
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Neuroscience
Fracionamento subcelular para o isolamento de componentes sinápticos do cérebro murino
Este protocolo apresenta um método robusto e detalhado para obter sinaptossomos, vesículas sinápticas e outras frações sinápticas altamente puras do cérebro de camundongos. Este método permite a avaliação de processos sinápticos, incluindo a análise bioquímica da localização e função de proteínas com resolução compartimental.
Os terminais sinápticos são os principais locais de comunicação neuronal. A disfunção sináptica é uma característica de muitos distúrbios neuropsiquiátricos e neurológicos. A caracterização de subcompartimentos sinápticos por isolamento bioquímico é, portanto, um método poderoso para elucidar as bases moleculares dos processos sinápticos, tanto na saúde quanto na doença. Este protocolo descreve o isolamento de terminais sinápticos e subcompartimentos sinápticos de cérebros de camundongos por fracionamento subcelular. Primeiro, estruturas terminais sinápticas seladas, conhecidas como sinaptossomos, são isoladas após a homogeneização do tecido cerebral. Os sinaptossomos são compartimentos neuronais pré e pós-sinápticos com membranas pinçadas e seladas. Essas estruturas retêm um estado metabolicamente ativo e são valiosas para o estudo da estrutura e função sináptica. Os sinaptossomos são então submetidos à lise hipotônica e ultracentrifugação para obtenção de subcompartimentos sinápticos enriquecidos para vesículas sinápticas, citosol sináptico e membrana plasmática sináptica. A pureza da fração é confirmada por microscopia eletrônica e análise de enriquecimento bioquímico para proteínas específicas de compartimentos subsinápticos. O método apresentado é uma ferramenta direta e valiosa para estudar as características estruturais e funcionais da sinapse e a etiologia molecular de vários distúrbios cerebrais.
Sinapses são as unidades computacionais básicas do cérebro através das quais os neurônios se comunicam e exercem funções diversas e requintadamente complexas. As sinapses são, portanto, fundamentais para a saúde do cérebro1; a disfunção sináptica está implicada como fonte ou resultado de muitos distúrbios2. As sinapses são constituídas por terminais pré e pós-sinápticos, extensões de dois neurônios diferentes que são estreitamente apostos e separados por uma fenda sináptica atravessada por moléculas de adesão sináptica. A informação flui do compartimento pré para o pós-sináptico na forma de mensageiros químicos chamados neurotransmissores1. Os processos moleculares envolvidos na neurotransmissão são áreas ativas de pesquisa 3,4,5. Compreender os processos patogênicos dentro dos terminais sinápticos e a resposta das sinapses à patologia em outros subcompartimentos neuronais são passos cruciais para o enfrentamento de distúrbios cerebrais 1,2. Diversos avanços metodológicos, predominantemente aplicados aos modelos murinos, têm avançado nessa busca6. O isolamento de frações sinápticas por centrifugação diferencial é um desses métodos de mudança de paradigma que permitiu a avaliação detalhada dos processos sinápticos na saúde e na doença.
O cérebro humano adulto consiste em 80-90 bilhões de neurônios 7,8. Entre as espécies murinas, o cérebro do rato contém aproximadamente ~ 200 milhões de neurônios, enquanto os ratos têm ~ 70 milhões 9,10. Cada neurônio forma milhares de conexões sinápticas específicas com uma rede de neurônios altamente polarizados misturados com células gliais e vasculatura densa. Em tecidos tão complexos e heterogêneos, já foi impensável isolar e estudar sinapses como um sistema independente. Na década de 1960, Victor Whittaker, Catherine Hebb e outros tornaram isso possível isolando terminais sinápticos intactos usando fracionamento subcelular11,12,13,14. Na tentativa de isolar vesículas sinápticas (SVs), homogeneizaram cérebros através de força de cisalhamento líquido em sacarose iso-osmótica (0,32 M) seguida de ultracentrifugação. Obtiveram terminais ou varicosidades nervosas intactas, pinçadas e fechadas na membrana plasmática, que denominaram partículas de terminação nervosa (NEPs)11,13. Como as características estruturais e funcionais da sinapse foram preservadas nessas estruturas, os NEPs foram posteriormente denominados "sinaptossomos" por congruência com outras organelas subcelulares13,15. Vale ressaltar que o trabalho de Eduardo de Robertis e colegas, que cunharam o termo "vesícula sináptica", sobrepôs-se ao de Whittaker e colegas e contribuiu para a validação do isolamento e caracterização dos "sinaptossomos"16,17,18.
Os sinaptossomos são estruturas fisiologicamente ativas que contêm todas as propriedades celulares e moleculares necessárias para o armazenamento, liberação e recaptação de neurotransmissores13,18. A preservação das principais características sinápticas in vitro e a ausência de componentes não sinápticos também contribuem para a utilidade deste método de isolamento. Os sinaptossomos têm contribuído imensamente para a compreensão das propriedades químicas e fisiológicas da neurotransmissão e agora estão sendo utilizados para estudar os processos moleculares sinápticos e suas alterações na doença 19,20,21,22,23. Os sinaptossomos também são o material de origem inicial para o isolamento de componentes sinápticos como SVs, vesículas revestidas de clatrina (CCVs), citosol sináptico, membrana plasmática sináptica, mitocôndrias sinápticas, moléculas de adesão sináptica e outros componentes de interesse, o que pode facilitar a compreensão dos mecanismos moleculares da função sináptica18,19,20,24,25,26, 27,28. Esses componentes subsinápticos podem ser obtidos pela lise osmótica dos sinaptossomos e ultracentrifugação do gradiente de densidade da sacarose15,29. Embora o método original de fracionamento subcelular do grupo de pesquisa de Whittaker seja conhecido por ser eficiente no isolamento de sinaptossomos de qualidade e SVs 13,30, otimizações recentes aumentam a pureza das frações subcelulares 22,23,31,32. Este artigo fornece uma versão altamente detalhada e acessível de um protocolo clássico para o fracionamento subcelular do tecido cerebral murino para isolar sinaptossomos, SVs e outros componentes subsinápticos.
Todos os experimentos com camundongos foram aprovados pelo Comitê Institucional de Cuidados e Uso de Animais (IACUC) da Universidade de Yale (Protocolo 2021-11117) e realizados em uma instalação credenciada pela Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Os cuidados com os animais e o alojamento estavam em conformidade com o Guia para o Cuidado e Uso de Animais de Laboratório33 e foram fornecidos pelo Yale Animal Resource Center (YARC). Os animais foram mantidos em um ciclo claro/escuro de 12 h com acesso ad libitum a alimentos e água. Cinco a oito camundongos ou dois a quatro ratos por genótipo ou condição são necessários para o seguinte protocolo. Menos ratos são necessários devido aos seus maiores volumes cerebrais. Do mesmo modo, a idade dos animais de experiência pode afectar o rendimento das fracções; camundongos adicionais podem ser necessários para idades inferiores a 2 meses. Caso contrário, os procedimentos descritos aplicam-se tanto a espécies murinas como a animais adultos saudáveis de qualquer idade. Os dados representativos apresentados neste estudo utilizaram camundongos do tipo selvagem (C57BL/6J) (idade = 2 meses; quatro machos e quatro fêmeas por repetição) obtidos de fonte comercial (ver Tabela de Materiais).
1. Preparação experimental
NOTA: Este protocolo requer ~11 h para que um único pesquisador seja concluído. É altamente recomendável concluir a configuração da bancada (Figura 1), a preparação do tampão (Tabela 1), o pré-resfriamento de centrífugas e rotores a 4 °C e a coleta e rotulagem dos materiais e equipamentos necessários (consulte Tabela de Materiais) no dia anterior à execução do protocolo, quando aplicável.
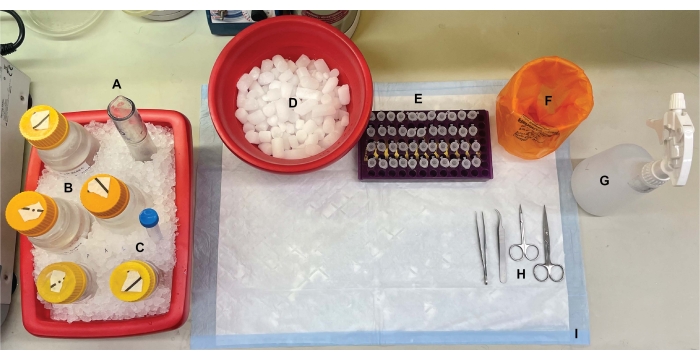
Figura 1: Configuração de bancada. Antes das dissecações cerebrais, (A) homogeneizadores de vidro de rejeição e (B) todos os tampões foram resfriados no gelo. (C) As soluções-mãe dos inibidores da protease foram descongeladas no gelo. Um segundo recipiente de gelo úmido para tubos de centrífuga, um Dewar de nitrogênio líquido (não mostrado) e (D) um recipiente de gelo seco para armazenamento de curto prazo das amostras congeladas em nitrogênio líquido foram obtidos. (E) Os tubos de microcentrífuga foram pré-marcados para todas as amostras, pois quatro alíquotas de cada amostra de fração subcelular por genótipo ou condição foram coletadas durante este procedimento (dica de economia de tempo: rotule cuidadosamente todos os tubos no dia anterior à realização do experimento). (F) Um recipiente apropriado de resíduos de risco biológico, (G) etanol a 70%, (H) ferramentas cirúrgicas e (I) uma almofada de superfície absorvente. Os tubos de centrífuga e descartáveis necessários foram reservados para acesso eficiente durante a implementação do protocolo (não mostrado). Por favor, clique aqui para ver uma versão maior desta figura.
- Prepare a bancada para a cirurgia e recolha a tesoura e a pinça necessárias para a excisão do cérebro (ver Tabela de Materiais). Pré-rotular tubos microcentrífugos de 1,5 mL para biópsias de cauda de camundongo e quatro tubos por fração coletada, conforme descrito na Figura 2.
- Obtenha dois recipientes de gelo úmido, um recipiente de gelo seco e um frasco Dewar de nitrogênio líquido de bancada.
- Descongelar as soluções de estoque de fenilmetilsulfonilo fluoreto (PMSF), pepstatina A, aprotinina e leupeptina no gelo (ver Tabela de Materiais). Prepare os buffers necessários (Tabela 1).
NOTA: As soluções de sacarose podem ser preparadas com antecedência e armazenadas a 4 °C. No entanto, os inibidores da protease (existências e comprimidos descongelados) devem ser adicionados frescos a todos os tampões no início da experiência devido à instabilidade destes reagentes em soluções aquosas. Além disso, todos os tampões devem ser preparados com vidro sem detergente e água sem detergente para permitir a recolha de sinaptossomas intactos. - Arrefecer todos os tampões e homogeneizadores de vidro Dounce (ver Tabela de Materiais) no gelo. Ajuste as centrífugas a 4 °C e arrefeça os rotores a 4 °C.
- Adicionar 14 mL de tampão A (Tabela 1) a um homogeneizador de rejeição sobre gelo.
Tabela 1: Composição dos tampões de fracionamento subcelular. Por favor, clique aqui para baixar esta Tabela.
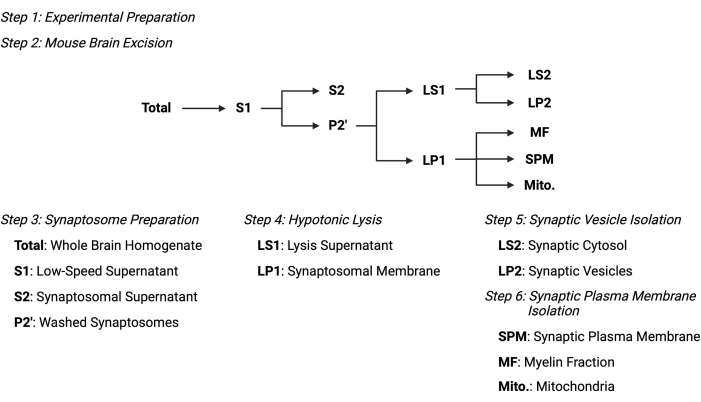
Figura 2: Visão geral do protocolo de fracionamento subcelular. Esquema resumido das etapas de fracionamento subcelular e amostras coletadas. Por favor, clique aqui para ver uma versão maior desta figura.
2. Excisão do cérebro do rato
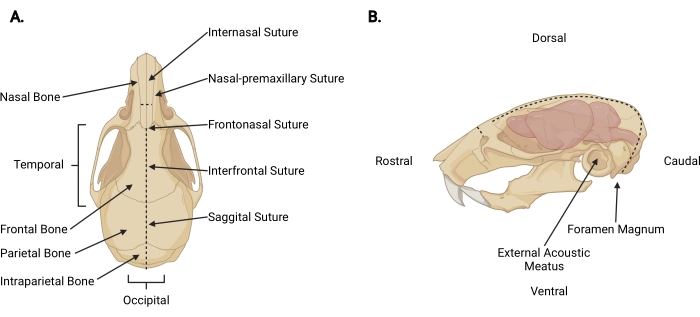
Figura 3: Anatomia craniofacial . (A) Vista dorsal de um crânio de rato com estruturas cranianas relevantes indicadas. (B) Vista lateral esquerda do crânio e do cérebro de um rato com estruturas cranianas relevantes e direções anatômicas indicadas. As linhas tracejadas representam os locais onde as incisões devem ser feitas. Por favor, clique aqui para ver uma versão maior desta figura.
- Anestesiar profundamente cada camundongo com 100% de isofluorano em uma câmara de anestesia localizada em um exaustor ou armário de biossegurança usando um método de gota aberta34. Sacrificar cada rato por luxação da coluna cervical seguida rapidamente de decapitação. Alternar entre genótipos ou grupos experimentais para cada sacrifício e dissecação12. Obter biópsias de cauda após a eutanásia extirpando 2 mm da ponta distal da cauda com tesoura fina. Armazenar o tecido para genotipagem.
- Pulverize a cabeça decapitada com etanol a 70% para evitar que o cabelo adira ao tecido e aos instrumentos cirúrgicos durante a dissecção.
- Inserir tesoura fina sob a pele na incisão de decapitação a uma profundidade pericraniana e fazer uma incisão sagital média até a sutura internasal (Figura 3A) para retrair o couro cabeludo do crânio.
- Trabalhando a partir da área occipital em direção a cada aspecto temporal, aparar a fáscia e o músculo para expor a superfície externa do crânio além de cada meato acústico externo (Figura 3B).
- Prenda o couro cabeludo e o aspecto rostral do crânio com a mão não dominante. Com o outro, insira uma tesoura fina de 2 mm no lado caudal do forame magno, onde a medula espinhal é visível saindo. Faça uma incisão na linha média até que a tesoura atinja a superfície interna do osso intraparietal (Figura 3; linhas tracejadas).
NOTA: Durante a incisão inicial, a tesoura deve ser paralela à medula espinhal com pressão aplicada em direção à superfície interna do crânio para evitar danos ao tronco encefálico e cerebelo. - Mude o ângulo da tesoura para que as lâminas corram paralelamente à superfície dorsal do crânio. Continue avançando a incisão sagital média rostralmente através dos ossos parietal e frontal, usando as suturas sagital e interfrontal como guia. Use pressão ascendente constante para evitar danos ao córtex. Terminar a incisão logo após a sutura internasal (Figura 3A).
- Faça uma pequena incisão perpendicular (~3 mm) ao osso nasal, rostral à sutura internasal, colocando a tesoura perpendicular ao crânio com cada lâmina posicionada em uma sutura nasal-pré-maxilar e fazendo um corte uniforme (Figura 3; linhas tracejadas).
NOTA: Este passo aumentará a facilidade de retração do crânio e será fundamental para a coleta do bulbo olfativo se essa área for de interesse. - Ao fixar o aspecto rostral, use um lado de um par de pinças texturizadas para levantar suavemente o crânio do cérebro, depois lateralmente e ventralmente. Repita ao longo da linha média, conforme necessário, em seguida, para o outro hemisfério até que toda a superfície do cérebro esteja exposta.
- Usando pinça curva ou uma espátula fina, levante suavemente o lado rostral do cérebro. Corte os nervos ópticos e cranianos para completar a excisão do crânio.
- Para cada condição, coletar cinco a oito cérebros de camundongos juntos no homogeneizador Dounce de vidro refrigerado contendo 14 mL de tampão A (Tabela 1).
3. Preparação do sinaptossoma
Observação : os esquemas desse procedimento são mostrados na Figura 4.
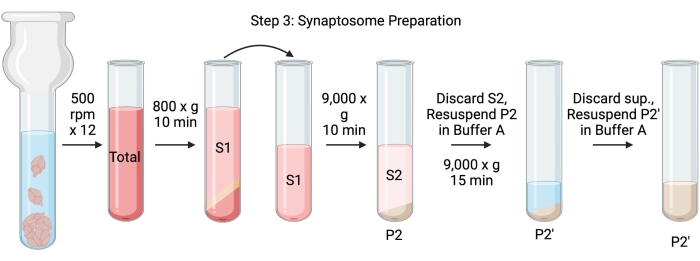
Figura 4: Preparação do sinaptossoma. Esquema do passo 3, a geração de sinaptossomos (P2'). Por favor, clique aqui para ver uma versão maior desta figura.
- Homogeneize os cérebros usando um homogeneizador Dounce de vidro em 12 passagens de cima para baixo a 500 rpm (total). Faça uma pausa breve a cada curso para baixo para garantir a homogeneização completa do tecido. Homogeneizar preferencialmente em um banho de gelo para evitar o aquecimento e a desnaturação de proteínas. Tomar alíquotas de 5 μL para a determinação da concentração de proteínas pelo ensaio de ácido bicinchonínico (BCA, ver Tabela de Materiais). Tome 100 μL de alíquotas de lisado de todo o cérebro para Western Blot (WB). Para esta e todas as amostras subsequentes (Figura 2), tomar duas alíquotas para BCA e duas alíquotas para WB. Congelar rapidamente todas as alíquotas coletadas em nitrogênio líquido e armazená-las a -80 °C.
- Girar o cérebro total homogeneizado num tubo de centrífuga de fundo redondo de alta velocidade (14 ml) (ver Tabela de Materiais) a 800 x g durante 10 min a 4 °C para obter o sobrenadante (S1). Transfira S1 para um novo tubo de centrífuga, deixando o pellet para trás (P1), que contém células e núcleos intactos. Evite pipetar o pellet fofo, branco, solto e superficial. Tome 2 x 5 μL de S1 para BCA e 2 x 100 μL de S1 para WB.
- Gire S1 a 9.000 x g por 15 min a 4 °C para obter o sobrenadante sinaptossomal (S2) e o pellet de sinaptossomo bruto (P2). Tome 2 x 10 μL de S2 para BCA e 2 x 500 μL de S2 para WB. Descarte o sobrenadante após a obtenção das alíquotas e prossiga para a próxima etapa com o pellet.
- Ressuspender P2 em 3 mL de tampão gelado A com inibidores de protease e centrífuga a 9.000 x g por 15 min a 4 °C para obtenção do sobrenadante (S2') e sinaptossomos lavados (P2'). Descarte o sobrenadante e mantenha o pellet.
- Ressuscite P2' em 3 mL de Tampão A. Evite ressuspender a porção vermelha escura no fundo do pellet, que contém principalmente mitocôndrias. Tome 2 x 20 μL de P2' para BCA e 2 x 100 μL de P2' para WB.
NOTA: Isto pode ser conseguido misturando suavemente as bordas e a superfície do pellet para ressuspender os sinaptossomos lavados de branco enquanto direciona a ponta da pipeta para longe do centro vermelho do pellet.
4. Lise hipotônica
NOTA: Os esquemas desse procedimento são mostrados na Figura 5.
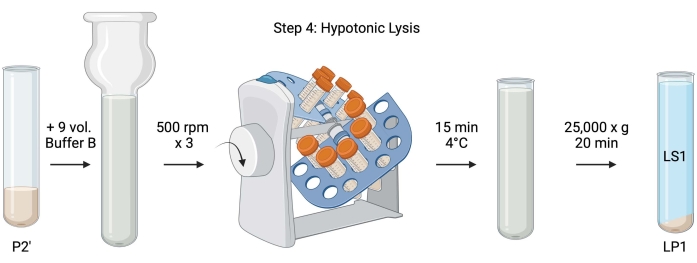
Figura 5: Lise hipotônica. Esquema do passo 4, a lise hipotônica dos sinaptossomos para gerar o sobrenadante de lise (LS1) e frações de membrana sinaptossomal (LP1). Por favor, clique aqui para ver uma versão maior desta figura.
- Para a lise hipotônica dos sinaptossomos lavados, adicionar 9 volumes de tampão B resfriado (Tabela 1) à ressuspensão de P2' (~27 mL). Homogeneizar os sinaptossomos em um homogeneizador de vidro Dounce (três passagens para cima e para baixo a 500 rpm).
- Transfira as amostras para tubos de centrífuga cônica tampados de 50 mL. Gire-os em um revólver de tubo em uma câmara fria de 4 °C por 15 min.
- Centrífuga lisada P2' a 25.000 x g por 20 min a 4 °C para obtenção do sobrenadante de lise (LS1) e do pellet de lise contendo membranas sinaptossomais (LP1). Tome 2 x 50 μL de LS1 para BCA e 2 x 400 μL de LS1 para WB. Transfira o LS1 para um tubo de centrífuga tampado para ultracentrifugação (ver Tabela de Materiais).
5. Isolamento da vesícula sináptica
NOTA: Os esquemas desse procedimento são mostrados na Figura 6.
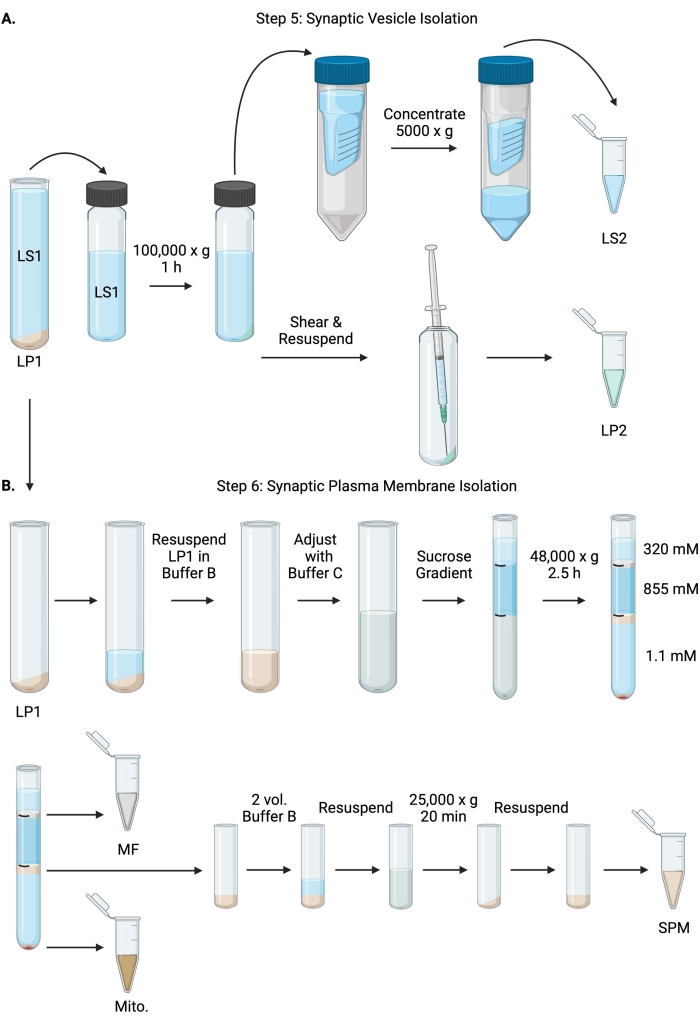
Figura 6: Isolamento da vesícula sináptica e isolamento da membrana plasmática sináptica. (A) Esquema da etapa 5, o isolamento das frações citosol sináptico (LS2) e vesícula sináptica (LP2) e (B) etapa 6, a geração de mielina (MF), membrana plasmática sináptica (SPM) e frações mitocondriais (Mito.) após a ultracentrifugação de gradientes de sacarose. Por favor, clique aqui para ver uma versão maior desta figura.
- Centrifugar LS1 num rotor ultracentrífugo de ângulo fixo (ver Tabela de Materiais) a 100 000 x g durante 60 min a 4 °C para obter sobrenadante de citosol sináptico (LS2) e pellet de vesícula sináptica (LP2). O LP2 será pequeno, translúcido e fortemente aderido ao lado do tubo de centrífuga.
- Ressuscite LP2 em 500 μL de tampão A. Usando uma agulha de 23 G e uma seringa de 1 mL, cisalhamento LP2 com trituração suave. Tome 2 x 10 μL de LP2 para BCA e 2 x 250 μL de LP2 para WB.
- Transfira LS2 (~30 mL) para unidades de filtro centrífugo com um ponto de corte de 10 kDa (ver Tabela de Materiais).
NOTA: Se proteínas menores que 10 kDa forem de interesse, unidades de filtro centrífugo de corte de 4 kDa estão disponíveis, mas resultarão em tempos de rotação mais longos. - Concentrar LS2 a aproximadamente 0,5 ml girando a 5000 x g até 1 h a 4 °C. Tome 2 x 10 μL de LS2 concentrado para BCA e 2 x 250 μL de LS2 concentrado para WB. Depois de iniciar o giro, prossiga diretamente para a etapa 6.1.
6. Isolamento da membrana plasmática sináptica
- Ressuspender LP1 (etapa 4.3) em 1 mL de Tampão B (Tabela 1). Tome 2 x 10 μL de LP1 para BCA e 2 x 50 μL de LP1 para WB. Ajustar o LP1 restante para um volume final de 7,5 mL e uma concentração final de sacarose de 1,1 M com tampão B e tampão C (Tabela 1).
- Transfira 7,5 mL de LP1 ressuspenso para um tubo ultracentrífugo de 14 mL (ver Tabela de Materiais). Sobreponha cuidadosamente o LP1 com 3,75 mL de Tampão D (Tabela 1) e, em seguida, sobreponha com 1,25 mL de Tampão A (ou um volume maior para preencher logo abaixo da parte superior do tubo de centrífuga). Evite pipetar para baixo o lado do tubo, o que irá interromper as interfaces de gradiente de sacarose. Depois de sobrepor cada fração de sacarose, marque a parte superior da solução com uma caneta. Equilibrar os tubos para ultracentrifugação em peso, não em volume, com a adição gota a gota de tampão A a menos de 10 mg. Centrífuga a 48.000 x g durante 2,5 h a 4 °C num rotor de ultracentrífuga de balde oscilante (ver Tabela de Materiais).
- Adquira imagens dos gradientes intactos após a ultracentrifugação para documentar a distinção de cada interface de sacarose e o sucesso do fracionamento.
- Remova cuidadosamente a camada superficial de sacarose 320 mM (tampão A). Recupere a fração de mielina (MF) na interface de sacarose de 320 mM/855 mM em um volume de 800 μL. Recuperar a fração da membrana plasmática sináptica (SPM) na interface de sacarose de 855 mM/1,1 M em um volume de 1.000 μL. Pipetar cada fração para cima da parede do tubo de forma circular para garantir que a fração completa seja coletada. Aspirar cuidadosamente a sacarose restante e recuperar o pellet mitocondrial (Mito.) ressuspendendo em 200 μL de tampão B. Tome 2 x 100 μL de MF para BCA e 2 x 10 μL de Mito. para BCA; dividir o restante de MF e Mito. amostras ao meio para WB.
- Diluir a fração SPM com 2 volumes de tampão B (~2 mL) e, em seguida, centrifugar em um rotor de ângulo fixo em um tubo de centrífuga de 3,5 mL (ver Tabela de Materiais) a 25.000 x g por 20 min a 4 °C. Descarte o sobrenadante e ressuspenda o pellet SPM no tampão A para um volume final de 250 μL. Pegue 2 x 5 μL de SPM para BCA e divida o SPM restante ao meio para WB.
- Realizar um BCA para determinar a concentração de proteína de cada amostra, contabilizando o volume de alíquota variável.
NOTA: Para a análise de WB, a concentração de proteína de trabalho sugerida para todas as frações subcelulares é de 2 μg/μL (ou tão alta quanto alcançável para LS1 e MF).
O método apresentado resulta em 11 frações subcelulares cerebrais que podem ser submetidas a uma purificação adicional e a várias formas de análise a jusante35,36. O método padrão-ouro para avaliar a qualidade dos sinaptossomos, SVs23 e outros componentes é a microscopia eletrônica (EM) (Figura 7). O immunoblotting quantitativo para proteínas presentes em frações subcelulares específicas também pode ser realizado para avaliar marcadores de pureza de fração (Figura 8). Por exemplo, a análise imunoblot das frações revela o enriquecimento da N-caderina (CDH2, nome UniProt) na fração da membrana plasmática sináptica (SPM), α-sinucleína (SYUA) no citosol sináptico (LS2), sinaptofisina (SYPH) na fração vesícula sináptica (LP2) e proteína básica de mielina (MBP) na fração de mielina (MF) quando comparada aos níveis de proteína no homogeneizado cerebral completo inicial (Total) (Figura 8 ). Uma vez que a pureza da fração tenha sido estabelecida (por exemplo, observe a ausência de CDH2 na fração LS2 ou o aumento de muitas vezes no SYPH na fração LP2), o immunoblotting quantitativo pode ser usado para determinar a localização de proteínas de interesse ou consultar diferenças na distribuição de proteínas entre genótipos ou tratamentos. Compreender a localização subcelular de proteínas sinápticas pode permitir a dissecção de funções proteicas não descritas anteriormente. Além disso, esse método pode elucidar defeitos de tráfico ou disfunção sináptica em estados de doença, especialmente quando emparelhado com ensaios funcionais. Por exemplo, nossa equipe usou esse método para identificar um pool de proteína palmitoila tioesterase 1 enzimaticamente ativa que é enriquecida no citosol sináptico19.
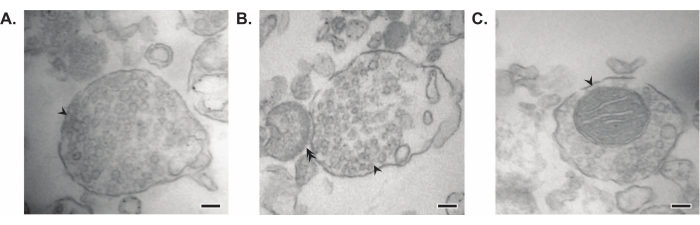
Figura 7: Microscopia eletrônica (EM) de sinaptossomos. (A) Imagem EM representativa de um sinaptossomo contendo vesículas sinápticas (seta). (B) Imagem EM representativa de um sinaptossomo com componentes pré (seta) e pós-sinápticos (seta dupla). (C) Imagem EM representativa de um sinaptossomo contendo vesículas sinápticas e uma mitocôndria (seta) (barras de escala = 100 nm). Por favor, clique aqui para ver uma versão maior desta figura.
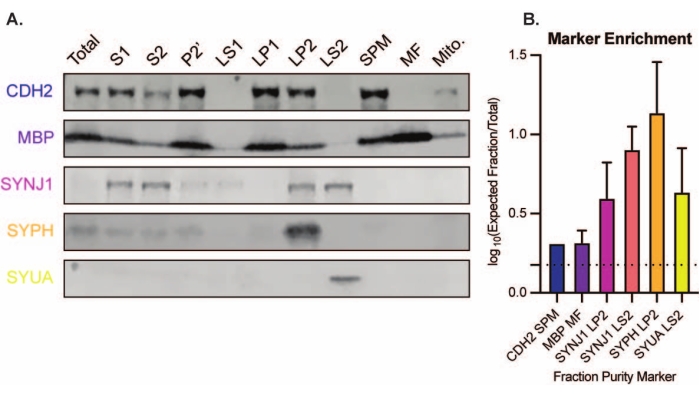
Figura 8: Análise de imunoblot das frações subcelulares. (A) Os marcadores de pureza da fração subcelular (indicados com a nomenclatura UniProt) estão adequadamente localizados em comparação com o homogeneizado do cérebro inteiro (total): N-caderina (CDH2) na fração da membrana plasmática sináptica (SPM), sinaptofisina 1 (SYPH) e sinaptojanina 1 (SYNJ1) na fração enriquecida da vesícula sináptica (LP2), α-sinucleína (SYUA) no citosol sináptico (LS2), e proteína básica de mielina (MBP) na fração de mielina (MF). (B) A análise de quantificação de immunoblot revela o enriquecimento (mudança de dobra em relação ao total) dos marcadores de pureza de fração. Os dados são representados como média ± desvio padrão em uma escala log10. A linha pontilhada indica uma mudança de 1,5 vezes (y = 0,176) (n = 3 replicar experimentos com 8 camundongos do tipo selvagem; idade = 2 meses; n = 4-5 manchas para SYPH, SYUA, MBP, com n = 3 valores plotados previamente publicados por Gorenberg et al.19; n = 5 para SYNJ1; n = 1 para CDH2). Por favor, clique aqui para ver uma versão maior desta figura.
Em seus estudos seminais, Whittaker e seus colegas usaram quatro critérios morfológicos para identificar sinaptossomos: (1) as estruturas têm uma membrana plasmática selada; (2) as estruturas contêm SVs semelhantes às dos terminais nervosos e varicosidades in situ em tamanho e número; (3) as estruturas possuem uma ou mais pequenas mitocôndrias; e (4) a membrana pré-sináptica é frequentemente aderida a um componente pós-sináptico11,12,13. Embora os dois primeiros critérios geralmente se apliquem a todos os métodos de isolamento, nos protocolos mais recentes descritos neste artigo, nem todos os sinaptossomos resultantes terão mitocôndrias e terminais pós-sinápticos ligados. Aproximadamente 60% dos sinaptossomos terão mitocôndrias, e estima-se que apenas até 15% tenham terminais pós-sinápticos anexados37. Se os componentes pós-sinápticos são de particular interesse, sabe-se que o uso de um tampão de homogeneização isotônico semelhante ao Krebs e filtração por pressão para enriquecimento produz altas concentrações de sinaptossomos com terminais pós-sinápticos (também denominados sinaptoneurossomas)22,38.
O método de sacrifício do animal pode afetar a qualidade dos sinaptossomos e subfrações sinápticas. Animais adultos sacrificados usando um método de eutanásia que não requer anestesia resultarão na melhor qualidade de fração. Além disso, os cérebros devem ser recém-dissecados, não congelados e homogeneizados usando uma proporção de 1:10 de tampão de homogeneização (peso/volume) para as frações sinápticas mais viáveis22. O cérebro tem uma população heterogênea de sinapses que podem ser diferenciadas pelo tipo de neurotransmissores que carregam. A formação de sinaptossomas geralmente não é afetada pelo tipo de sinapse ou conteúdo de neurotransmissores13. Uma exceção são as fibras musgosas no cerebelo, que são conhecidas por serem interrompidas em condições ótimas para a obtenção de sinaptossomos do resto do cérebro39,40. Assim, recomenda-se a remoção do cerebelo antes da homogeneização cerebral se a exclusão dessa região não afetar o objetivo experimental. Se estiver interessado em isolar sinaptossomos de um caráter neurotransmissor particular, áreas do cérebro que são enriquecidas para neurônios contendo o neurotransmissor de interesse podem primeiro ser isoladas. No entanto, essa abordagem imporá limitações ao rendimento da fração final, dependendo do tamanho da região de interesse (a idade dos animais também é, portanto, uma consideração). Existem métodos imunoquímicos para o isolamento de sinaptossomos neurotransmissores específicos, mas a viabilidade e o rendimento serão significativamente comprometidos22. Se a avaliação da viabilidade metabólica dos sinaptossomos for importante, a medida da liberação de neurotransmissores41,42 ou certos ensaios enzimáticos 43 pode ser empregada.
Contaminantes comuns em preparações de sinaptossomos incluem microssomos, mitocôndrias livres, SVs e membranas neuronais e gliais. A contaminação pode ser reduzida aumentando o número de lavagens nas frações P1 e P222 e evitando a ressuspensão do pellet mitocondrial vermelho nas etapas subsequentes. Em experimentos em que a viabilidade metabólica e o tempo são cruciais, a redução do número de lavagens e o uso de gradientes de Ficoll ou Percoll sobre gradientes de sacarose serão úteis 44,45,46. Esses métodos também reduzem significativamente a contaminação. O protocolo original de Whittaker produziu SVs de alta qualidade. A otimização adicional por Nagy et al.23, incluída nesse método, produz VS com notável homogeneidade e pureza, sem comprometer significativamente o rendimento36. Se subtipos específicos de VS forem de interesse, como SVs glutamatérgicos (contendo VGLUT-1) ou GABAérgicos (contendo VGAT-1), o imunoisolamento com anticorpos específicos pode ser realizado47,48. Métodos alternativos também estão disponíveis para isolar CCVs de sinaptossomos, que, devido à densidade diferencial, podem não estar presentes na mesma interface que as SVs obtidas com esse método20,49,50.
No geral, o presente protocolo para isolar componentes sinápticos pode ser ainda mais otimizado para obter frações com melhor homogeneidade e viabilidade com base na qualidade e quantidade do tecido cerebral de origem e nos objetivos experimentais. Para maiores detalhes sobre a solução de problemas, deve-se consultar os capítulos de livros de Dunkley e Robinson22 e Ganzella et al.36.
Os autores não têm nada a revelar.
Gostaríamos de agradecer a P. Colosi pela preparação da imagem EM. Este trabalho foi apoiado pelos Institutos Nacionais de Saúde (R01 NS064963, SSC; R01 NS110354, SSC; R01 NS083846, SSC; R21 NS094971, SSC; T32 NS007224, SMT; T32 NS041228, SMT), do Departamento de Defesa dos Estados Unidos (W81XWH-17-1-0564, SSC; W81XWH-19-1-0264, VDJ), Aligning Science Across Parkinson's (ASAP) Collaborative Research Network (SSC) e o Michael J. Fox Foundation Target Advancement Program (MJFF-020160, SSC & VDJ). Criamos ilustrações gráficas usando BioRender.com.
| Name | Company | Catalog Number | Comments |
| 1 mL TB Syringe | BD | 309649 | |
| 1.5 mL Eppendorf Tubes | USA Scientific | 1415-2500 | |
| 14 mL, Open-Top Thinwall Ultra-Clear Tube | Beckman Coulter | 344060 | Compatible with SW 40 Ti |
| 23 Gauge Precision Glide Hypodermic Needle | BD | 305145 | |
| 26.3 mL, Polycarbonate Bottle with Cap Assembly | Beckman Coulter | 355618 | Compatible with Ti70 |
| 3.5 mL, Open-Top Thickwall Polypropylene Tube | Beckman Coulter | 349623 | Compatible with TLA-100.3 |
| 50 mL Falcon Tubes | Fisher Scientific | 14-432-22 | |
| Amicon Ultra-15 Centrifugal Filter Unit | Millipore Sigma | UFC901024 | |
| Aprotinin | Sigma-Aldrich | A6279 | 1 mg/mL in diH2O |
| Avanti J-26 XP Centrifuge | Beckman Coulter | B22984 | <26,000 rpm |
| Benchtop HDPE Dewar Flask | Thermo Scientific | 5028U19 | |
| C57BL/6J Mice | The Jackson Labs | 000664 | |
| Centrifuge 5810R | Eppendorf | EP022628168 | <14,000 rpm |
| complete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets | Roche | 11873580001 | Add 1 tablet per 50 mL of solution |
| Curved Forceps | Fine Science Tools | 11273-20 | |
| Fine Surgical Scissors | Fine Science Tools | 8r | |
| Glas-Col Tissue Homogenizing System | Cole-Parmer | UX-04369-15 | |
| Graefe Forceps | Fine Science Tools | 11650-10 | |
| High-Speed Polycarbonate Round Bottom Centrifuge Tubes | ThermoFisher | 3117-0500 | Compatible with JA20 |
| Isofluorane | Henry Schein Animal Health | NDC 11695-6776-2 | |
| JA-20 Rotor | Beckman Coulter | 334831 | |
| Leupeptin | American Bio | AB01108 | 1 mg/mL in diH2O |
| N-[2-Hydroxyethyl] piperazine-N’-[2-ethanesulfonic acid] (HEPES) | American Bio | AB00892 | |
| Optima L-80 XP Ultracentrifuge | Beckman Coulter | <100,000 rpm | |
| Optima TLX Ultracentrifuge | Beckman Coulter | <120,000 rpm | |
| Pepstatin A | Thermo Scientific | 78436 | 1 mg/mL in DMSO |
| Phenylmethylsulfonyl fluoride (PMSF) | American Bio | AB01620 | |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23335 | For determination of protein concentration |
| Pipette Tips | |||
| Serological Pipettes | |||
| Sucrose | Sigma-Aldrich | S0389 | |
| Surgical Scissors | Fine Science Tools | 14002-12 | |
| SW 40 Ti Swinging-Bucket Rotor | Beckman Coulter | 331301 | |
| Teflon-Coated Pestle and Mortar Tissue Grinder | Thomas Scientific | 3431D94 | |
| Ti70 Rotor | Beckman Coulter | 337922 | |
| TLA-100.3 Rotor | Beckman Coulter | 349490 | |
| Tube Revolver | Dot Scientific | DTR-02VS |
- Kandel, E. R., Schwartz, J. H., Jessell, T. M., Siegelbaum, S. A., Hudspeth, A. J., Education, A. J. Synaptic Transmission. Principles of Neural Science, Fifth Edition. , (2014).
- Lepeta, K., et al. Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. Journal of Neurochemistry. 138 (6), 785-805 (2016).
- Südhof, T. C., Malenka, R. C. Understanding synapses: Past, present, and future. Neuron. 60 (3), 469-476 (2008).
- Südhof, T. C. The molecular machinery of neurotransmitter release (Nobel lecture). Angewandte Chemie International Edition. 53 (47), 12696-12717 (2014).
- Jahn, R., Boyken, J., Pfaff, D. W. Molecular Regulation of Synaptic Release. Neuroscience in the 21st Century: From Basic to Clinical. , 351-401 (2013).
- Xiong, H., Gendelman, H. E. . Current Laboratory Methods in Neuroscience Research. , (2014).
- Azevedo, F. A., et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. Journal of Comparative Neurology. 513 (5), 532-541 (2009).
- Herculano-Houzel, S. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proceedings of the National Academy of Sciences of the United States of America. 109, 10661-10668 (2012).
- Herculano-Houzel, S., Lent, R. Isotropic fractionator: A simple, rapid method for the quantification of total cell and neuron numbers in the brain. Journal of Neuroscience. 25 (10), 2518-2521 (2005).
- Herculano-Houzel, S., Mota, B., Lent, R. Cellular scaling rules for rodent brains. Proceedings of the National Academy of Sciences of the United States of America. 103 (32), 12138-12143 (2006).
- Gray, E. G., Whittaker, V. P. The isolation of nerve endings from brain: An electron-microscopic study of cell fragments derived by homogenization and centrifugation. Journal of Anatomy. 96, 79-88 (1962).
- Gray, E. G., Whittaker, V. P. The isolation of synaptic vesicles from the central nervous system. Journal of Physiology. 153, 35-37 (1960).
- Whittaker, V. P. Thirty years of synaptosome research. Journal of Neurocytology. 22 (9), 735-742 (1993).
- Jahn, R., Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 490, 201-207 (2012).
- Whittaker, V. P., Michaelson, I. A., Kirkland, R. J. The separation of synaptic vesicles from nerve-ending particles ('synaptosomes). Biochemical Journal. 90 (2), 293-303 (1964).
- De Robertis, E., Rodriguez De Lores Arnaiz, G., Pellegrino De Iraldi, A. Isolation of synaptic vesicles from nerve endings of the rat brain. Nature. 194, 794-795 (1962).
- De Robertis, E., Pellegrino De Iraldi, A., Rodriguez, G., Gomez, C. J. On the isolation of nerve endings and synaptic vesicles. The Journal of Biophysical and Biochemical Cytology. 9 (1), 229-235 (1961).
- Zimmermann, H., Whittaker, V. P., Murphy, K. M. The Discovery of the Synaptosome and Its Implications. Synaptosomes. , 9-26 (2018).
- Gorenberg, E. L., et al. Identification of substrates of palmitoyl protein thioesterase 1 highlights roles of depalmitoylation in disulfide bond formation and synaptic function. PLoS Biology. 20 (3), 3001590 (2022).
- Vidyadhara, D. J., et al. Dopamine transporter and synaptic vesicle sorting defects initiate auxilin-linked Parkinson's disease. bioRxiv. , (2022).
- Schrimpf, S. P., et al. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics. 5 (10), 2531-2541 (2005).
- Dunkley, P. R., Robinson, P. J., Murphy, K. M. Synaptosome Preparations: Which Procedure Should I Use. Synaptosomes. , 27-53 (2018).
- Nagy, A., Baker, R. R., Morris, S. J., Whittaker, V. P. The preparation and characterization of synaptic vesicles of high purity. Brain Research. 109 (2), 285-309 (1976).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127 (4), 831-846 (2006).
- Wagner, J. A., Kelly, R. B. Topological organization of proteins in an intracellular secretory organelle: the synaptic vesicle. Proceedings of the National Academy of Sciences of the United States of America. 76 (8), 4126-4130 (1979).
- Jahn, R., Schiebler, W., Ouimet, C., Greengard, P. A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America. 82 (12), 4137-4141 (1985).
- Binotti, B., Jahn, R., Pérez-Lara, &. #. 1. 9. 3. ;. An overview of the synaptic vesicle lipid composition. Archives of Biochemistry and Biophysics. 709, 108966 (2021).
- Siegel, D. P., Ware, B. R. Electrokinetic properties of synaptic vesicles and synaptosomal membranes. Biophysical Journal. 30 (1), 159-172 (1980).
- Whittaker, V. P., Michaelson, I. A., Kirkland, R. J. The separation of synaptic vesicles from disrupted nervending particles. Biochemical Pharmacology. 12 (3), 300-302 (1963).
- Clementi, F., Whittaker, V. P., Sheridan, M. N. The yield of synaptosomes from the cerebral cortex of guinea pigs estimated by a polystyrene bead "tagging" procedure. Zeitschrift für Zellforschung und Mikroskopische Anatomie. 72, 126-138 (1966).
- Carlson, S. S., Wagner, J. A., Kelly, R. B. Purification of synaptic vesicles from elasmobranch electric organ and the use of biophysical criteria to demonstrate purity. Biochemistry. 17 (7), 1188-1199 (1978).
- Huttner, W. B., Schiebler, W., Greengard, P., De Camilli, P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. Journal of Cell Biology. 96 (5), 1374-1388 (1983).
- Hawkins, P., et al. A guide to defining and implementing protocols for the welfare assessment of laboratory animals: eleventh report of the BVAAWF/FRAME/RSPCA/UFAW Joint Working Group on Refinement. Laboratory Animals. 45 (1), 1-13 (2011).
- Risling, T. E., Caulkett, N. A., Florence, D. Open-drop anesthesia for small laboratory animals. Canadian Veterinary Journal. 53 (3), 299-302 (2012).
- Deutsch, C., Drown, C., Rafalowska, U., Silver, I. A. Synaptosomes from rat brain: Morphology, compartmentation, and transmembrane pH and electrical gradients. Journal of Neurochemistry. 36 (6), 2063-2072 (1981).
- Ganzella, M., Ninov, M., Riedel, D., Jahn, R. Isolation of synaptic vesicles from mammalian brain. Methods in Molecular Biology. 2417, 131-145 (2022).
- Dunkley, P. R., et al. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: homogeneity and morphology of subcellular fractions. Brain Research. 441 (1-2), 59-71 (1988).
- Schwartz, R. D., Skolnick, P., Hollingsworth, E. B., Paul, S. M. Barbiturate and picrotoxin-sensitive chloride efflux in rat cerebral cortical synaptoneurosomes. FEBS Letters. 175 (1), 193-196 (1984).
- Pittaluga, A., Thellung, S., Maura, G., Raiteri, M. Characterization of two central AMPA-preferring receptors having distinct location, function and pharmacology. Naunyn-Schmiedeberg's Archives of Pharmacology. 349 (6), 555-558 (1994).
- Israël, M., Whittaker, V. P. The isolation of mossy fibre endings from the granular layer of the cerebellar cortex. Experientia. 21 (6), 325-326 (1965).
- Khvotchev, M., Lonart, G., Südhof, T. C. Role of calcium in neurotransmitter release evoked by alpha-latrotoxin or hypertonic sucrose. Neuroscience. 101 (3), 793-802 (2000).
- Lonart, G., Janz, R., Johnson, K. M., Südhof, T. C. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 21 (5), 1141-1150 (1998).
- Nicholls, D. G., Sihra, T. S. Synaptosomes possess an exocytotic pool of glutamate. Nature. 321 (6072), 772-773 (1986).
- Dunkley, P. R., Jarvie, P. E., Robinson, P. J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nature Protocols. 3 (11), 1718-1728 (2008).
- Cotman, C. W., Matthews, D. A. Synaptic plasma membranes from rat brain synaptosomes: Isolation and partial characterization. Biochimica et Biophysica Acta. 249 (2), 380-394 (1971).
- Booth, R. F., Clark, J. B. A rapid method for the preparation of relatively pure metabolically competent synaptosomes from rat brain. Biochemical Journal. 176 (2), 365-370 (1978).
- Takamori, S., Riedel, D., Jahn, R. Immunoisolation of GABA-specific synaptic vesicles defines a functionally distinct subset of synaptic vesicles. Journal of Neuroscience. 20 (3), 4904-4911 (2000).
- Burger, P. M., et al. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron. 3 (6), 715-720 (1989).
- Blondeau, F., et al. Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proceedings of the National Academy of Sciences of the United States of America. 101 (11), 3833-3838 (2004).
- Maycox, P. R., Link, E., Reetz, A., Morris, S. A., Jahn, R. Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. Journal of Cell Biology. 118 (6), 1379-1388 (1992).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved