
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Subcellulær fraksjonering for isolering av synaptiske komponenter fra Murine Brain
Denne protokollen presenterer en robust, detaljert metode for å oppnå svært rene synaptosomer, synaptiske vesikler og andre synaptiske fraksjoner fra musehjernen. Denne metoden muliggjør evaluering av synaptiske prosesser, inkludert biokjemisk analyse av proteinlokalisering og funksjon med kompartmentoppløsning.
Synaptiske terminaler er de primære stedene for nevronkommunikasjon. Synaptisk dysfunksjon er et kjennetegn på mange nevropsykiatriske og nevrologiske lidelser. Karakteriseringen av synaptiske underavdelinger ved biokjemisk isolasjon er derfor en kraftig metode for å belyse molekylære baser av synaptiske prosesser, både i helse og sykdom. Denne protokollen beskriver isoleringen av synaptiske terminaler og synaptiske underrom fra musehjerner ved subcellulær fraksjonering. Først isoleres forseglede synaptiske terminalstrukturer, kjent som synaptosomer, etter homogenisering av hjernevev. Synaptosomer er nevronale pre- og postsynaptiske rom med avklemte og forseglede membraner. Disse strukturene beholder en metabolsk aktiv tilstand og er verdifulle for å studere synaptisk struktur og funksjon. Synaptosomene blir deretter utsatt for hypotonisk lyse og ultracentrifugation for å oppnå synaptiske underrom beriket for synaptiske vesikler, synaptisk cytosol og synaptisk plasmamembran. Fraksjonsrenhet bekreftes ved elektronmikroskopi og biokjemisk anrikningsanalyse for proteiner som er spesifikke for subsynaptiske rom. Den presenterte metoden er et enkelt og verdifullt verktøy for å studere de strukturelle og funksjonelle egenskapene til synapsen og den molekylære etiologien til ulike hjernesykdommer.
Synapser er de grunnleggende beregningsenhetene i hjernen gjennom hvilke nevroner kommuniserer og utøver ulike og utsøkt komplekse funksjoner. Synapser er dermed grunnleggende for hjernens helse1; Synaptisk dysfunksjon er implisert som en kilde eller et resultat av mange lidelser2. Synapser utgjøres av pre- og postsynaptiske terminaler, utvidelser av to forskjellige nevroner som er tett apposed og separert av en synaptisk kløft krysset av synaptiske adhesjonsmolekyler. Informasjon flyter fra pre- til postsynaptisk rom i form av kjemiske budbringere kalt nevrotransmittere1. De molekylære prosessene som er involvert i nevrotransmisjon er aktive forskningsområder 3,4,5. Å forstå de patogene prosessene i synaptiske terminaler og synapsens respons på patologi i andre nevronale underrom er avgjørende skritt for å håndtere forstyrrelser i hjernen 1,2. Flere metodologiske fremskritt, hovedsakelig brukt på murine modeller, har avansert denne jakten6. Isoleringen av synaptiske fraksjoner ved differensiell sentrifugering er en slik paradigmeskiftende metode som har muliggjort detaljert evaluering av synaptiske prosesser i helse og sykdom.
Den voksne menneskehjernen består av 80-90 milliarder nevroner 7,8. Blant murine arter inneholder rottehjernen omtrent ~ 200 millioner nevroner, mens mus har ~ 70 millioner 9,10. Hvert nevron danner tusenvis av spesifikke synaptiske forbindelser med et nettverk av svært polariserte nevroner blandet med gliaceller og tett vaskulatur. I et slikt komplekst og heterogent vev var det en gang utenkelig å isolere og studere synapser som et selvstendig system. På 1960-tallet gjorde Victor Whittaker, Catherine Hebb og andre dette mulig ved å isolere intakte synaptiske terminaler ved hjelp av subcellulær fraksjonering11,12,13,14. I et forsøk på å isolere synaptiske vesikler (SVs), homogeniserte de hjerner gjennom flytende skjærkraft i iso-osmotisk (0,32 M) sukrose etterfulgt av ultracentrifugation. De oppnådde klemte, plasmamembranlukkede, intakte nerveterminaler eller varicositeter, som de kalte nerveendende partikler (NEPs) 11,13. Ettersom synapsens strukturelle og funksjonelle egenskaper ble bevart i disse strukturene, ble NEPs senere kalt "synaptosomer" for kongruens med andre subcellulære organeller13,15. Det er verdt å merke seg at arbeidet til Eduardo de Robertis og kolleger, som laget begrepet "synaptisk vesikkel", overlappet med Whittaker og kolleger og bidro til validering av "synaptosom" isolasjon og karakterisering16,17,18.
Synaptosomer er fysiologisk aktive strukturer som inneholder alle de cellulære og molekylære egenskapene som kreves for lagring, frigjøring og gjenopptak av nevrotransmittere13,18. Bevaring av viktige synaptiske egenskaper in vitro og frihet fra ikke-synaptiske komponenter bidrar også til nytten av denne isolasjonsmetoden. Synaptosomer har bidratt enormt til forståelsen av de kjemiske og fysiologiske egenskapene til nevrotransmisjon og brukes nå til å studere synaptiske molekylære prosesser og deres endringer i sykdom 19,20,21,22,23. Synaptosomer er også det første kildematerialet for isolering av synaptiske komponenter som SVer, clathrinbelagte vesikler (CCV), synaptisk cytosol, synaptisk plasmamembran, synaptiske mitokondrier, synaptiske adhesjonsmolekyler og andre komponenter av interesse, noe som kan lette forståelsen av de molekylære mekanismene for synaptisk funksjon 18,19,20,24,25,26, 27,28. Disse subsynaptiske komponentene kan oppnås ved osmotisk lysis av synaptosomer og sukrosedensitetsgradient ultracentrifugation15,29. Selv om den opprinnelige subcellulære fraksjoneringsmetoden av Whittakers forskningsgruppe er kjent for å være effektiv i å isolere kvalitetssynaptosomer og SVs 13,30, forbedrer nyere optimaliseringer renheten til de subcellulære fraksjonene 22,23,31,32. Denne artikkelen gir en svært detaljert og tilgjengelig versjon av en klassisk protokoll for subcellulær fraksjonering av murine hjernevev for å isolere synaptosomer, SVer og andre subsynaptiske komponenter.
Alle eksperimenter med mus ble godkjent av Institutional Animal Care and Use Committee (IACUC) ved Yale University (protokoll 2021-11117) og utført i et anlegg akkreditert av Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Dyrepleie og boliger overholdt veiledningen for pleie og bruk av forsøksdyr33 og ble levert av Yale Animal Resource Center (YARC). Dyrene ble opprettholdt i en 12 timers lys / mørk syklus med ad libitum tilgang til mat og vann. Fem til åtte mus eller to til fire rotter per genotype eller tilstand er nødvendig for følgende protokoll. Færre rotter er nødvendige på grunn av deres større hjernevolum. På samme måte kan alderen på forsøksdyrene påvirke fraksjonsutbyttet; Ytterligere mus kan være nødvendig i aldre mindre enn 2 måneder. Ellers gjelder de skisserte prosedyrene både murine arter og friske voksne dyr i alle aldre. De representative dataene som ble presentert i denne studien benyttet villtype (C57BL / 6J) mus (alder = 2 måneder, fire hanner og fire kvinner per replikasjon) hentet fra en kommersiell kilde (se materialtabell).
1. Eksperimentell forberedelse
MERK: Denne protokollen krever ~ 11 timer for en enkelt forsker å fullføre. Det anbefales på det sterkeste å fullføre oppsett på benken (figur 1), klargjøring av buffere (tabell 1), forkjøling av sentrifuger og rotorer til 4 °C, og innsamling og merking av nødvendige materialer og utstyr (se materialtabell) dagen før protokollutførelse, der det er aktuelt.
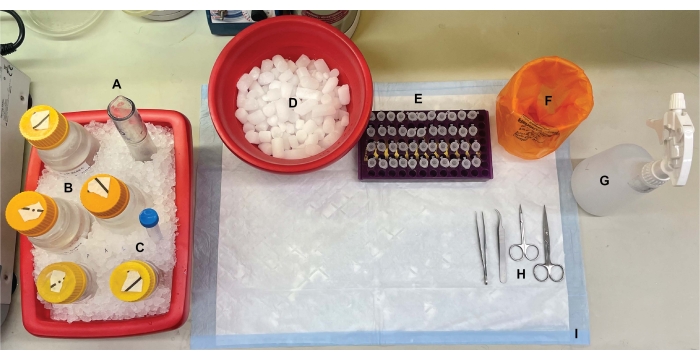
Figur 1: Oppsett på benken. Før hjernedisseksjoner ble (A) Dounce glasshomogenisatorer og (B) alle buffere kjølt på is. (C) Proteasehemmere stamløsninger ble tint på is. En annen beholder med våt is for sentrifugerør, en Dewar av flytende nitrogen (ikke vist) og (D) en beholder med tørris for kortvarig lagring av prøvene flashfrosset i flytende nitrogen ble oppnådd. (E) Mikrosentrifugerør ble forhåndsmerket for alle prøver, da fire aliquots av hver subcellulær fraksjonsprøve per genotype eller tilstand ble samlet inn under denne prosedyren (tidsbesparende tips: Merk grundig alle rørene dagen før eksperimentet utføres). (F) En egnet biohazard avfallsbeholder, (G) 70% etanol, (H) kirurgiske verktøy og (I) en absorberende overflatepute. De nødvendige sentrifugerørene og engangsmaterialene ble satt til side for effektiv tilgang under protokollimplementering (ikke vist). Vennligst klikk her for å se en større versjon av denne figuren.
- Forbered benkeplaten for kirurgi og samle saks og tang som kreves for hjerneeksisjon (se materialtabell). Pre-label 1,5 ml mikrosentrifugerør for musehalebiopsier og fire rør per innsamlet fraksjon, som beskrevet i figur 2.
- Få to beholdere med våt is, en beholder med tørris og en benk flytende nitrogen Dewar kolbe.
- Tine fenylmetylsulfonylfluorid (PMSF), pepstatin A, aprotinin og leupeptin lagerløsninger på is (se tabell over materialer). Forbered de nødvendige bufferne (tabell 1).
MERK: Sukroseoppløsninger kan tilberedes på forhånd og oppbevares ved 4 °C. Imidlertid må proteasehemmere (tinte lagre og tabletter) tilsettes friske til alle bufferne ved starten av forsøket på grunn av ustabiliteten til disse reagensene i vandige løsninger. Videre må alle bufferne tilberedes med vaskemiddelfritt glass og vaskemiddelfritt vann for å muliggjøre oppsamling av intakte synaptosomer. - Avkjøl alle buffere og glass Dounce homogenisatorer (se tabell over materialer) på is. Sett sentrifugene til 4 °C og avkjøl rotorene til 4 °C.
- Tilsett 14 ml buffer A (tabell 1) til en Dounce homogenisator på is.
Tabell 1: Sammensetning av de subcellulære fraksjoneringsbufferne. Klikk her for å laste ned denne tabellen.
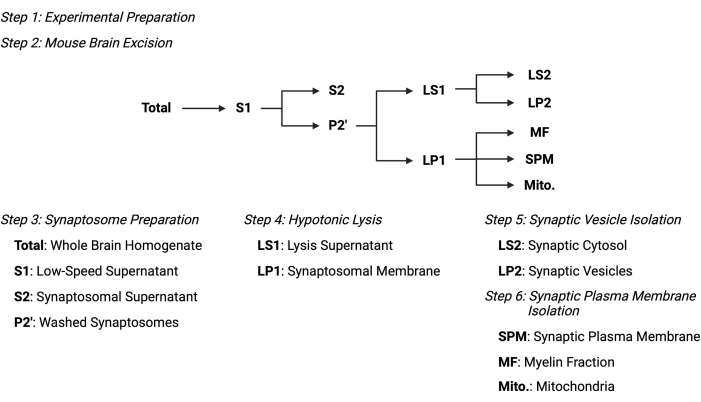
Figur 2: Oversikt over den subcellulære fraksjoneringsprotokollen. Sammendragsskjema for de subcellulære fraksjoneringstrinnene og innsamlede prøver. Vennligst klikk her for å se en større versjon av denne figuren.
2. Eksisjon av musehjerne
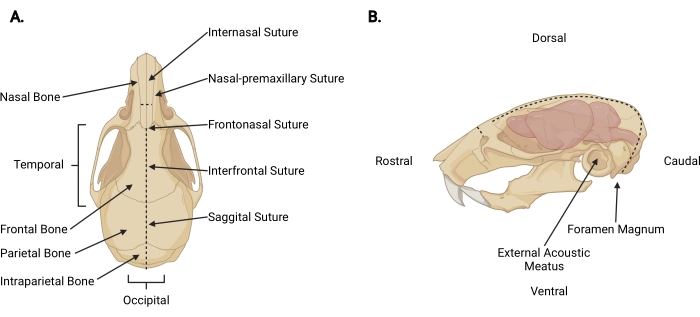
Figur 3: Kraniofacial anatomi . (A) Dorsal visning av en museskalle med relevante kraniale strukturer angitt. (B) Venstre sidevisning av en museskalle og hjerne med relevante kraniale strukturer og anatomiske retninger angitt. De stiplede linjene representerer stedene der snitt skal gjøres. Vennligst klikk her for å se en større versjon av denne figuren.
- Bedøv hver mus dypt med 100% isofluoran i et anestesikammer plassert i en avtrekkshette eller biosikkerhetsskap ved hjelp av en åpen dråpemetode34. Ofre hver mus ved cervikal ryggradsdislokasjon etterfulgt raskt av halshugging. Veksle mellom genotyper eller eksperimentelle grupper for hvert offer og disseksjon12. Få halebiopsier etter eutanasi ved å fjerne 2 mm av den distale halespissen med fin saks. Oppbevar vevet for genotyping.
- Spray det halshuggede hodet med 70% etanol for å forhindre at håret fester seg til vevet og kirurgiske instrumenter under disseksjon.
- Sett inn fin saks under huden ved halshuggingssnittet til en perikraniell dybde og gjør et midsagittalt snitt opp til internasal sutur (figur 3A) for å trekke hodebunnen fra skallen.
- Arbeid fra oksipitalt område mot hvert temporalt aspekt, trim fascia og muskel for å eksponere den ytre overflaten av skallen utover hver ekstern akustisk meatus (figur 3B).
- Fest hodebunnen og rostral aspektet av skallen med den ikke-dominerende hånden. Med den andre, sett fin saks 2 mm inn i kaudalsiden av foramen magnum, hvor ryggmargen er synlig ut. Lag et midtlinjesnitt til saksen når den indre overflaten av det intraparietale beinet (figur 3; stiplede linjer).
NOTAT: Under det første snittet må saksen være parallell med ryggmargen med trykk påført mot den indre overflaten av skallen for å forhindre skade på hjernestammen og lillehjernen. - Endre saksens vinkel slik at bladene løper parallelt med skallens dorsale overflate. Fortsett å fremme midsagittalinnsnittet rostralalt gjennom parietale og frontale bein, ved å bruke sagittal og interfrontal suturer som en guide. Bruk konstant trykk oppover for å unngå skade på cortex. Avslutt snittet like utenfor den internasale suturen (figur 3A).
- Lag et lite vinkelrett snitt (~ 3 mm) til nesebenet, rostral til internasal suturen, ved å plassere saksen vinkelrett på skallen med hvert blad plassert ved en nese-premaxillær sutur og gjør en jevn kutt (figur 3; stiplede linjer).
MERK: Dette trinnet vil øke det enkle å trekke tilbake skallen og vil være avgjørende for å samle olfaktorisk pære hvis dette området er av interesse. - Mens du sikrer rostralaspektet, bruk den ene siden av et par teksturerte tang for å forsiktig løfte skallen opp fra hjernen, deretter lateralt og ventralt. Gjenta langs midtlinjen etter behov, deretter for den andre halvkule til hele hjerneoverflaten er eksponert.
- Ved hjelp av buede tang eller en fin spatel, løft forsiktig rostralsiden av hjernen. Klipp optiske og kraniale nerver for å fullføre eksisjonen fra skallen.
- For hver tilstand, samle fem til åtte musehjerner sammen i det kjølte glasset Dounce homogenisator som inneholder 14 ml buffer A (tabell 1).
3. Synaptosom forberedelse
MERK: Skjemaene for denne prosedyren er vist i figur 4.
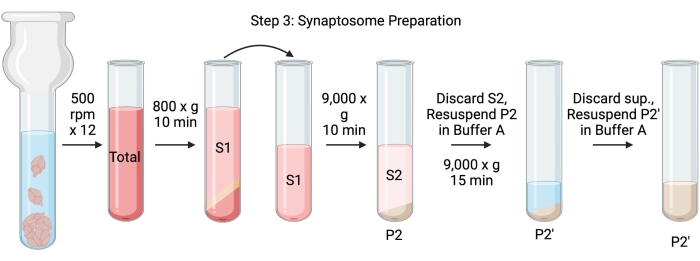
Figur 4: Synaptosompreparat. Skjematisk av trinn 3, generering av synaptosomer (P2'). Vennligst klikk her for å se en større versjon av denne figuren.
- Homogeniser hjernen ved hjelp av et glass Dounce homogenisator i 12 opp-ned passerer ved 500 rpm (totalt). Pause kort ved hvert nedslag for å sikre grundig homogenisering av vevet. Homogeniser fortrinnsvis i et isbad for å unngå oppvarming og proteindenaturering. Ta 5 μL aliquots for proteinkonsentrasjonsbestemmelse ved bicinchoninsyreanalysen (BCA, se materialtabell). Ta 100 μL av hele hjernen lysat aliquots for western blot (WB). For dette og alle påfølgende prøver (figur 2), ta to aliquots for BCA og to aliquots for WB. Flash-frys alle oppsamlede aliquots i flytende nitrogen og oppbevar dem ved -80 ° C.
- Spinn det totale hjernehomogenatet i et høyhastighets rundt bunnsentrifugerør (14 ml) (se materialtabell) ved 800 x g i 10 minutter ved 4 ° C for å oppnå supernatanten (S1). Overfør S1 til et nytt sentrifugerør, og la pelleten ligge igjen (P1), som inneholder intakte celler og kjerner. Unngå å pipettere opp den luftige, hvite, løse, overfladiske pelleten. Ta 2 x 5 μL S1 for BCA og 2 x 100 μL S1 for WB.
- Spinn S1 ved 9000 x g i 15 minutter ved 4 °C for å oppnå synaptosomal supernatant (S2) og rå synaptosompellet (P2). Ta 2 x 10 μL av S2 for BCA og 2 x 500 μL av S2 for WB. Kast supernatanten etter å ha fått aliquots og fortsett til neste trinn med pelleten.
- Resuspend P2 i 3 ml iskald buffer A med proteasehemmere og sentrifuge ved 9000 x g i 15 minutter ved 4 °C for å oppnå supernatanten (S2') og vaskede synaptosomer (P2'). Kast supernatanten og behold pelleten.
- Resuspend P2' i 3 ml buffer A. Unngå å resuspendere den mørkerøde delen i bunnen av pelleten, som hovedsakelig inneholder mitokondrier. Ta 2 x 20 μL av P2 'for BCA og 2 x 100 μL av P2' for WB.
NOTAT: Dette kan oppnås ved å forsiktig blande kantene og overflaten av pelleten for å resuspendere de hvitkalkede synaptosomene mens du leder pipettespissen bort fra pelletens røde senter.
4. Hypotonisk lysis
MERK: Skjemaene for denne prosedyren er vist i figur 5.
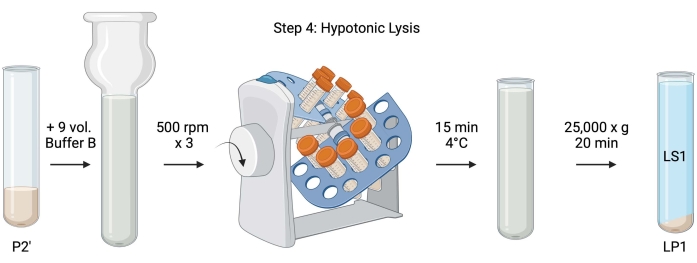
Figur 5: Hypotonisk lysis. Skjematisk av trinn 4, hypotonisk lysis av synaptosomer for å generere lysis supernatant (LS1) og synaptosomale membranfraksjoner (LP1). Vennligst klikk her for å se en større versjon av denne figuren.
- For hypotonisk lyse av vasket synaptosomer, tilsett 9 volumer kjølt buffer B (tabell 1) til resuspended P2' (~27 ml). Homogeniser synaptosomene i et glass Dounce homogenisator (tre opp-ned passerer ved 500 rpm).
- Overfør prøvene til 50 ml kappede koniske sentrifugerør. Roter dem på en rørrevolver i et 4 °C kjølerom i 15 minutter.
- Sentrifugen lyste P2' ved 25 000 x g i 20 minutter ved 4 °C for å oppnå lysis supernatant (LS1) og lysispelleten som inneholder synaptosomale membraner (LP1). Ta 2 x 50 μL LS1 for BCA og 2 x 400 μL LS1 for WB. Overfør LS1 til et avkortet sentrifugerrør for ultracentrifugering (se materialtabell).
5. Synaptisk vesikkel isolasjon
MERK: Skjemaene for denne prosedyren er vist i figur 6.
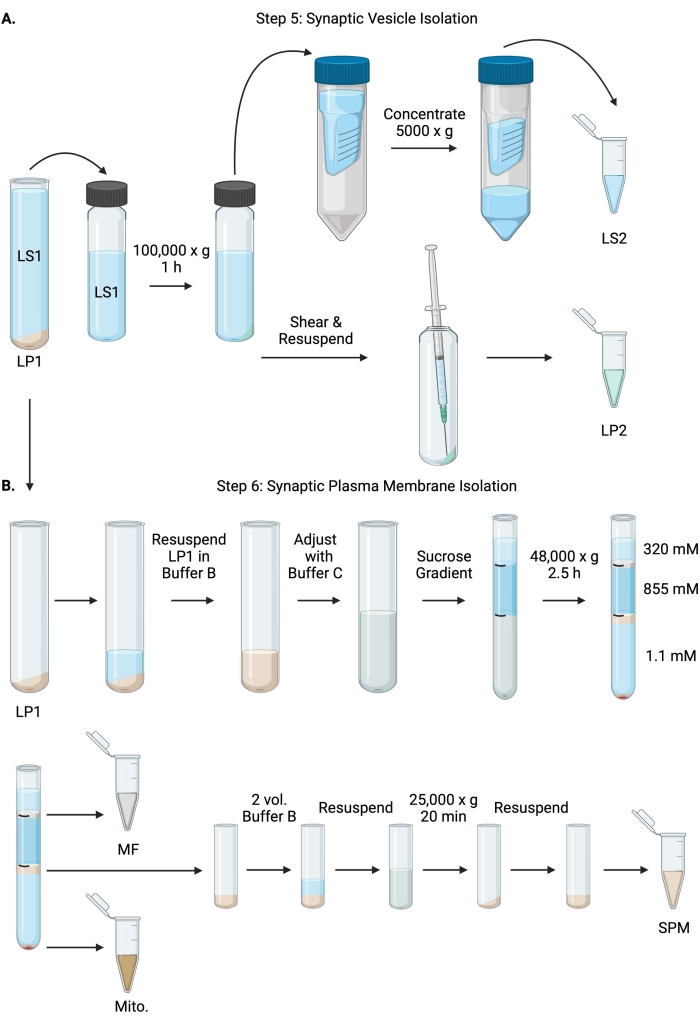
Figur 6: Synaptisk vesikkelisolasjon og synaptisk plasmamembranisolering. (A) Skjematisk for trinn 5, isolering av synaptisk cytosol (LS2) og synaptisk vesikkel (LP2) fraksjoner, og (B) trinn 6, generering av myelin (MF), synaptisk plasmamembran (SPM) og mitokondrielle (Mito.) fraksjoner etter ultracentrifugering av sukrosegradienter. Vennligst klikk her for å se en større versjon av denne figuren.
- Sentrifuge LS1 i en fast vinkel ultracentrifuge rotor (se materialtabell) ved 100 000 x g i 60 minutter ved 4 ° C for å oppnå synaptisk cytosol supernatant (LS2) og synaptisk vesikkepellets (LP2). LP2 vil være liten, gjennomsiktig og sterkt festet til siden av sentrifugerøret.
- Resuspend LP2 i 500 μL buffer A. Bruk en 23 G nål og en 1 ml sprøyte, skjær LP2 med mild triturasjon. Ta 2 x 10 μL LP2 for BCA og 2 x 250 μL LP2 for WB.
- Overfør LS2 (~ 30 ml) til sentrifugalfilterenheter med en 10 kDa-avskjæring (se materialtabell).
MERK: Hvis proteiner mindre enn 10 kDa er av interesse, er 4 kDa cutoff sentrifugalfilterenheter tilgjengelige, men vil resultere i lengre spinntider. - Konsentrer LS2 til ca. 0,5 ml ved å spinne ved 5000 x g i opptil 1 time ved 4 °C. Ta 2 x 10 μL konsentrert LS2 for BCA og 2 x 250 μL konsentrert LS2 for WB. Etter å ha startet spinnet, fortsett direkte til trinn 6.1.
6. Synaptisk plasmamembranisolasjon
- Resuspend LP1 (trinn 4.3) i 1 ml buffer B (tabell 1). Ta 2 x 10 μL LP1 for BCA og 2 x 50 μL LP1 for WB. Juster gjenværende LP1 til et endelig volum på 7,5 ml og en endelig sukrosekonsentrasjon på 1,1 M med buffer B og buffer C (tabell 1).
- Overfør 7,5 ml resuspendert LP1 til et 14 ml ultrasentrifugerrør (se materialtabell). Legg LP1 forsiktig over med 3,75 ml buffer D (tabell 1), og overlegg deretter med 1,25 ml buffer A (eller et større volum for å fylle like under toppen av sentrifugerøret). Unngå pipettering ned på siden av røret, noe som vil forstyrre sukrosegradientgrensesnittene. Etter å ha lagt over hver sukrosefraksjon, merk toppen av løsningen med en penn. Balanser rørene for ultracentrifugation etter vekt, ikke volum, med dråpevis tilsetning av Buffer A til innen 10 mg. Sentrifuge ved 48 000 x g i 2,5 timer ved 4 °C i en svingbar bøtte ultrasentrifugerotor (se materialtabell).
- Skaff bilder av intakte gradienter etter ultracentrifugation for å dokumentere distinktheten til hvert sukrosegrensesnitt og suksessen til fraksjonering.
- Fjern forsiktig det overfladiske laget av 320 mM sukrose (Buffer A). Gjenopprett myelinfraksjonen (MF) ved 320 mM/855 mM sukrosegrensesnitt i et volum på 800 μL. Gjenopprett den synaptiske plasmamembranfraksjonen (SPM) ved 855 mM / 1,1 M sukrosegrensesnitt i et volum på 1000 μL. Pipetter hver fraksjon opp fra rørveggen på en sirkulær måte for å sikre at hele fraksjonen samles inn. Aspirer forsiktig av den gjenværende sukrose og gjenopprett mitokondriell pellet (Mito.) ved å resuspendere i 200 μL buffer B. Ta 2 x 100 μL MF for BCA og 2 x 10 μL Mito. for BCA; dele resten av MF og Mito. prøver i halvparten for WB.
- Fortynn SPM-fraksjonen med 2 volumer buffer B (~ 2 ml), og sentrifuger deretter i en fast vinkelrotor i et sentrifugerør på 3,5 ml (se materialtabell) ved 25 000 x g i 20 minutter ved 4 ° C. Kast supernatanten og resuspendere SPM-pelleten i buffer A for et endelig volum på 250 μL. Ta 2 x 5 μL SPM for BCA og del den gjenværende SPM i to for WB.
- Utfør en BCA for å bestemme proteinkonsentrasjonen til hver prøve, med hensyn til variabelt aliquotvolum.
MERK: For WB-analyse er den foreslåtte arbeidsproteinkonsentrasjonen for alle subcellulære fraksjoner 2 μg / μL (eller så høy som oppnåelig for LS1 og MF).
Den presenterte metoden resulterer i 11 hjerne subcellulære fraksjoner som kan underkastes ytterligere rensing og ulike former for nedstrøms analyse35,36. Gullstandardmetoden for å vurdere kvaliteten på synaptosomer, SVs23 og andre komponenter er elektronmikroskopi (EM) (figur 7). Kvantitativ immunblotting for proteiner som er tilstede i spesifikke subcellulære fraksjoner kan også utføres for å vurdere markører for fraksjonsrenhet (figur 8). For eksempel avslører immunoblotanalyse av fraksjoner anrikning av N-cadherin (CDH2, UniProto-navn) i synaptisk plasmamembranfraksjon (SPM), α-synuclein (SYUA) i synaptisk cytosol (LS2), synaptofysin (SYPH) i synaptisk vesikkelfraksjon (LP2) og myelin basisprotein (MBP) i myelinfraksjonen (MF) sammenlignet med proteinnivåer i det opprinnelige hele hjernehomogenatet (Totalt) (figur 8 ). Når fraksjonsrenhet er etablert (for eksempel merk fraværet av CDH2 i LS2-fraksjonen eller den mange ganger økningen i SYPH i LP2-fraksjonen), kan kvantitativ immunoblotting brukes til å bestemme lokalisering av proteiner av interesse eller spørringsforskjeller i proteinfordeling mellom genotyper eller behandlinger. Forståelse av subcellulær lokalisering av synaptiske proteiner kan muliggjøre disseksjon av tidligere ubeskrevne proteinfunksjoner. Videre kan denne metoden belyse menneskehandelsdefekter eller synaptisk dysfunksjon i sykdomstilstander, spesielt når de er parret med funksjonelle analyser. For eksempel har teamet vårt brukt denne metoden til å identifisere et basseng av enzymatisk aktivt palmitoylprotein tioesterase 1 som er beriket i synaptisk cytosol19.
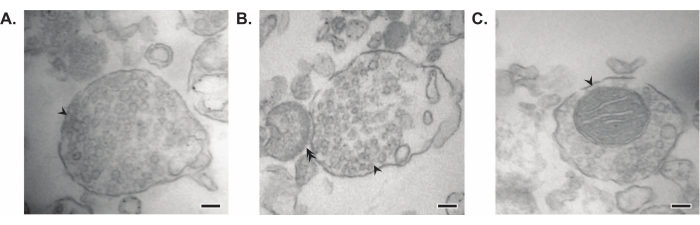
Figur 7: Elektronmikroskopi (EM) av synaptosomer. (A) Representativt EM-bilde av et synaptosom som inneholder synaptiske vesikler (pil). (B) Representativt EM-bilde av et synaptosom med både pre- (pil) og postsynaptiske komponenter (dobbeltpil). (C) Representativt EM-bilde av et synaptosom som inneholder synaptiske vesikler og en mitokondrion (pil) (skalafelt = 100 nm). Vennligst klikk her for å se en større versjon av denne figuren.
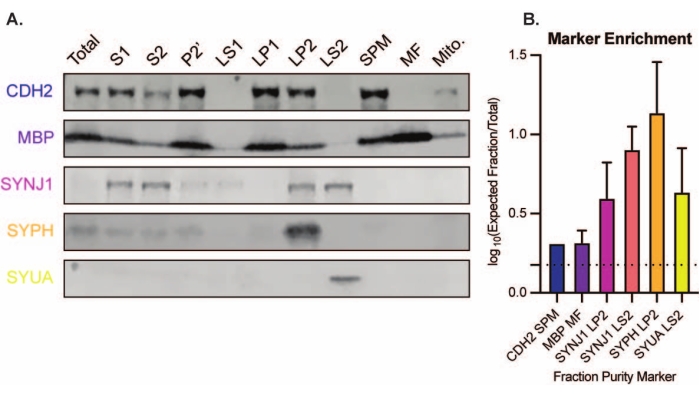
Figur 8: Immunoblotanalyse av subcellulære fraksjoner. (A) Markører for subcellulær fraksjonsrenhet (indikert med UniProt-nomenklatur) er hensiktsmessig lokalisert sammenlignet med hele hjernehomogenatet (totalt): N-cadherin (CDH2) i synaptisk plasmamembranfraksjon (SPM), synaptofysin 1 (SYPH) og synaptojanin 1 (SYNJ1) i den synaptiske vesikkelberikede fraksjonen (LP2), α-synuclein (SYUA) i synaptisk cytosol (LS2), og myelin basisk protein (MBP) i myelinfraksjonen (MF). (B) Immunoblot-kvantifiseringsanalyse avslører anrikningen (foldeendring fra totalt) av fraksjonsrenhetsmarkører. Data representeres som gjennomsnittlig ± standardavvik på en log10-skala. Den stiplede linjen indikerer en endring på 1,5 ganger (y = 0,176) (n = 3 replikasjonseksperimenter med 8 villtypemus; alder = 2 måneder; n = 4-5 flekker for SYPH, SYUA, MBP, med n = 3 plottede verdier tidligere publisert av Gorenberg et al.19; n = 5 for SYNJ1; n = 1 for CDH2). Vennligst klikk her for å se en større versjon av denne figuren.
I sine banebrytende studier brukte Whittaker og kolleger fire morfologiske kriterier for å identifisere synaptosomer: (1) strukturene har en forseglet plasmamembran; (2) strukturene inneholder SVer som ligner de i nerveterminaler og varicosities in situ i størrelse og antall; (3) strukturene har en eller flere små mitokondrier; og (4) den presynaptiske membranen blir ofte festet til en postsynaptisk komponent11,12,13. Selv om de to første kriteriene generelt gjelder for hver isolasjonsmetode, vil ikke alle resulterende synaptosomer ha mitokondrier og vedlagte postsynaptiske terminaler i de nyeste protokollene som er beskrevet i denne artikkelen. Omtrent 60% av synaptosomene vil ha mitokondrier, og bare opptil 15% anslås å ha festet postsynaptiske terminaler37. Hvis postsynaptiske komponenter er av spesiell interesse, er bruken av en isotonisk Krebs-lignende homogeniseringsbuffer og trykkfiltrering for anrikning kjent for å gi høye konsentrasjoner av synaptosomer med postsynaptiske terminaler (også kalt synaptoneurosomer)22,38.
Metoden for å ofre dyret kan påvirke kvaliteten på synaptosomer og synaptiske subfraksjoner. Voksne dyr ofret ved hjelp av en eutanasimetode som ikke krever anestesi, vil resultere i den beste brøkkvaliteten. Videre bør hjernen være nydissekert, ikke frosset og homogenisert ved hjelp av et 1:10-forhold mellom homogeniseringsbuffer (vekt / volum) for de mest levedyktige synaptiske fraksjonene22. Hjernen har en heterogen populasjon av synapser som kan differensieres etter hvilken type nevrotransmittere de bærer. Synaptosomdannelse er generelt upåvirket av synapsetype eller nevrotransmitterinnhold13. Et unntak er mosete fibre i lillehjernen, som er kjent for å bli forstyrret under optimale forhold for å oppnå synaptosomer fra resten av hjernen39,40. Dermed anbefales fjerning av cerebellum før hjernehomogenisering dersom utelukkelsen av denne regionen ikke påvirker det eksperimentelle målet. Hvis du er interessert i å isolere synaptosomer av en bestemt nevrotransmitterkarakter, kan områder av hjernen som er beriket for nevroner som inneholder nevrotransmitteren av interesse, først isoleres. Denne tilnærmingen vil imidlertid pålegge begrensninger på det endelige brøkdelutbyttet, avhengig av størrelsen på interesseområdet (dyrets alder er derfor også en vurdering). Det finnes immunokjemiske metoder for isolering av nevrotransmitterspesifikke synaptosomer, men levedyktigheten og utbyttet vil bli betydelig kompromittert22. Hvis vurdering av synaptosom metabolsk levedyktighet er viktig, kan måling av nevrotransmitterfrigivelse 41,42 eller visse enzymatiske analyser43 brukes.
Vanlige forurensninger i synaptosompreparater inkluderer mikrosomer, frie mitokondrier, SV og nevron- og glialmembraner. Forurensning kan reduseres ved å øke antall vasker ved P1- og P2-fraksjonene22 og unngå resuspendering av den røde mitokondrielle pelleten i påfølgende trinn. I eksperimenter der metabolsk levedyktighet og tid er avgjørende, vil det være nyttig 44,45,46 å redusere antall vasker og bruke Ficoll- eller Percoll-gradienter over sukrosegradienter. Disse metodene reduserer også forurensning betydelig. Whittakers opprinnelige protokoll ga SV-er av høy kvalitet. Ytterligere optimalisering av Nagy et al.23, inkludert i denne metoden, produserer SVer med bemerkelsesverdig homogenitet og renhet uten å gå vesentlig på kompromiss med utbytte36. Hvis spesifikke SV-subtyper er av interesse, for eksempel glutamatergiske (VGLUT-1-inneholdende) eller GABAergic (VGAT-1-inneholdende) SVer, kan immunisolering ved bruk av spesifikke antistoffer utføres47,48. Alternative metoder er også tilgjengelige for å isolere CCVer fra synaptosomer, som på grunn av differensiell tetthet kanskje ikke er til stede i samme grensesnitt som SVer oppnådd med denne metoden20,49,50.
Samlet sett kan den nåværende protokollen for å isolere synaptiske komponenter optimaliseres ytterligere for å oppnå fraksjoner med forbedret homogenitet og levedyktighet basert på kvaliteten og mengden av kildehjernevevet og de eksperimentelle målene. For ytterligere feilsøkingsdetaljer, bør man se bokkapitler av Dunkley og Robinson22 og Ganzella et al.36.
Forfatterne har ingenting å avsløre.
Vi vil gjerne takke P. Colosi for EM-bildeforberedelse. Dette arbeidet ble støttet av National Institutes of Health (R01 NS064963, SSC; R01 NS110354, SSC; R01 NS083846, SSC; R21 NS094971, SSC; T32 NS007224, SMT; T32 NS041228, SMT), USAs forsvarsdepartement (W81XWH-17-1-0564, SSC; W81XWH-19-1-0264, VDJ), Aligning Science Across Parkinson's (ASAP) Collaborative Research Network (SSC), og Michael J. Fox Foundation Target Advancement Program (MJFF-020160, SSC & VDJ). Vi laget grafiske illustrasjoner ved hjelp av BioRender.com.
| Name | Company | Catalog Number | Comments |
| 1 mL TB Syringe | BD | 309649 | |
| 1.5 mL Eppendorf Tubes | USA Scientific | 1415-2500 | |
| 14 mL, Open-Top Thinwall Ultra-Clear Tube | Beckman Coulter | 344060 | Compatible with SW 40 Ti |
| 23 Gauge Precision Glide Hypodermic Needle | BD | 305145 | |
| 26.3 mL, Polycarbonate Bottle with Cap Assembly | Beckman Coulter | 355618 | Compatible with Ti70 |
| 3.5 mL, Open-Top Thickwall Polypropylene Tube | Beckman Coulter | 349623 | Compatible with TLA-100.3 |
| 50 mL Falcon Tubes | Fisher Scientific | 14-432-22 | |
| Amicon Ultra-15 Centrifugal Filter Unit | Millipore Sigma | UFC901024 | |
| Aprotinin | Sigma-Aldrich | A6279 | 1 mg/mL in diH2O |
| Avanti J-26 XP Centrifuge | Beckman Coulter | B22984 | <26,000 rpm |
| Benchtop HDPE Dewar Flask | Thermo Scientific | 5028U19 | |
| C57BL/6J Mice | The Jackson Labs | 000664 | |
| Centrifuge 5810R | Eppendorf | EP022628168 | <14,000 rpm |
| complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets | Roche | 11873580001 | Add 1 tablet per 50 mL of solution |
| Curved Forceps | Fine Science Tools | 11273-20 | |
| Fine Surgical Scissors | Fine Science Tools | 8r | |
| Glas-Col Tissue Homogenizing System | Cole-Parmer | UX-04369-15 | |
| Graefe Forceps | Fine Science Tools | 11650-10 | |
| High-Speed Polycarbonate Round Bottom Centrifuge Tubes | ThermoFisher | 3117-0500 | Compatible with JA20 |
| Isofluorane | Henry Schein Animal Health | NDC 11695-6776-2 | |
| JA-20 Rotor | Beckman Coulter | 334831 | |
| Leupeptin | American Bio | AB01108 | 1 mg/mL in diH2O |
| N-[2-Hydroxyethyl] piperazine-N’-[2-ethanesulfonic acid] (HEPES) | American Bio | AB00892 | |
| Optima L-80 XP Ultracentrifuge | Beckman Coulter | <100,000 rpm | |
| Optima TLX Ultracentrifuge | Beckman Coulter | <120,000 rpm | |
| Pepstatin A | Thermo Scientific | 78436 | 1 mg/mL in DMSO |
| Phenylmethylsulfonyl fluoride (PMSF) | American Bio | AB01620 | |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23335 | For determination of protein concentration |
| Pipette Tips | |||
| Serological Pipettes | |||
| Sucrose | Sigma-Aldrich | S0389 | |
| Surgical Scissors | Fine Science Tools | 14002-12 | |
| SW 40 Ti Swinging-Bucket Rotor | Beckman Coulter | 331301 | |
| Teflon-Coated Pestle and Mortar Tissue Grinder | Thomas Scientific | 3431D94 | |
| Ti70 Rotor | Beckman Coulter | 337922 | |
| TLA-100.3 Rotor | Beckman Coulter | 349490 | |
| Tube Revolver | Dot Scientific | DTR-02VS |
- Kandel, E. R., Schwartz, J. H., Jessell, T. M., Siegelbaum, S. A., Hudspeth, A. J., Education, A. J. Synaptic Transmission. Principles of Neural Science, Fifth Edition. , (2014).
- Lepeta, K., et al. Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. Journal of Neurochemistry. 138 (6), 785-805 (2016).
- Südhof, T. C., Malenka, R. C. Understanding synapses: Past, present, and future. Neuron. 60 (3), 469-476 (2008).
- Südhof, T. C. The molecular machinery of neurotransmitter release (Nobel lecture). Angewandte Chemie International Edition. 53 (47), 12696-12717 (2014).
- Jahn, R., Boyken, J., Pfaff, D. W. Molecular Regulation of Synaptic Release. Neuroscience in the 21st Century: From Basic to Clinical. , 351-401 (2013).
- Xiong, H., Gendelman, H. E. . Current Laboratory Methods in Neuroscience Research. , (2014).
- Azevedo, F. A., et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. Journal of Comparative Neurology. 513 (5), 532-541 (2009).
- Herculano-Houzel, S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proceedings of the National Academy of Sciences of the United States of America. 109, 10661-10668 (2012).
- Herculano-Houzel, S., Lent, R. Isotropic fractionator: A simple, rapid method for the quantification of total cell and neuron numbers in the brain. Journal of Neuroscience. 25 (10), 2518-2521 (2005).
- Herculano-Houzel, S., Mota, B., Lent, R. Cellular scaling rules for rodent brains. Proceedings of the National Academy of Sciences of the United States of America. 103 (32), 12138-12143 (2006).
- Gray, E. G., Whittaker, V. P. The isolation of nerve endings from brain: An electron-microscopic study of cell fragments derived by homogenization and centrifugation. Journal of Anatomy. 96, 79-88 (1962).
- Gray, E. G., Whittaker, V. P. The isolation of synaptic vesicles from the central nervous system. Journal of Physiology. 153, 35-37 (1960).
- Whittaker, V. P. Thirty years of synaptosome research. Journal of Neurocytology. 22 (9), 735-742 (1993).
- Jahn, R., Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 490, 201-207 (2012).
- Whittaker, V. P., Michaelson, I. A., Kirkland, R. J. The separation of synaptic vesicles from nerve-ending particles ('synaptosomes). Biochemical Journal. 90 (2), 293-303 (1964).
- De Robertis, E., Rodriguez De Lores Arnaiz, G., Pellegrino De Iraldi, A. Isolation of synaptic vesicles from nerve endings of the rat brain. Nature. 194, 794-795 (1962).
- De Robertis, E., Pellegrino De Iraldi, A., Rodriguez, G., Gomez, C. J. On the isolation of nerve endings and synaptic vesicles. The Journal of Biophysical and Biochemical Cytology. 9 (1), 229-235 (1961).
- Zimmermann, H., Whittaker, V. P., Murphy, K. M. The Discovery of the Synaptosome and Its Implications. Synaptosomes. , 9-26 (2018).
- Gorenberg, E. L., et al. Identification of substrates of palmitoyl protein thioesterase 1 highlights roles of depalmitoylation in disulfide bond formation and synaptic function. PLoS Biology. 20 (3), 3001590 (2022).
- Vidyadhara, D. J., et al. Dopamine transporter and synaptic vesicle sorting defects initiate auxilin-linked Parkinson's disease. bioRxiv. , (2022).
- Schrimpf, S. P., et al. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics. 5 (10), 2531-2541 (2005).
- Dunkley, P. R., Robinson, P. J., Murphy, K. M. Synaptosome Preparations: Which Procedure Should I Use. Synaptosomes. , 27-53 (2018).
- Nagy, A., Baker, R. R., Morris, S. J., Whittaker, V. P. The preparation and characterization of synaptic vesicles of high purity. Brain Research. 109 (2), 285-309 (1976).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127 (4), 831-846 (2006).
- Wagner, J. A., Kelly, R. B. Topological organization of proteins in an intracellular secretory organelle: the synaptic vesicle. Proceedings of the National Academy of Sciences of the United States of America. 76 (8), 4126-4130 (1979).
- Jahn, R., Schiebler, W., Ouimet, C., Greengard, P. A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America. 82 (12), 4137-4141 (1985).
- Binotti, B., Jahn, R., Pérez-Lara, &. #. 1. 9. 3. ;. An overview of the synaptic vesicle lipid composition. Archives of Biochemistry and Biophysics. 709, 108966 (2021).
- Siegel, D. P., Ware, B. R. Electrokinetic properties of synaptic vesicles and synaptosomal membranes. Biophysical Journal. 30 (1), 159-172 (1980).
- Whittaker, V. P., Michaelson, I. A., Kirkland, R. J. The separation of synaptic vesicles from disrupted nervending particles. Biochemical Pharmacology. 12 (3), 300-302 (1963).
- Clementi, F., Whittaker, V. P., Sheridan, M. N. The yield of synaptosomes from the cerebral cortex of guinea pigs estimated by a polystyrene bead "tagging" procedure. Zeitschrift für Zellforschung und Mikroskopische Anatomie. 72, 126-138 (1966).
- Carlson, S. S., Wagner, J. A., Kelly, R. B. Purification of synaptic vesicles from elasmobranch electric organ and the use of biophysical criteria to demonstrate purity. Biochemistry. 17 (7), 1188-1199 (1978).
- Huttner, W. B., Schiebler, W., Greengard, P., De Camilli, P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. Journal of Cell Biology. 96 (5), 1374-1388 (1983).
- Hawkins, P., et al. A guide to defining and implementing protocols for the welfare assessment of laboratory animals: eleventh report of the BVAAWF/FRAME/RSPCA/UFAW Joint Working Group on Refinement. Laboratory Animals. 45 (1), 1-13 (2011).
- Risling, T. E., Caulkett, N. A., Florence, D. Open-drop anesthesia for small laboratory animals. Canadian Veterinary Journal. 53 (3), 299-302 (2012).
- Deutsch, C., Drown, C., Rafalowska, U., Silver, I. A. Synaptosomes from rat brain: Morphology, compartmentation, and transmembrane pH and electrical gradients. Journal of Neurochemistry. 36 (6), 2063-2072 (1981).
- Ganzella, M., Ninov, M., Riedel, D., Jahn, R. Isolation of synaptic vesicles from mammalian brain. Methods in Molecular Biology. 2417, 131-145 (2022).
- Dunkley, P. R., et al. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: homogeneity and morphology of subcellular fractions. Brain Research. 441 (1-2), 59-71 (1988).
- Schwartz, R. D., Skolnick, P., Hollingsworth, E. B., Paul, S. M. Barbiturate and picrotoxin-sensitive chloride efflux in rat cerebral cortical synaptoneurosomes. FEBS Letters. 175 (1), 193-196 (1984).
- Pittaluga, A., Thellung, S., Maura, G., Raiteri, M. Characterization of two central AMPA-preferring receptors having distinct location, function and pharmacology. Naunyn-Schmiedeberg's Archives of Pharmacology. 349 (6), 555-558 (1994).
- Israël, M., Whittaker, V. P. The isolation of mossy fibre endings from the granular layer of the cerebellar cortex. Experientia. 21 (6), 325-326 (1965).
- Khvotchev, M., Lonart, G., Südhof, T. C. Role of calcium in neurotransmitter release evoked by alpha-latrotoxin or hypertonic sucrose. Neuroscience. 101 (3), 793-802 (2000).
- Lonart, G., Janz, R., Johnson, K. M., Südhof, T. C. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 21 (5), 1141-1150 (1998).
- Nicholls, D. G., Sihra, T. S. Synaptosomes possess an exocytotic pool of glutamate. Nature. 321 (6072), 772-773 (1986).
- Dunkley, P. R., Jarvie, P. E., Robinson, P. J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nature Protocols. 3 (11), 1718-1728 (2008).
- Cotman, C. W., Matthews, D. A. Synaptic plasma membranes from rat brain synaptosomes: Isolation and partial characterization. Biochimica et Biophysica Acta. 249 (2), 380-394 (1971).
- Booth, R. F., Clark, J. B. A rapid method for the preparation of relatively pure metabolically competent synaptosomes from rat brain. Biochemical Journal. 176 (2), 365-370 (1978).
- Takamori, S., Riedel, D., Jahn, R. Immunoisolation of GABA-specific synaptic vesicles defines a functionally distinct subset of synaptic vesicles. Journal of Neuroscience. 20 (3), 4904-4911 (2000).
- Burger, P. M., et al. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron. 3 (6), 715-720 (1989).
- Blondeau, F., et al. Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proceedings of the National Academy of Sciences of the United States of America. 101 (11), 3833-3838 (2004).
- Maycox, P. R., Link, E., Reetz, A., Morris, S. A., Jahn, R. Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. Journal of Cell Biology. 118 (6), 1379-1388 (1992).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved