them unsuitable for use with snRNA-seq according to recommendations for single nuclei RNA-seq using microfluidics (Supplementary Figure 1).
In addition to nuclei isolation, we demonstrate how to assess nuclei quality and yield by microscopy and flow cytometry. This article provides examples of both optimal and suboptimal results and discusses troubleshooting. This protocol was designed and optimized for frozen killifish brains but can also be used without major modifications on freshly dissected killifish samples. Killifish brain nuclei isolated using this method have been optimized for use in single nucleus RNA-seq (snRNA-seq) as a downstream application, but should also be amenable for use in snATAC-seq and bulk ATAC-seq.
Protocol
Animal care and animal experimentation were performed in accordance with the University of Southern California IACUC under approved protocols #21215. For any work using this protocol, it is necessary to obtain approval from the institution's IACUC prior to starting any research work on vertebrate animals.
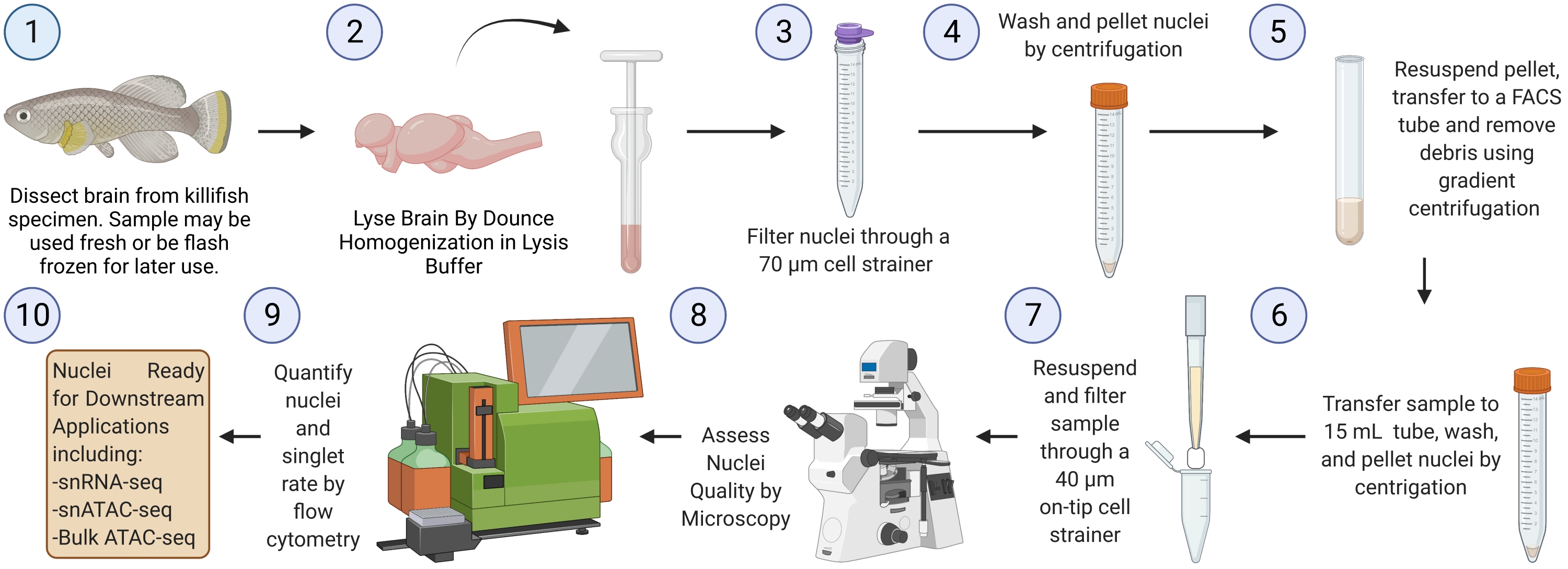
NOTE: A complete run through the protocol starting from flash-frozen brain tissue (starting from step 3) should take ~2.5 h for six samples (Figure 1).
1. Dissect killifish brains
- Humanely euthanize killifish using a solution of 1.5 g/L of methanosulfonate/tricaine (MS-222) in the system water.
- Use sharp scissors to rapidly decapitate killifish after all body and gill movement has ceased.
- Dissect the decapitated head on a clean Petri dish as previously described19.
- Place the dish under a dissecting microscope (using an 8x-35x magnification range). Turn the head such that the branchiostegal rays are in focus
- Using scissors, cut through the dentary such that the branchiostegal rays and the cranium are revealed.
- Using forceps, hold down the branchiostegal rays on either side of the cranium. Use another pair of forceps to remove any remaining tissue fragments from the cranium. The V-shaped optical chiasm that connects the brain and the eyes should become visible.
- Cut the optical chiasm using scissors to detach both eyes from the brain. Use forceps to gently free the cranium.
- Invert the cranium over and use forceps to scrape the muscle from the dorsal side of the cranium.
- Use one pair of forceps to hold the cranium in place and another pair of forceps to gently remove the bones of the cranium, revealing the brain.
- The brain appears white and soft. Once visible, it is not required to remove all cranial bones. Remove the brain with clean forceps by gently lifting and scraping.
- Flash freeze the brain by placing it into a microcentrifuge tube stored on dry ice.
NOTE: The frozen brain tissue can be stored at -80 °C until all samples to be processed together have been collected to limit batch effects when processing for library generation. While samples stored at -80 °C cannot be stored indefinitely, this protocol has been successfully performed on killifish brains stored for up to 12 months with no notable drop in quality.
2. Prepare fresh buffers
- Prepare nuclei wash buffer (2% bovine serum albumin [BSA] in 1x PBS, phosphate-buffered saline pH 7.4, and 0.2 U/µL RNase inhibitor) and store on ice. Prepare 250 mL of nuclei wash buffer for up to eight samples. For 250 mL, use 50 mL of 10% BSA stock solution, 25 mL of a 10x PBS stock solution, 1.25 mL of the 40 U/µL RNase inhibitor stock, and then bring volume to 250 mL with RNAse-free ddH2O.
- Prepare nuclei lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.01875% v/v NP-40, and 0.2 U/µL RNase inhibitor) and store on ice. Prepare 10 mL of nuclei lysis buffer for up to eight samples. For 10 mL, use 100 µL of 1 M Tris-HCl pH 7.4 stock solution, 20 µL of 5 M NaCl stock solution, 30 µL of 1 M MgCl2 stock solution, 18.75 µL of a 10% NP-40 stock solution, 50 µL of the 40 U/µL RNase inhibitor stock, and then bring the volume to 10 mL with RNAse-free ddH2O.
3. Isolate nuclei
- If using frozen samples, thaw them on ice for 10 min. If using freshly dissected brains, proceed directly to step 3.2
NOTE: Although this protocol can work on single brains, smaller samples from young fish (5-6 weeks old) will have a poorer yield. Sufficient yields can always be obtained by processing two brains as one sample, regardless of the fish's age and sex (Table 1). This protocol has successfully isolated high-quality nuclei from fish aged 5-26 weeks, including animals from both GRZ and ZMZ1001 strains. There are no issues expected in applying this protocol to other strains. - Dounce the brain in 1 mL of ice-cold nuclei lysis buffer in a 2 mL Dounce homogenizer. Keep the homogenizer on ice and avoid generating bubbles while lysing the sample. Dounce at an approximate rate of 0.5 s/downstroke and 0.5 s/upstroke with a 180° twist per cycle. Dounce using the loose pestle for 15 strokes, or until the tissue/lysis buffer mixture appears homogeneous if additional strokes are needed.
NOTE: Dounce homogenizers should be washed, rinsed with distilled water, and sterilized with standard dry autoclave sterilization cycles between uses.- Dounce using the tight pestle for 30 strokes with small breaks, 30 s to 1 min, every 10 strokes. Dounce at an approximate rate of 0.5 s/downstroke and 0.5 s/upstroke with a 180° twist per cycle. Let the sample rest on ice in the Dounce homogenizer for 2 min.
NOTE: This incubation time ensures adequate lysis but should be shortened if nuclear overlysis is observed.
- Dounce using the tight pestle for 30 strokes with small breaks, 30 s to 1 min, every 10 strokes. Dounce at an approximate rate of 0.5 s/downstroke and 0.5 s/upstroke with a 180° twist per cycle. Let the sample rest on ice in the Dounce homogenizer for 2 min.
- Strain the sample through a 70 µm cell filter into a 15 mL conical tube precoated in 5% BSA.
- Add 4 mL of nuclei wash buffer to the sample by pipetting the wash buffer over the 70 µm cell filter from step 3.3 and gently mix by inversion five times.
NOTE: This allows for the recovery of nuclei that may be trapped on the filter and improves yield. - Centrifuge using a swinging bucket rotor at 500 x g, 4 °C, for 10 min. Discard the supernatant by gently pouring into a liquid waste receptacle. Return the sample to an upright position and remove the remaining wash buffer using a P1000 pipette.
NOTE: The nuclei pellet is likely to be visible when starting from two brains in one sample. However, when processing single brains from young animals (5-6 weeks old), a lower yield is expected, and pellets may not always be visible. In this case, the supernatant should be carefully removed while assuming the position of the pellet at the bottom of the tube. - Immediately resuspend the nuclei in 1 mL of nuclei wash buffer with a wide bore P1000 pipette tip.
- Bring the sample to 5 mL by adding 4 mL of nuclei wash buffer and gently mix by inversion five times. Centrifuge using a swinging bucket rotor at 500 x g, 4 °C, for 10 min, and discard the supernatant.
- Resuspend the nuclei in 1 mL of nuclei wash buffer with a wide bore pipette tip and transfer to a flow cytometry (FACS) tube.
- Add 300 µL of debris removal solution (Table of Materials) and mix thoroughly by pipetting with a wide bore tip until the mixture is homogeneous.
- Gently overlay 1 mL of nuclei wash buffer on top of the nuclei solution prepared in step 3.9 using a P1000 pipette and holding the tube at a slight angle (Supplementary Figure 2). The debris removal solution fraction and the nuclei wash buffer fraction should form a clear interface if layered correctly.
- Centrifuge in a swinging bucket rotor at 3000 x g, 4 °C, for 10 min. Completely discard the top two phases using a P1000 pipette.
NOTE: The clear FACS tube will permit the visualization of the three distinct phases following centrifugation (top, interphase, bottom). To facilitate maximal debris removal, it is advised to be more aggressive than conservative in removing the top two phases (i.e., removing a small amount of the bottom phase is preferable to carrying over any of the top phases; Supplementary Figure 2). Additionally, the interphase layer may be too thin to visualize. In this case, remove the top (apparent) layer, which contains the interphase layer. The bottom layer that is retained should contain ~1 mL total volume. - Transfer the bottom layer to a 15 mL conical tube precoated with 5% BSA.
- In the conical tube, bring the volume to 15 mL with nuclei wash buffer and invert three times to mix.
- Centrifuge in a swinging bucket rotor at 1000 x g, 4 °C, for 10 min. Remove the supernatant carefully.
- Immediately resuspend in 150 µL of nuclei wash buffer using a wide bore pipette tip.
- Switch to a standard bore pipette tip set to ~300 µL. Uptake the entire sample and then attach a 40 µm on-tip filter to the end of the pipette tip, taking care not to expel the sample.
NOTE: On-tip filters are necessary to obtain smaller volumes, which are needed to maintain sufficient nuclei concentration for downstream applications. For instance, for snRNA-seq using microfluidics, a minimum concentration of >300 nuclei/µL is recommended, although the optimal range is 700-1200 nuclei/µL18. Higher concentrations of nuclei can also be easily diluted if necessary. - Filter the sample by forcefully expelling the sample through the filter into a low DNA binding 1.5 mL microcentrifuge tube. The final elution volume should be 120-150 µL.
NOTE: Some frothing may result from filtration. This does not appear to affect the nuclei quality.
4. Assess nuclei quality by microscopy
- In a separate microcentrifuge tube, mix 10 µL of the filtered sample with 10 µL of 0.4% trypan blue solution by gentle pipetting. Samples require no extended incubation time and are ready for visualization immediately after mixing with trypan blue solution.
- Deposit 10 µL of the stained nuclei into a chamber of a counting chamber slide.
NOTE: Alternatively, deposit 10 µL of the stained nuclei sample onto a hemocytometer or microscopy slide and cover with a cover slip. - Assess the nuclei quality by light microscopy.
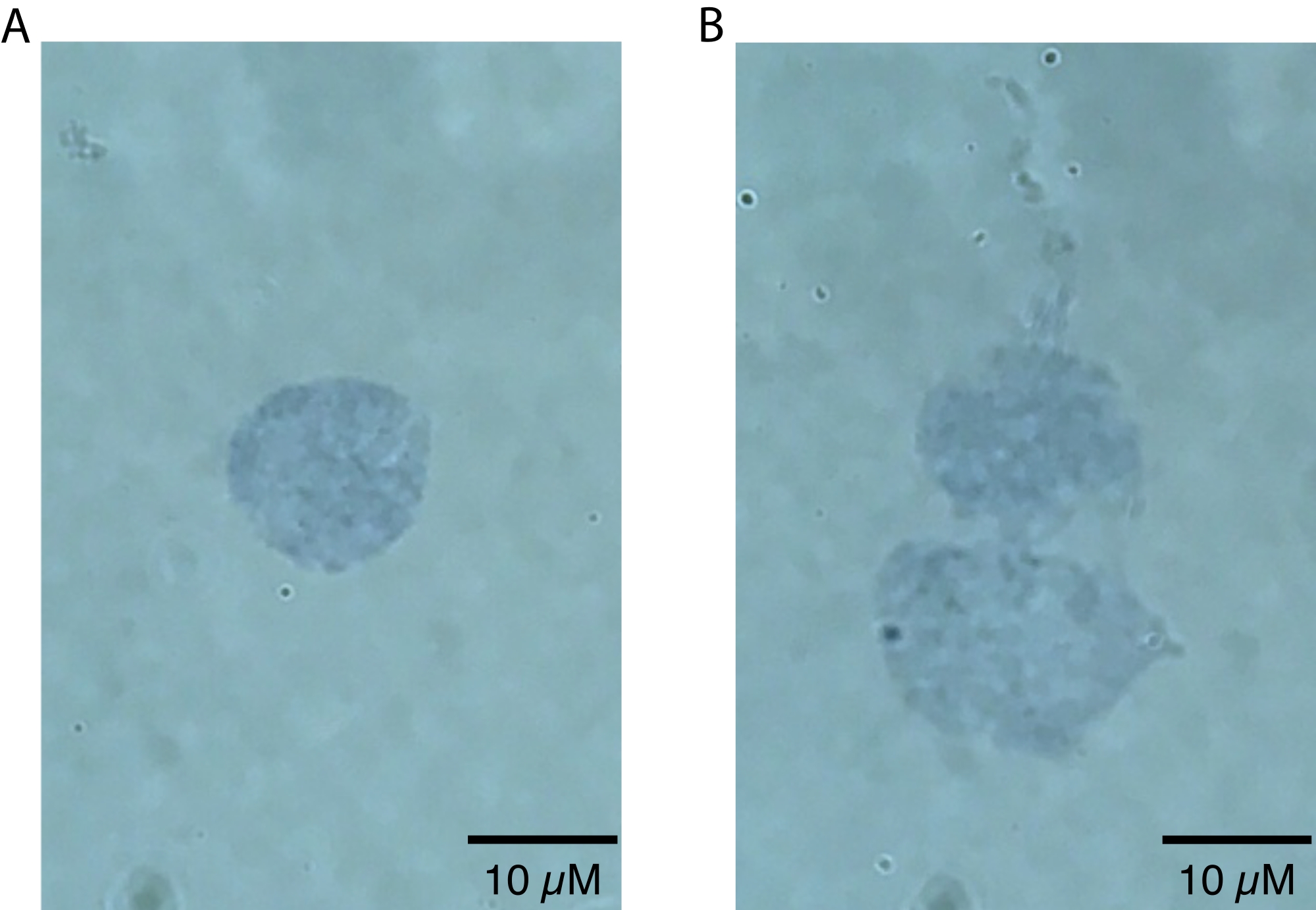
NOTE: Lower magnifications should only be used to zoom in on nuclei, since nuclei defects will only be visually apparent at higher magnifications (i.e., 60x or higher). This step can be done quickly to assess the success of nuclei preparation and does not require secondary analysis beyond visual inspection. High quality, intact nuclei will have a sharply defined border20 (Figure 2A). Poor quality nuclei will have disrupted nuclear membranes, patchy trypan blue staining near the nuclear membrane indicative of potential nucleic acid leakage, and/or evidence of blebbing20 (Figure 2B). Healthy nuclei are expected to be 10-15 µm in diameter.
5. Quantify singlet nuclei, debris, and multiplet proportion by flow cytometry
NOTE: The specific terminology and interface of the flow cytometer software may differ based on the brand of the machine, but these steps should be easily adapted to other systems if required.
- In a FACS tube, resuspend 10 µL of the filtered nuclei sample in 90 µL of the buffer recommended for flow cytometry for a 1:10 dilution. For this purpose, 0.22 µm filtered sterilized PBS, pH 7.2, with 2 mM EDTA and 0.5% BSA is used in this study.
NOTE: Although this dilution will work for most nuclei preps, a lower dilution (e.g., 1:5) may be needed to sample enough nuclei for quality assessment in the case of a lower yield sample (i.e., <30 nuclei/µL in the diluted sample). - Stain the samples with propidium iodide (PI) at a final concentration of 1 µg/mL. No incubation time is required, and nuclei can be analyzed immediately.
NOTE: Alternatively, stain the samples with a final concentration of 0.1 µg/mL DAPI. Note that DAPI should be read on a different fluorescence channel than PI (Vioblue-A V1-A channel; excitation at 405 nm, emission at 450/50 nm). - Analyze the samples on a flow cytometer as follows:
- Set up a workspace:
- Have the flow cytometer in Acquisition mode. Click on File, then select New Workspace from the drop-down menu. Under the Experiment tab in the left panel, enter the Name of Project and Sample ID into their respective fields.
- In the upper left toolbar, click on the New Analysis Window button (scatter plot icon). Click the box with three plots (Plot 4t) from the pop-up menu. Three plots will appear on the screen. Click on the Analysis Mode button in the upper left toolbar (A icon) so that it is gray and not orange.
- Set up analysis plots:
- Change the axes of the three plots as follows, ordered as X-axis and Y-axis, respectively:
Plot 1: X-axis: FSC-A, Y-axis: SSC-A
Plot 2: X-axis: HDR-T, Y-axis: SSC-A
Plot 3: X-axis: PerCP-Vio700-A B3-A, Y-axis: SSC-A - Change the axes by clicking the axis label and selecting the desired channel from the drop-down menu.
- Set the plots to be density color-coded by clicking the square gray "i" icon that appears next to the upper right corner of each plot. A Properties window opens with a panel of buttons on the left. In the Properties window, click on the Scatterplot icon (second from the top on the left of the panel). Click on OK.
NOTE: The PerCP-Vio700-A B3-A fluorescence channel corresponds to an excitation at 488 nm, and emission in the 650-730 nm range.
- Change the axes of the three plots as follows, ordered as X-axis and Y-axis, respectively:
- Set up instrument settings:
- Click on the Channels tab in the left panel. A list of all channels will appear with three boxes to the right. Set B3, FSC-A, and SSC-A to Hlog by clicking the down arrow on their respective first boxes on the right and selecting Hlog from the drop-down menu. Set the voltage of SSC to 640 V, FSC to 300 V, and B3 to 480 V by typing in these numbers in their respective middle boxes to the right and pressing the Enter key. Note the third box to the right is a toggle that can be used alternately for typing.
- Under the Trigger header, set the FSC trigger to 4 and the SSC and B3 trigger to 0 by clicking the down arrow, selecting them from the drop-down menu on the left, typing in the number in the box on the right, and then pressing the Enter key. Note that there is a toggle box on the right that can be used alternatively to typing.
NOTE: PMT voltages need to be optimized for each particular flow cytometry machine, but the trigger for nuclei must be lower than what is typically used for cells. Furthermore, the axis scale should be set to h-log.
- Type 100 µL in the text box Sample Volume and 50 µL in the text box Uptake Volume on the left side of the flow software window. Verify that the Mix Sample parameter is set to Mix Gentle to avoid lysing nuclei at this step. Press the triangle within a circle (Play icon) on the bottom right corner to start flow.
- Verify flow quality:
- Use side scatter area (SSC-A) versus HDR-T to check for air bubbles or clumps in the sample. Ensure that this plot is a smooth continuum of points. Air bubbles or clumps will result in blank regions in the SSC-A versus HDR-T plot.
- Check the side scatter (SSC-A) versus forward scatter (FSC-A) plot to verify the voltages used are correct for the sample.
NOTE: Unlike cells, nuclei will not fully resolve from debris in a side scatter vs. forward scatter plot. However, there should be a high-density cluster of events toward the middle of the plot corresponding to nuclei (warmer colored) with a lower density of events (cooler colored) in the background corresponding to debris.
- Analyze nuclei count and quality:
- Double click the SSC-A vs. PerCp-Vio-700A plot for an enlarged view. Click the button on the upper left toolbar with perpendicular lines (Quadrant) to select a quadrant gate. Then, click anywhere in the SSC-A vs. PerCp-Vio-700A plot and quadrants will appear inside the plot.
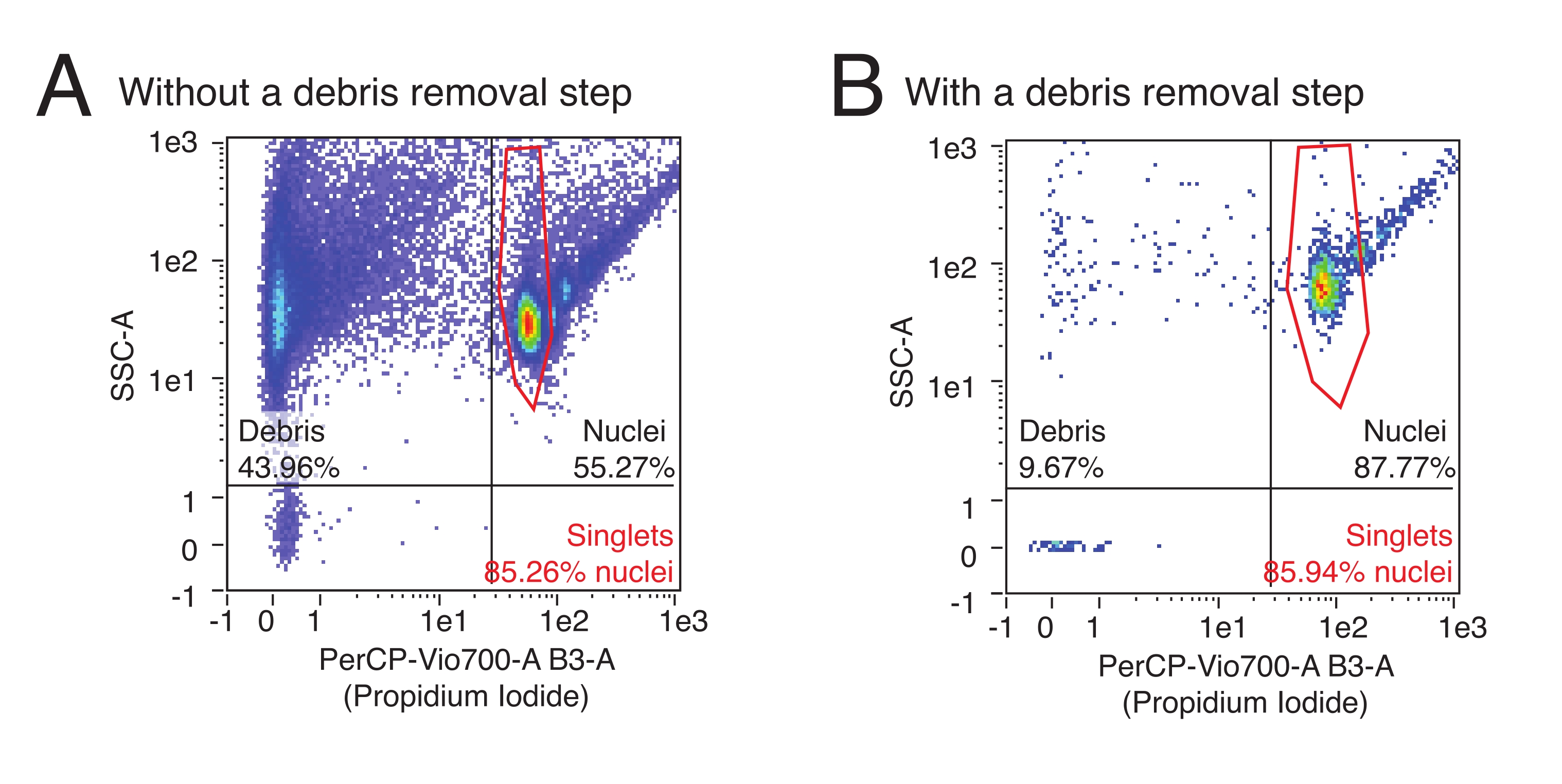
- Click anywhere on the quadrant dividing lines and slide the cursor to modify the quadrant placement. Place the quadrants such that the upper left quadrant contains the far left population (debris), and the upper right quadrant contains several teardrop-shaped clusters (nuclei) (see Figure 3 for gate setup and examples of Flow results for typical results with preps including or not including a debris removal step).
- Debris versus nuclei counts:
- Click on the square gray "i" icon in the upper right corner of the plot to open the Properties window. Click on the Region Functions tab; lists of plot regions and functions will appear.
- Under the Regions list, ensure a green check mark next to the top item, indicating the entire plot is selected. Under the Functions list, click on Count/µL to produce a green check mark. Click on OK, and a counts/µL metric will appear in each quadrant.
- To view raw counts and percentages, select Count and %-T from the Region Functions tab if desired. The counts/µL in the upper left and right quadrants correspond with the debris and nuclei yield, respectively.
- Singlet counts:
- The left-most teardrop-shaped cluster in the lower portion of the upper right quadrant represents singlets. Draw a polygonal gate around this population by clicking the polygon button (Polygon) on the upper left toolbar.
- Click a single time, slide the mouse to start drawing, and click again to finish a linear segment of the gate and begin the next one. Double click to finish the gate. Click the border of the gate and slide the mouse to reposition the gate.
- Click the vertices of the gate to modify the shape. Counts/µL of the gated population (singlets) will appear. This is the concentration of the nuclei prep with the initial dilution.
NOTE: The forward scatter area versus height 1:1 linear relationship, commonly used to determine singlet rate when flowing cells, does not apply to nuclei and can be disregarded.
- Using the concentration of the diluted nuclei prep and the dilution factor, back-calculate the concentration of the initial prep (e.g., for a 1:10 dilution, multiply the observed concentration by 10). Concentrations from >300 nuclei/µL are suitable for snRNA-seq.
- Set up a workspace:
Representative Results
The killifish brain nuclei isolation protocol described here is optimized specifically for the killifish and is summarized in Figure 1. In addition to nuclei extraction, the protocol details different methods for assessing the quality and quantity of isolated nuclei. Figure 2 shows examples of both healthy (Figure 2A) and unhealthy (Figure 2B) nuclei as assessed by light microscopy. Healthy nuclei suitable for downstream analysis (Figure 2A) present as singlet nuclei with intact membranes. As shown in Figure 2B, the most common failure mode of this protocol (and other nuclei isolation protocols) is the generation of overlysed nuclei, which are characterized by damaged nuclear membranes. This often leads to nuclei clumping and can contribute to background signals in downstream applications due to the leakage of nucleic acids from the ruptured nuclei. If excessive clumping is observed, we recommend being gentler during the dounce steps and/or reducing the incubation times in lysis buffer.
This protocol uses flow cytometry to quantify the number of isolated nuclei and determine the relative proportion of multiplet nuclei in a sample. Additionally, flow cytometry can be used to assess the relative content of contaminating debris, often ruptured nuclei fragments and other cellular debris that can detrimentally affect downstream assays in the nuclei sample. Representative flow data can be seen in Figure 3. A high-quality nuclei isolation procedure will produce: 1) a small debris fraction, and 2) a high fraction of singlet versus multiplet nuclei (> 80% of the nuclei fraction). The use of PI staining, which stains nucleic acids, allows singlet nuclei to be segregated from debris and multiplet nuclei. Figure 3A is an example of a prep contaminated by debris, whereas Figure 3B is an example of a high-quality experiment.

Figure 1: Killifish brain nuclei isolation workflow. Experimental workflow for nuclei isolation from frozen or fresh killifish brains. Once isolated and assessed, nuclei can be used for various downstream "omics" analyses. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Assessment of nuclei quality by microscopy. Nuclei were mixed 1:1 in a solution of 0.4% Trypan Blue and visualized on an Echo Revolve microscope by brightfield imaging at 60x. (A) An example of a high-quality nucleus. The nucleus is present as a singlet and the nuclear membrane is intact. (B) An example of poor-quality nuclei. The nuclear membranes are damaged, and nuclei are clumping into a doublet, likely due to leakage of sticky DNA, which can be seen leaking from the top of the topmost nucleus. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Nuclei quantification and purity assessment by flow cytometry. Nuclei samples were stained with a 1:100 dilution of PI and run on a flow cytometer. Nuclei are present in singlet and multiplet forms in the upper right quadrant of (A,B) with singlets as the lowest cloud of events followed by doublets, triplets, etc. in ascending order. Debris is marked by the events in the leftmost two quadrants. (A) An example of a low-quality nuclei prep, where the debris removal step was omitted and debris makes up >40% of events. (B) An example of a high-quality nuclei sample, with the included debris removal step. In this example, debris makes up <10% of all events registered by the flow cytometer. Please click here to view a larger version of this figure.
{kind=link}
| Sample type (2 Brains) | Average Yield (4 independent preps) |
| 5-week Male Brains | 3.59 ± 1.76 x 105 |
| 10-week Male Brains | 6.41 ± 1.33 x 105 |
| 15-week Male Brains | 14.59 ± 2.05 x 105 |
| 5-week Female Brains | 2.54 ± 0.75 x 105 |
| 10-week Female Brains | 4.66 ± 1.29 x 105 |
| 15-week Female Brains | 7.95 ± 3.51 x 105 |
Table 1: Average expected yields from two brains of African turquoise killifish across sex and age. Average yields expressed as 105 nuclei ± standard error of the mean over four independent nuclei preparations in each category.
Supplementary Figure 1: Comparison of nuclei isolation quality using standard protocols and our optimized protocol on frozen killifish brains. (A) Nuclei quality assessment by microscopy. Nuclei were mixed 1:1 in a solution of 0.4% Trypan Blue and visualized on a microscope by brightfield imaging at 60x. (B) Nuclei and debris load quantification by flow cytometry. Nuclei samples were stained with a 1:100 dilution of PI and run on a flow cytometer. (C) Summary of quality assessment metrics by microscopy and flow cytometry with the different benchmarked protocols. Please click here to download this File.
Supplementary Figure 2: Schematic representation of the debris removal steps. (A) Scheme of debris removal layering pre-centrifugation. (B) Scheme of supernatant removal after centrifugation. Please click here to download this File.
Discussion
The protocol presented here can be used to reproducibly generate high-quality nuclei from killifish brains. This protocol had to be specifically designed for the killifish brain as typical mammalian-based brain nuclei isolation protocols applied to killifish brains consistently resulted in poor nuclei quality in our hands. We suspect that this is due to the lower relative myelin content of the killifish brain compared to their mammalian counterparts, which would lyse and clump in response to the harsh conditions required for mammalian brain cell lysis. This protocol is an advancement in the aging and killifish fields as it facilitates the exploration of brain aging at the single-cell level in a cost and time-effective model of vertebrate brain aging.
This protocol is robust to fresh or frozen samples, though one must consider the downstream applications when using fresh or frozen tissue. Frozen tissue is often convenient as it can be collected and stored for months while samples are collected. Such samples can be safely used for applications such as snRNA-seq. However, freezing samples may disrupt the nuclear structure and thus the ability to accurately measure the chromatin landscape by ATAC-seq21. Thus, for downstream applications such as bulk ATAC-seq or snATAC-seq, it is recommended to use freshly dissected brains instead of frozen brains. In addition, because all steps after brain homogenization can be performed in parallel, this protocol is amenable to running multiple samples in a reasonable timeframe, thus limiting RNA degradation caused by prolonged incubation on ice.
Furthermore, it is imperative to prepare buffers fresh (within hours) of performing nuclei extraction. We found that detergents as well as BSA must be added to buffers immediately prior to beginning the protocol. Buffers containing only salts (PBS, NaCl, etc.) may be made as concentrates, filter sterilized (0.22 µm), and stored indefinitely at room temperature. BSA stock solutions may be prepared, sterilized, and stored at 4 °C within days of the nuclei extraction (if prepared from a powder) but should always be added to the buffers used in this protocol immediately before undertaking the protocol. However, we recommend preparing BSA solutions on the day of the protocol. If using premade BSA solutions from a third party, it is advised to use fresh, unopened bottles. Using fresh BSA generally leads to lower debris content in nuclei preps.
Whether fresh or frozen samples are used as input, it is important to assess nuclei quality following nuclei isolation. Though this protocol is specifically designed to avoid overlysis, this is the most common cause of nuclei quality loss. Overlysis may result from too much time spent in the lysis buffer, overly rough handling of the nuclei such as excessive pipetting with a standard bore pipette tip, or an excessive amount of time spent between nuclei isolation and downstream applications (>1 h). Overlysed nuclei will often have damaged nuclear peripheries, which leak DNA and cause clumping (Figure 2B). This will lead to an increased number of multiplets and contribute background nucleic acids that will interfere with downstream applications, especially snRNA-seq. Both qualities can be assessed by microscopy following nuclei isolation. If excessive nuclear clumping is observed, we recommend trying to shorten the lysis step incubation to reduce the chance of overlysis. As an alternative method for increasing nuclei singlets, fluorescence-assisted cell sorting (FACS) may be used to enrich for singlets downstream of this protocol. However, we note that, when working with already fragile nuclei from frozen tissue, the shear stress occurring during sorting may lead to increased nuclear rupture and thus increased ambient RNA/DNA. In addition, we note that the time required to run a FACS yield sort for nuclei when processing multiple samples would require all nuclei samples to remain on ice for hours, while other samples are being sorted. Thus, increased wait times when processing multiple samples in parallel with the FACS approach could also likely lead to overall reduced nuclei quality and increase the risk of RNA degradation. Thus, if FACS is desired for reduced doublet rate, we recommend that debris content should be checked again after the yield sort and that possible reduced RNA quality be taken into account for single cell RNA-seq applications as a potential caveat.
An accurate estimate of nuclei counts and singlet proportion is essential for nearly all downstream "omics" applications and is of the utmost importance. Due to the ability to easily gate and count nuclei by size, flow cytometry is the most accurate method of counting nuclei that we have assessed. Alternatively, one may quantify nuclei using cell counters such as Invitrogen's Countess 2 FL Automated Cell Counter or the DeNovix CellDrop Automated Cell Counter. To note, Invitrogen's Countess 2 FL Automated Cell Counter, and to a much lesser extent the DeNovix, tend to overestimate nuclei counts by counting debris as nuclei, which means that manual size gating may be required. Furthermore, the flow cytometer allows one to easily assess the purity of the nuclei. One can discern the relative proportion of singlet versus multiplet nuclei in a quantitative manner that is difficult by microscopy. This is vital for snRNA-seq and snATAC-seq, since those protocols will suffer from a surplus of multiplet samples, which must be excluded from downstream analyses. In addition to multiplets, the relative proportion of "debris" (fragmented nuclei, cellular debris) can be quantified by flow cytometry and must be relatively low, since this material often contains contaminating nucleic acids that can contribute a background signal and corrupt single nucleus "omics" data.
As with previously described nuclei isolation protocols, the proportions of cell types in the original tissue may not be recapitulated faithfully in the nuclei prep19, and thus should be interpreted with caution. To note, like all teleosts, African turquoise killifish have nucleated erythrocytes, which are also expected to be represented in the nuclei prep. These nuclei can be identified in snRNA-seq and snATAC-seq datasets by the higher expression/accessibility of hemoglobin genes and may be excluded computationally if desired.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Some panels were generated with BioRender.com. This work in our laboratory was supported by the NIA T32 AG052374 Postdoctoral Training Grant to B.T., a grant from the Simons Foundation as part of the Simons Collaboration for Plasticity in the Aging Brain, a pilot grant from the Navigage Foundation, and a Hanson-Thorell Family award to B.A.B.
Materials
| Name | Company | Catalog Number | Comments |
| 0.4% Trypan Blue solution | Gibco | 15250061 | |
| 10x PBS | Bioland | PBS01-03 | |
| 15 mL Conicle Centrifuge Tube | VWR | 89039-664 | |
| 2 mL Tissue Grinder | Kimble | 885300-0002 | Dounce Homogenizer |
| 5 mL Polystyrene Round-Bottom Tube | Falcon | 352054 | |
| 5M Sodium Chloride, Molecular Biology Grade | Promega | V4221 | |
| autoMACS Rinsing Solution | Miltenyi Biotec | 130-091-222 | This corresponds to 1x PBS pH 7.2, 2 mM EDTA (used for flow cytometry) |
| Debris Removal Solution | Miltenyi Biotec | 130-109-398 | |
| DNA LoBInd Tube | Eppendorf | 22431021 | |
| Echo Revolve microscope (fitted with 60x objective) | Echo | NA | No catalog number; Used to visually inspect nuclei. |
| Falcon Round-Bottom Polystyrene Tubes, 5 mL | Falcon | 352052 | FACS tube |
| FLOWMI Cell Strainers, 40 μM | SP Bel-Art | 136800040 | Referred to as on-tip filters in Protocol |
| Hydrochloric Acid | Sigma-Alrdich | 258146-500 mL | Used to lower TRIS pH from 8.0 to 7.4 |
| Leica EZ4 dissecting scope | Leica | NA | No catalog number |
| MACS BSA stock solution | Miltenyi Biotec | 130091376 | |
| MACS SmartStrainers (70 μM) | Miltenyi Biotec | 130-110-916 | |
| MACSQuant Analyzer 10 Flow Cytometer | Miltenyi Biotec | NA | No catalog number |
| Magnesium Chloride, Hexahydrate, Molecular Biology Grade (Powder) | Millipore | 442611 | |
| Megafuge 16R Centrifuge | ThermoScientific | 75003629 | |
| Micro Cover Glass | VWR | 48393081 | |
| Micro Slides Superfrost Plus | VWR | 48311-703 | |
| Nonidet P-40 Substitute | Roche | (Roche) 11332473001/ Catalog: 983739 P Code: -102368106 (Sigma Aldrich) | |
| NP-40 Surfact-Amps Detergent Solution | ThermoFisher | 85124 | |
| Nuclease-Free HyPure Molecular Biology Grade Water | HyClone | SH30538.02 | |
| NxGen RNase Inhibitor (50,000 U) | Lucigen | 30281-2 | |
| Propidium Iodide solution | MBL | FP00010020 | |
| PureBlu DAPI Nuclear Staining Dye | Biorad | 1351303 | |
| TipOne RPT Pipette Tips (Ultra low retention, filtered) in 10 µL, 20 µL, 200 µL, and 1000 µL sizes | USA Scientific | #1181-3810; #1180-1810; #1180-8810; #1182-1830 | |
| Tricaine-S (MS 222) | Syndel | Tricaine10G | Syndel is an FDA-approved provider for pharmaceutical grade Tricaine |
| TRIS, 1 M, pH 8.0 | VWR | E199-500 mL | |
| Wide Bore Pipet Tips | Axygen | T-1005-WB-C |
References
- Yankner, B. A., Lu, T., Loerch, P. The aging brain. Annual Review of Pathology. 3 (1), 41-66 (2008).
- Hu, C. K., Brunet, A. The African turquoise killifish: A research organism to study vertebrate aging and diapause. Aging Cell. 17 (3), 12757 (2018).
- McKay, A., et al. An automated feeding system for the African killifish reveals effects of dietary restriction on lifespan and allows scalable assessment of associative learning. bioRxiv. , (2021).
- Valenzano, D. R., Terzibasi, E., Cattaneo, A., Domenici, L., Cellerino, A. Temperature affects longevity and age-related locomotor and cognitive decay in the short-lived fish Nothobranchius furzeri. Aging Cell. 5 (3), 275-278 (2006).
- Vanhunsel, S., et al. The killifish visual system as an in vivo model to study brain aging and rejuvenation. NPJ Aging and Mechanisms of Disease. 7 (1), 22 (2021).
- Armand, E. J., Li, J., Xie, F., Luo, C., Mukamel, E. A. Single-cell sequencing of brain cell transcriptomes and epigenomes. Neuron. 109 (1), 11-26 (2021).
- Bakken, T. E., et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 13 (12), 0209648 (2018).
- Ziffra, R. S., et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 598 (7879), 205-213 (2021).
- Grindberg, R. V., et al. RNA-sequencing from single nuclei. Proceedings of the National Academy of Sciences. 110 (49), 19802-19807 (2013).
- Kalish, B. T., et al. Single-nucleus RNA sequencing of mouse auditory cortex reveals critical period triggers and brakes. Proceedings of the National Academy of Sciences. 117 (21), 11744-11752 (2020).
- Slyper, M., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine. 26 (5), 792-802 (2020).
- Ayhan, F., Douglas, C., Lega, B. C., Konopka, G. Nuclei isolation from surgically resected human hippocampus. STAR Protocols. 2 (4), 100844 (2021).
- Gaublomme, J. T., et al. Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nature Communications. 10 (1), 2907 (2019).
- Narayanan, A., et al. Nuclei isolation from fresh frozen brain tumors for single-nucleus RNA-seq and ATAC-seq. Journal of Visualized Experiments. (162), e61542 (2020).
- Saunders, A., et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 174 (4), 1015-1030 (2018).
- . Isolation of Nuclei for Single Cell RNA Sequencing & Tissues for Single Cell RNA Sequencing Available from: https://support.10xgenomics.com/permalink/1dlB6Z91VqClgUmSC2OM8k (2021)
- Martin, C., et al. Frozen tissue nuclei extraction (for 10xV3 snSEQ) V.2. protocols.io. , (2020).
- Nuclei Isolation from Adult Mouse Brain Tissue for Single Cell RNA Sequencing. 10xGenomics Available from: https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/sample-prep/nuclei-isolation-from-adult-mouse-brain-tissue-for-single-cell-ma-sequencing (2022)
- Denisenko, E., et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology. 21 (1), 130 (2020).
- What are the best practices for working with nuclei samples for 3' single-cell gene expression. 10xGenomics Available from: https://kb.10xgenomics.com/hc/en-us/articles/360050780051-What-are-the-best-practices-for-working-with-nuclei-samples-for-3-single-cell-gene-expression (2022)
- Rocks, D., et al. Cell type-specific chromatin accessibility analysis in the mouse and human brain. Epigenetics. 17 (2), 202-219 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved