Robust Tissue Fabrication for Long-Term Culture of iPSC-Derived Brain Organoids for Aging Research
In This Article
Summary
The present protocol provides a step-by-step procedure for the reproducible generation, maintenance, and aging of cerebral organoids derived from human-induced pluripotent stem cells (iPSCs). This method enables culturing and maturing cerebral organoids for extended periods, which facilitates the modeling of processes involved in brain aging and age-related pathogenesis.
Abstract
The currently available animal and cellular models do not fully recapitulate the complexity of changes that take place in the aging human brain. A recent development of procedures describing the generation of human cerebral organoids, derived from human induced pluripotent stem cells (iPSCs), has the potential to fundamentally transform the ability to model and understand the aging of the human brain and related pathogenic processes. Here, an optimized protocol for generating, maintaining, aging, and characterizing human iPSC-derived cerebral organoids is presented. This protocol can be implemented to generate brain organoids in a reproducible manner and serves as a step-by-step guide, incorporating the latest techniques that result in improved organoid maturation and aging in culture. Specific issues related to organoid maturation, necrosis, variability, and batch effects are being addressed. Taken together, these technological advances will allow the modeling of brain aging in organoids derived from a variety of young and aged human donors, as well as individuals afflicted with age-related brain disorders, allowing the identification of physiologic and pathogenic mechanisms of human brain aging.
Introduction
Aging disease models have become increasingly relevant as human life expectancy continues to rise. Large-scale genomic studies have uncovered aged populations with dysregulation of molecular processes and genetic changes affecting the quality of life1. The aging process is characterized by a general loss of functionality of the organism, including loss of cognitive function, increased risk for neurodegenerative disorders, and a host of chronic diseases2.
Current cell culture techniques do not appropriately represent the multifactorial nature of aging, as these dysfunctions cannot be properly replicated by using mutations, toxins, or infections3. Animal models that explore the process of aging are often associated with long experimental times and high costs, but also bring ethical considerations with them. Using induced pluripotent stem cells (iPSCs) from patients can elucidate the molecular mechanisms that underlie disease progression as iPSCs allow for the natural development of cells into mature tissue3. iPSCs have become the working horse of many labs investigating neurodegenerative disorders, as the cellular reprogramming of harvested cells does not seem to erase the disease or aging imprints of the donors4. These imprints recapitulate cellular phenotypes that have been demonstrated in human and animal models, making iPSCs suitable for examining individual cellular deterioration of the highly dense brain tissue5,6. IPSC-derived organoids have become the preeminent model for three-dimensional culturing of tissue, enabling more complex cell-to-cell interactions and an improved developmental recapitulation. Although primarily used for developmental data, organoids have been increasingly applied toward disease modeling, specifically models for inflammation, neurodegeneration, and aging7. Building upon previous iPSC studies, organoids retain disease phenotypes and cellular phenotypes within the physiological context of tissue-like network connections8,9. However, culturing three-dimensional tissue of certain dimensions can be challenging, especially for extended periods of time.
This work presents a detailed method for the reproducible generation of cerebral organoids that allows the tissue to mature substantially in size for longer periods. Cerebral organoid creation has remained relatively standardized, adopting methods from several prominent protocols10,11. However, several modifications have been suggested for improved differentiation and maintenance. These alternate methods include using neurogenic factors to enhance neural differentiation12, additional scaffolds for improved nutrient exchange promoting cellular longevity13, and low-sheer stress agitation for prolonged culture and growth14. These improvements have been incorporated into this method to develop mature organoids capable of expressing neurodegenerative and aging phenotypes.
Protocol
Patient studies were approved by the Institutional Review Board. All participants signed written informed consent and repository consent to allow their data and biospecimen to be repurposed. iPSC lines were generated following IRB and institutional guidelines. Figure 1 illustrates a schematic overview of the workflow of this protocol.
1. iPSC reprogramming and maintenance
- Collect 8 mL of the subject's blood (aged or disease patient; ages 65 and older) into cell preparation tubes (CPT) with sodium citrate or into EDTA or heparinized tubes.
- Centrifuge at 1,800 x g for 30 min at room temperature (RT) to collect pellet containing only peripheral blood and no serum15.
- Use specialized reprogramming vectors according to the manufacturer's protocol (see Table of Materials) to obtain iPSCs16.
- Culture iPSCs in feeder-free, lactose dehydrogenase elevating virus (LDEV)-free reduced growth factor basement membrane matrix coated 6-well culture plates in Essential 8 (E8) media (see Table of Materials) at 37 °C in a humidified atmosphere with 5% CO2.
- Maintain iPSCs in E8 media for 3-4 days in order to avoid overcrowding or spontaneous differentiation.
- Validate the pluripotency of the iPSC lines using immunofluorescent markers (step 2) and test for mycoplasma (step 3).
2. Pluripotency staining
- To conserve solutions and antibodies, seed the cells on coated (step 1.4) 24-well culture plates 3-4 days prior to analysis. Aspirate the medium with a pipette and fix the cells with 4% formaldehyde in phosphate-buffered saline (1x PBS) for 15-20 minutes at RT.
- Wash the cells 3x with 1x PBS and permeabilize with 0.1% Triton-X 100 in 1% bovine serum albumin (BSA) in PBS for at least 15 min, but no more than 1 h at RT.
- Wash 3x with 1x PBS and block with 5% BSA in PBS for 30 min at RT.
- Wash again 3x with 1x PBS and add the antibodies Sox2 and Oct3/4 (1:100 dilution; see Table of Materials), and the nucleic stain DAPI in 1% BSA (1:4,000 dilution) overnight at 4 °C . Wrap in aluminum foil to protect from light.
- Wash 3x with 1x PBS and leave the cells in PBS. Image with a fluorescence microscope at a 10-20x magnification. Ensure that the pluripotency markers Sox2 and Oct3/4 are localized in the nuclei of the cells17,18.
3. Mycoplasma testing
NOTE: Refer to the detection kit's protocol (see Table of Materials) for detailed assay execution and analysis steps. The detection kit provides the reagent, the substrate, and the assay buffer for mycoplasma testing.
- Prior to passaging cells or refreshing medium, collect 2-3 mL of cell culture medium in a centrifuge tube and pellet any cells or debris at 200 x g for 5 min at RT. Store the supernatant at 4 °C for ≤5 days. Incubate the cells with the medium for at least 24 h to ensure a detectable signal.
- Add 100 µL of cell supernatant to a fresh tube or well of a white-walled 96-well plate (opaque bottom recommended, see Table of Materials). Reconstitute the reagent and substrate in the assay buffer and equilibrate for 15 min at RT.
- Add 100 µL of the reagent to the sample and incubate for 5 min at RT. Measure the luminescence with a luminometer (measurement #1).
- Add 100 µL of the substrate to the sample and incubate for 10 min at RT. Measure the luminescence (measurement #2).
- Determine the mycoplasma contamination by the ratio of measurement #2 to #1. Refer to the kit's manual for interpretation of the results.
4. Microfilament preparation
- Begin the preparation of the poly (lactic-co-glycolic acid) (PLGA, see Table of Materials) microfilaments by fraying the suture strand with the blunt end of a scalpel. Spay the frayed fiber lightly with 70% ethanol.
- Under the microscope and using a ruler, begin cutting the PLGA fiber into fragments of 500 µm to 1 mm long strands. Cut about 25 mm of the fiber in total. Keep the filaments in a 15 mL tube with a 1 mL antibiotic-antimycotic solution.
- In the hood, dilute the fiber solution with 10 mL of DMEM/F-12 (Table 1). Vortex well to mix the solution.
NOTE: Work in a cell culture flow hood in a sterile environment.
- In the hood, dilute the fiber solution with 10 mL of DMEM/F-12 (Table 1). Vortex well to mix the solution.
- Add 20 µL of the fiber solution to three embryoid body (EB) forming wells of a 96-well plate. Under brightfield microscopy, count and average the fibers per well. Dilute or concentrate to average 5-10 PLGA microfilaments per well. Prepare each well in this manner.
- The wells are now ready to be seeded with cells. Store the plate at room temperature until needed or at 4 ˚C for the following day.
5. Embryoid Body (EB) formation
NOTE: All media and solutions must be warmed to RT.
- Once the iPSCs have reached 70%-80% confluency (Figure 2A), they are ready to be passaged and used for EB formation. Check the cells with a microscope at 10x-20x magnification. Ensure that the colonies display minimal (<10%) areas of spontaneous differentiation.
- Aspirate the medium with a pipette and wash the cells once with DPBS. Dissociate the colonies by adding 500 µL of cell detachment solution (see Table of Materials) or 0.5 mM of ethylenediaminetetraacetic acid (EDTA) and incubate for 3-5 min at 37 °C.
NOTE: Work in a cell culture flow hood in a sterile environment. - Collect the released cells by adding 1 mL of fresh E8 media to each well and pipette gently until all cells are detached.
- Transfer 1.5 mL of cell suspension into a 15 mL tube, and add another 1 mL of fresh E8 media to reach a total volume of 2.5 mL.
- Centrifuge at 290 x g for 3 min at RT.
- Aspirate the supernatant with a pipette, resuspend the cell pellet in 1 mL of Essential 6 (E6, see Table of Materials) media supplemented with 50 µM ROCK inhibitor, and count the cells using a hemocytometer19.
- Prepare a cell suspension of 60,000-90,000 cells/mL, depending on the desired seeding density, in E6 media supplemented with 50 µM of ROCK inhibitor (see Table of Materials).
- Add 150 µL of the cell suspension into each well of a 96-well ULA plate (or a prepared concave plate20). Seed 9,000-11,000 cells per well.
- Centrifuge the plate to force-aggregate the cells at 290 x g for 1 min at RT. Place the plate in the incubator at 37 °C in a humidified atmosphere with 5% CO2.
6. Neuroepithelial induction
- After 24 h, carefully aspirate 120 µL of the media with a pipette. Ensure not to aspirate the EB by lowering the pipette tip too far into the well.
- Add 150 µL of E6 media (at RT) supplemented with 2 µM of XAV939 and the SMAD inhibitors: 10 µM of SB431542 and 500 nM of LDN 193189 per well (see Table of Materials).
- Change the medium daily with freshly prepared E6 media supplemented with 2 µM of XAV939, 10 µM of SB431542, and 500 nM of LDN 193189.
NOTE: By day 6 (DIV6), the EBs should have a diameter of 550-600 µm and be ready for further differentiation.
7. Organoid differentiation and maturation
NOTE: All media needs to be warmed to RT.
- At approximately DIV7, check whether all the EBs have reached a diameter of 550-600 µm and display a smooth and clear edge (Figure 2B); at this stage, they are ready to be embedded in an extracellular matrix (ECM).
NOTE: Work in a sterile environment. - Prepare dimpled embedding sheets from the thermoplastic sealing film (see Table of Materials) by placing a film sheet (about 4 in long) on an empty P200 box. Using a 15 mL conical tube or a 500 µL microcentrifuge tube, gently press down the film sheet into the holes to make 12 dimples. Spray the film sheet with 70% ethanol and let it dry inside the flow hood with the UV light switched on for at least 30 min.
- Thaw a sufficient quantity of basement membrane matrix (Matrigel, see Table of Materials) on ice and place it inside the flow hood.
NOTE: Per EB, approximately 30 µL of undiluted membrane matrix is needed. Always keep the membrane matrix below 4 °C to prevent it from gelling. Pre-chilled pipette tips are also recommended as they decelerate the matrix from polymerizing in the tip during pipetting, thereby reducing material loss. - Using a wide-bore P200 tip, transfer one EB to each dimple, and remove as much media as possible with a normal pipette tip. Be mindful not to let the EBs dry. Using a regular P200 tip, add ~30 µL of undiluted membrane matrix to each organoid, ensuring that the EB is at the center of the droplet.
- Once all EBs are embedded in the matrix, place the film sheet containing the EBs in a sterile Petri dish.
NOTE: Optionally, a small Petri dish filled with sterile water can be placed in the larger Petri dish next to the film sheet to prevent evaporation.- Transfer the dish to an incubator and incubate at 37 ˚C in a humidified atmosphere with 5% CO2 for approximately 10 min to let the membrane matrix solidify.
- For each set of 12 embedded organoids, prepare 5 mL of differentiation media with B27 without vitamin A (Table 1) in one well of a ULA 6-well plate. Pre-warm the plate to 37 °C in an incubator.
- Once the incubation time is finished, transfer the embedded EBs to the ULA 6-well plate by taking the film sheet and pushing out the dimples from the back of the sheet. If necessary, take 1 mL from the well and pipette it onto the sheet to help the droplets detach from the film.
NOTE: Important morphological changes can be seen 1 day after embedding; EBs go from having smooth edges to bulging protrusions forming buds (Figure 2C). - After 2 days (DIV9), perform a half-media change. Be careful not to aspirate or damage the membrane matrix droplets in the process.
- After 2 more days (DIV11), perform a full media change, supplementing the media with 3 µM of CHIR99021 (see Table of Materials).
- At DIV14, change the media to differentiation media with B27 with vitamin A (Table 1) for a gradual increase in organoid size.
- At DIV16, place the well-plate on an orbital shaker at 90 rpm inside an incubator. Change the media every 2 days.
- Every 40 DIV, dilute 500 µL of the membrane matrix for every 50 mL of media for additional nutrients in the media.
Representative Results
Integrating PLGA fibers, dimple embedding, and agitation leads to a robust generation of cerebral organoids that enables iPSC-derived cultures to be maintained for extended periods of time (Figure 1).

Figure 1: Schematic illustration of the workflow and timeline of this method. Please click here to view a larger version of this figure.
{kind=link}
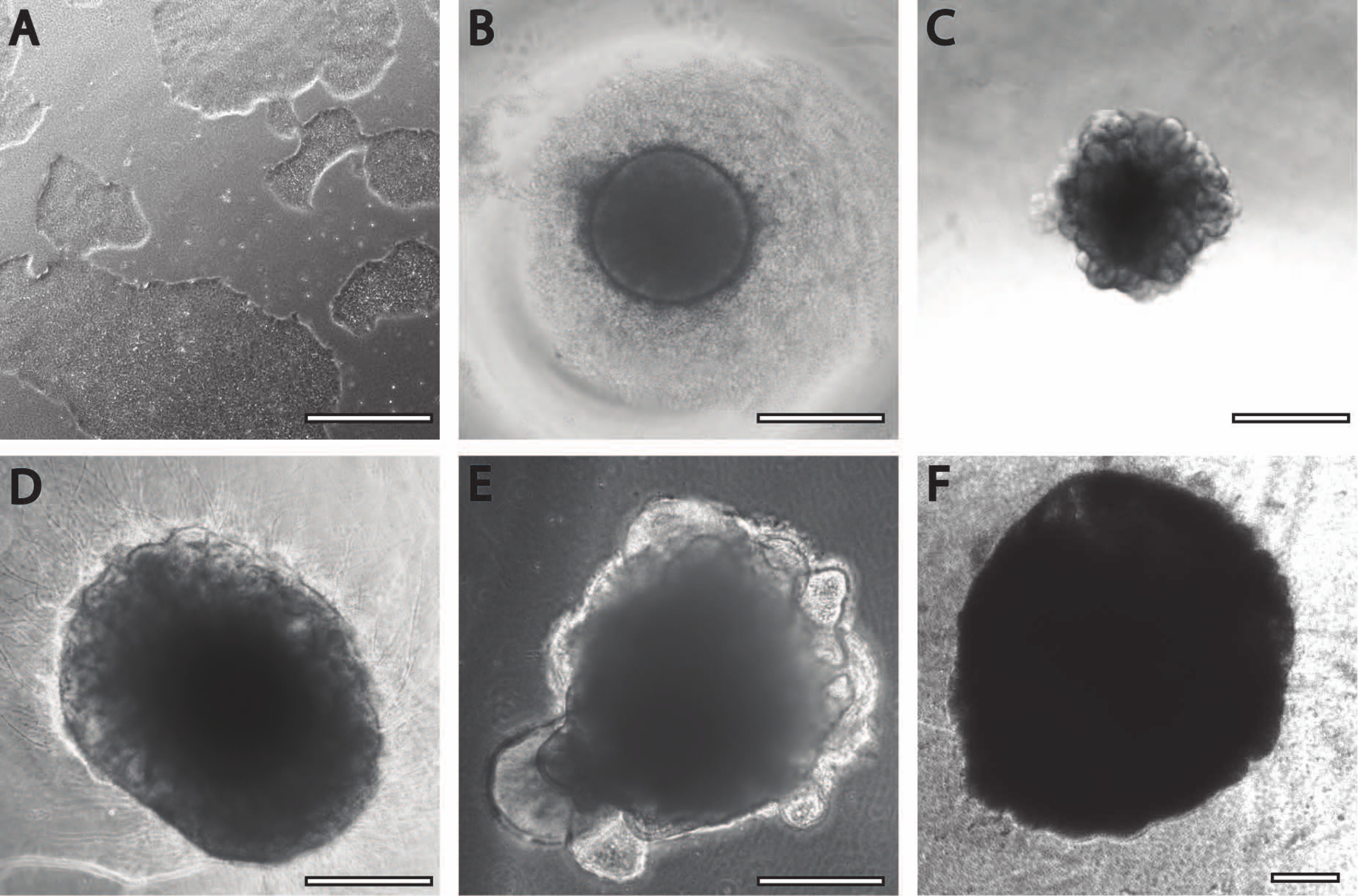
The initial neural induction period is comparable to previously published procedures11. The embryoid bodies (EB) begin as circular aggregates (Figure 2B) with white or transparent edges. As the EB is changed from neural induction media to neural maintenance media at DIV7, protrusions and buddings emerge within 24 h from the circular tissue (Figure 2C). Furthermore, as the organoid continues to grow, the surface area of the PLGA fiber helps the organoid to elongate (Figure 2D). During maturation, the edges of the organoid need to remain intact, as it is a good sign of healthy cells and development; otherwise, additional nutrients must be provided13 (Figure 2E). Growth of the organoids is further facilitated by the agitation of the organoid culture, as this improves perfusion with nutrients.

Figure 2: Differentiation and maturation of cerebral organoids. (A) Representative image of an iPSC culture at 70%-80% confluency. (B) Embryoid bodies (EBs) were generated and neuroepithelial formation was induced until DIV7. EBs were then embedded in the membrane matrix and further differentiated toward cerebral organoids. (C) Organoids at DIV10 displaying distinct budding formations. Organoids can be further matured in the membrane matrix using either (D) dimple or (E) sandwich embedding, here shown at DIV30. (F) Long-term culture shows cerebral organoids growing to significant sizes (DIV70). Scale bar = 500 µm. Please click here to view a larger version of this figure.
{kind=link}
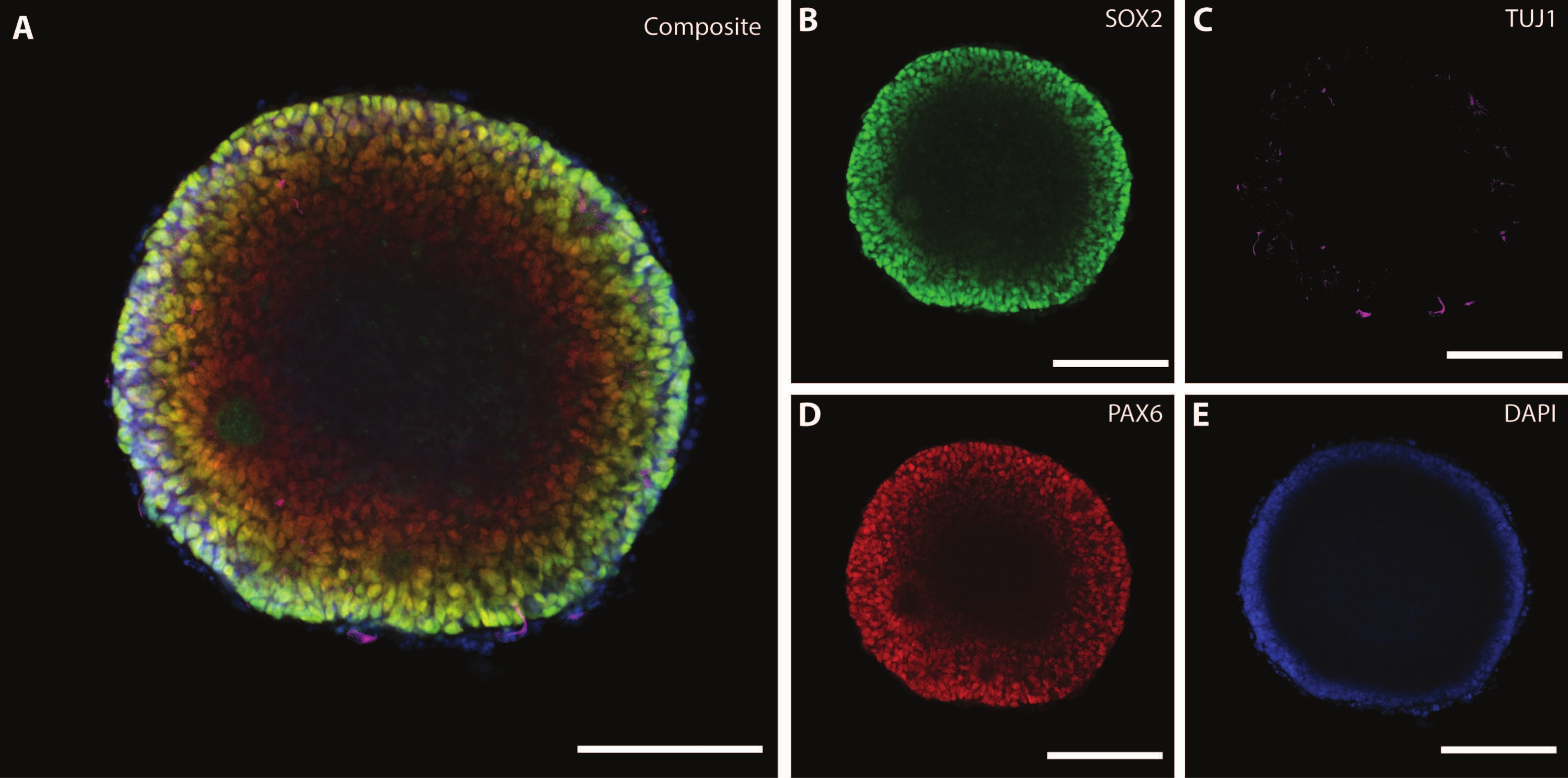
The dimensions and external morphology of the organoid are complemented by a complex architecture inside of the organoid. After fixing the tissue at DIV7 after the first stage of differentiation, cells of the EB express SOX2, an HMG box transcription factor that acts as a marker for multipotent neural stem cells21, as well as paired protein box Pax-6, indicating neural progenitor cells (Figure 3). Immature neurons marked by TuJ1 (class III β-tubulin)22 can already be seen scattered throughout the tissue.
At this stage, an example of the self-organization that these organoids go through becomes apparent. The organization of radial structures, called rosettes, is analogous to the neural tube, with SOX2+ cells in the rosette center21 and PAX6 towards the rosette periphery. These rosettes give rise to neurons as they migrate out. These radiating cells are initially double positive for the neural stem/progenitor cell marker Nestin23 and glial fibrillary acidic protein (GFAP), similar to the radial glia found in neurogenic areas of the in vivo brain. As these migrating neurons mature, the cytoskeletal markers reflect this change22. The early-stage neural differentiation marker TuJ122 is visible in the inner circle of the rosette and shifts to microtubule-associated protein 2 (MAP2)24, a neuron-specific maturation marker at the periphery.

Figure 3: Embryoid bodies at DIV7 show structural organization and immature characterization. Whole mount immunohistochemistry image of an EB at DIV7. (A) A composite image of the EB displaying (B) SOX2+ neural rosettes, (C) immature neurons (TUJ1) scattered throughout the EB, (D) neural progenitor cells (PAX6), and (E) nuclei visualized by DAPI. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
As the organoids age, the organization and markers of the developing neurons start replicating physiological conditions. At DIV30, many rosettes giving rise to neurogenic regions equivalent to the developing brain can be observed25. By DIV60, these SOX2+ neurogenic regions are non-existent and are replaced by mature MAP2 and NeuN26, a neuron differentiation marker, and positive neurons (Figure 4 and Figure 5).

Figure 4: Organoids at DIV120 show mature neuronal characterization. Immunohistochemistry image of an organoid section at DIV 120. (A) A composite image of the section showing mature neuronal markers of (B) MAP2 (purple) and (C) NeuN (green). (D) Nuclei were visualized by DAPI. Scale bar = 20 µm. Please click here to view a larger version of this figure.
{kind=link}

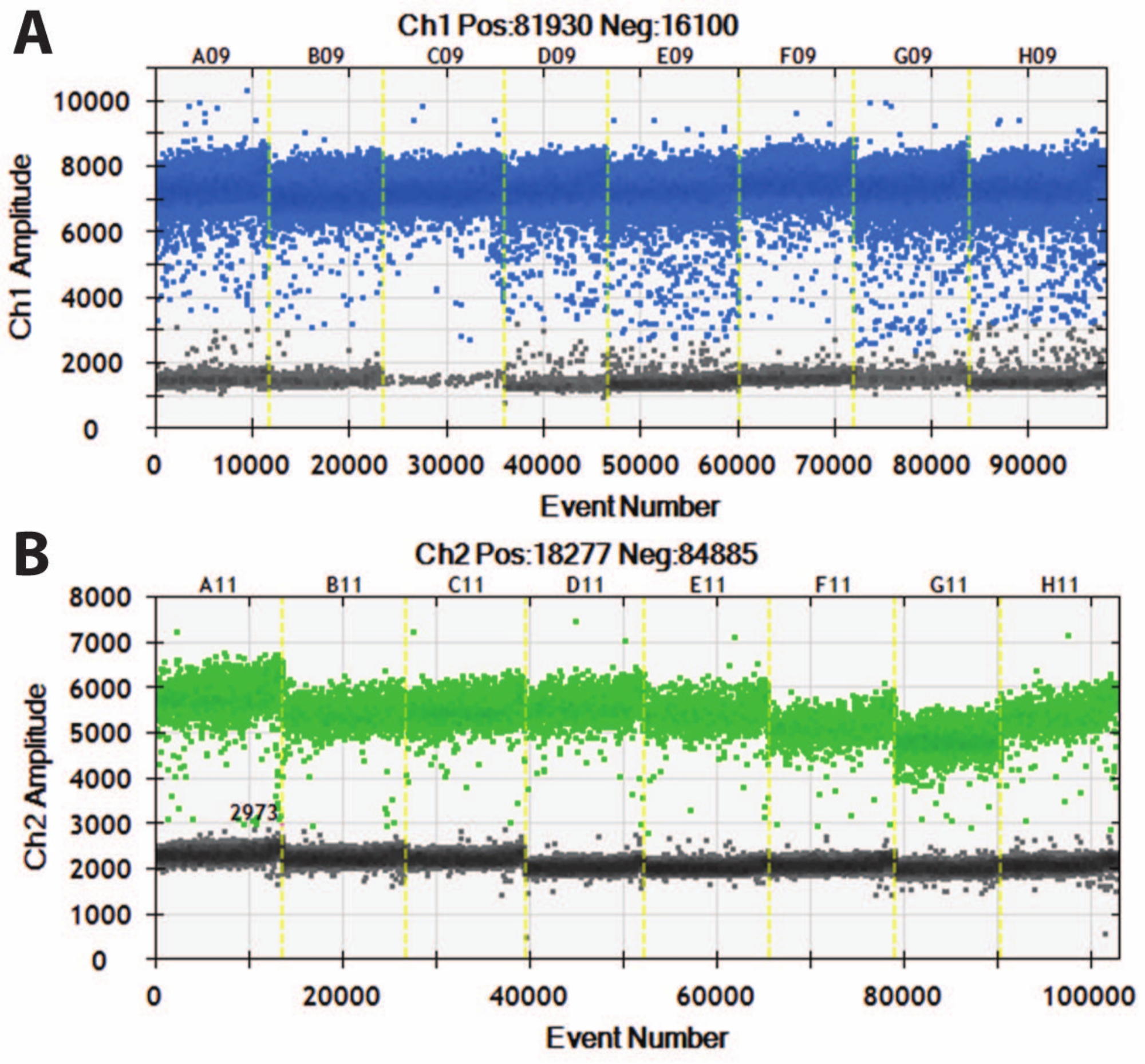
Figure 5: Digital droplet PCR of DIV120 organoids. Digital droplet PCR graphs showing the absolute expression value of (A) MAP2 (top, blue) and (B) NeuN (bottom, green). Slashed yellow lines separate different iPSC lines (A, B, C, etc.), and organoids have been batched together. N = 5. Please click here to view a larger version of this figure.
{kind=link}
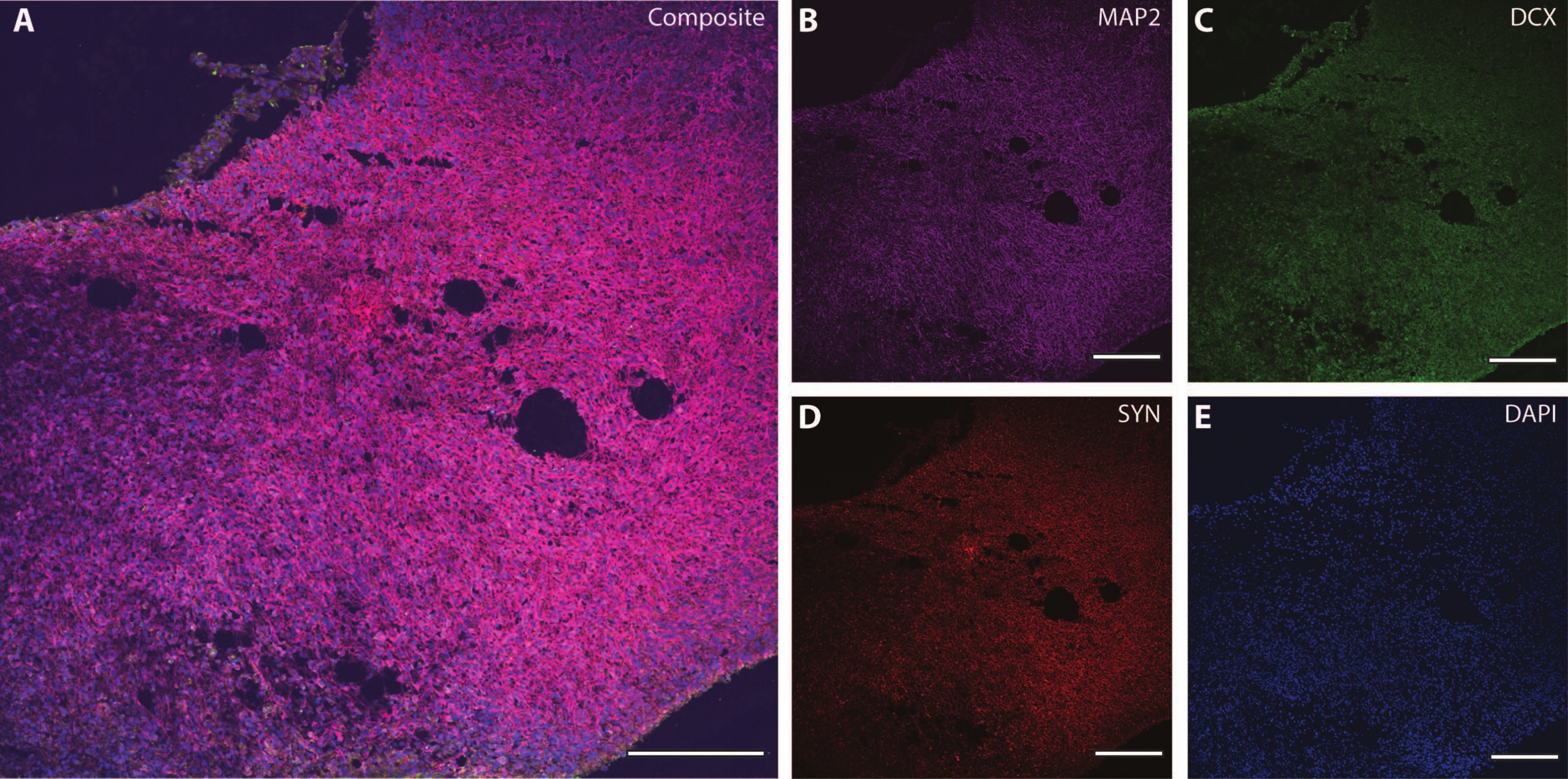
These cytoskeletal markers can be used in conjunction with other post-mitotic markers (like doublecortin27 and synapsin28) to probe for synaptic plasticity and other age-related declines (Figure 6), as well as additional brain tissue like astrocytes and glia (Figure 7).

Figure 6: Example of synaptic terminal analysis. Immunohistochemistry image of a section of the organoid at DIV 120. (A) A composite image of the section stained for (B) MAP2, (C) doublecortin (DCX), and (D) synapsin I (SYN). (E) Nuclei were visualized by DAPI. Scale bar = 200 µm. Please click here to view a larger version of this figure.
{kind=link}

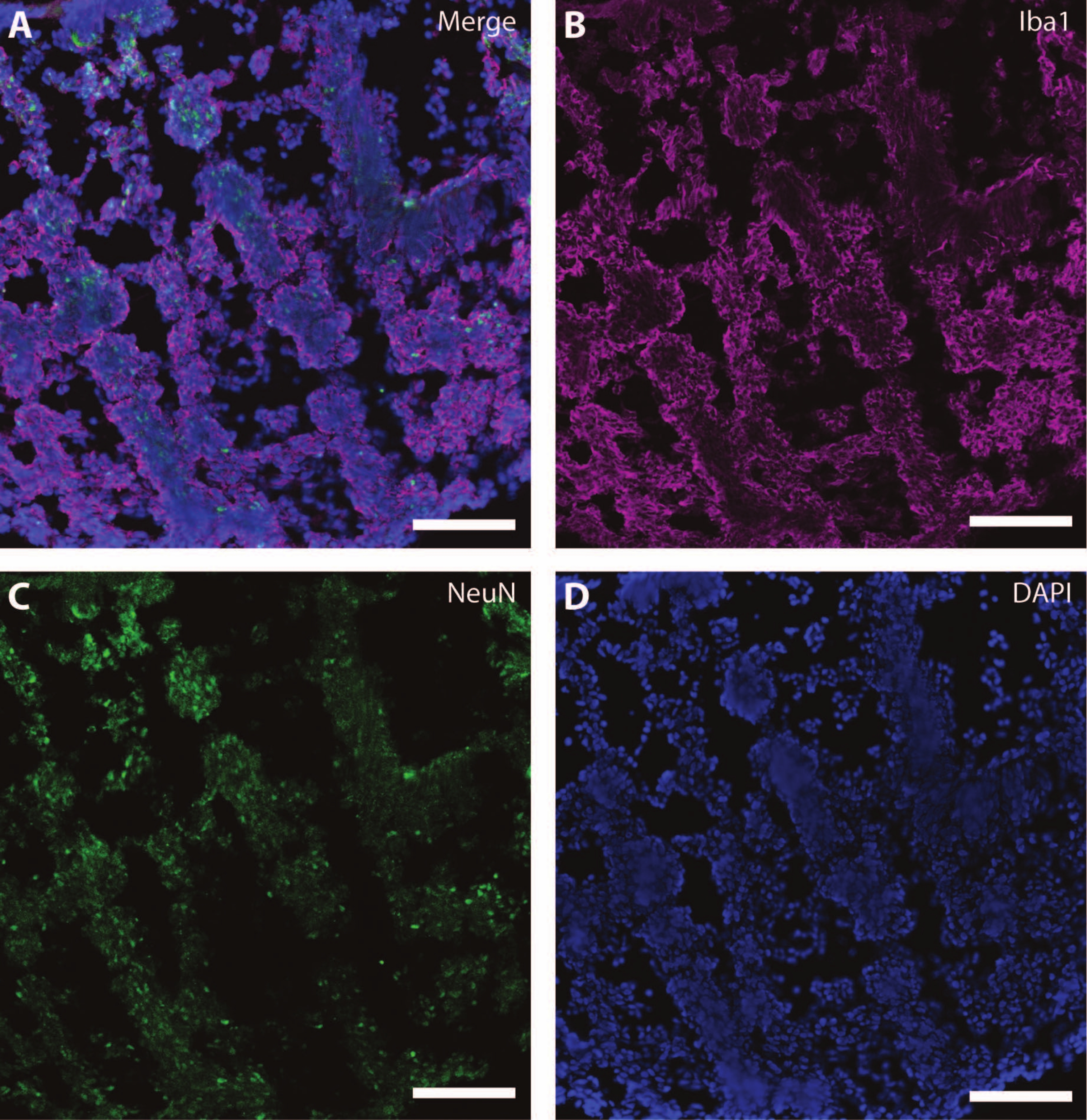
Figure 7: Evidence of glial development. Immunohistochemistry image of a section of the organoid at DIV 120. (A) A composite image of the section stained for (B) ionized calcium-binding adaptor molecule 1 (Iba1) and (C) NeuN. (D) Nuclei were visualized by DAPI. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Final Concentration | Volume (50 mL total) |
| DMEM-F12 | 50% | 25 mL |
| Neurobasal Medium | 50% | 25 mL |
| N2 Supplement (100x) | 1x | 0.25 mL |
| B27 Supplement -/+ Vitamin A (50x) | 0.5x | 0.5 mL |
| Insulin | 0.25% | 12.5 µL |
| GlutaMAX (100x) | 1x | 0.5 mL |
| MEM-NEAA (100x) | 0.5x | 0.25 mL |
| HEPES (1 M) | 10 mM | 0.5 mL |
| Antibiotic/Antimycotic (100x) | 1x | 0.5 mL |
| 2-β-mercaptoethanol | 50 µM | 17.5 µL |
| *NOTE: some DMEM-F12 already contains GlutaMAX, no need to add additional. | ||
Table 1: Composition of the differentiation media used in the present study.
Discussion
The standardized formation of EBs is a critical step in the reproducible conversion of pluripotent stem cells into brain organoids. The force aggregation of stem cells into singular tissues can vary depending on the geometry of the concave wells, seeding density, and well treatment. Although the current method cites a diameter range of 500-600 µm after 6 days, these diameters do not exclude other diameters of proper organoid formation, as many other diameters have proven successful. However, varying diameters have been shown to influence differentiation rates and success29. For reproducibility purposes, diameter variations below 50 µm are highly recommended20. Additionally, the number of neural induction days can be extended from 6 to 10 days to allow for the EBs to grow in diameter, as the use of SMAD inhibitors has been shown to efficiently produce and maintain neuroepithelia formation after only 5 days of exposure30. In the absence of SMAD inhibitors, neural induction can yield inconsistent results, requiring longer induction periods. SMAD inhibitors cited in this work are the most effective, but other dorsomorphin and TGF-β small molecules can be used at their effective concentration.
The basement membrane matrix provides an excellent substrate for growth and can be administered differently. Early uses of the matrix included the basal membrane as a well coating31. However, the use of the matrix for embedding tissue showed to improve differentiation and maturation in these tissues32. For organoids, the use of the matrix has been shown to improve EB differentiation into organoids, promote maturation, and extend culture periods33. While organoids can still be formed without the matrix, as many groups have strived to accomplish a matrix-free tissue culture34,35, studies have found that the matrix-embedded organoids have a higher chance of prolonged culture times and require less maintenance36. The dimple method of embedding provides equal coverage of the organoid surface, ensuring similar diffusion, access to nutrients, and reproducible differentiation. Alternatively, dome embedding can be employed to fully encapsulate the organoids37.
The transfer of the EBs into the membrane matrix dimples is a critical step. Although a 1 mL pipette tip has a wide enough opening to transfer the EBs, a 200 µL tip is preferred for ease of transfer. The 200 µL tips need to be cut to create a large enough opening, ensuring a smooth edge to reduce shear stress when pipetting. Alternatively, wide-bore 200 µL tips exist for ease of transfer. Pipetting needs to be done slowly, as fast pipetting might disrupt the organoid periphery and hinder proper growth. Special attention must be taken to ensure sufficient medium is transferred to provide nutrients. Too little medium runs the risk of drying out the organoid, causing necrosis. Transferring the EB with too much culture media can cause the matrix to be too diluted and fail to encapsulate the organoid securely. Ideally, the matrix must not be diluted by more than 50% to ensure its polymerization and function as an ECM for the EBs. If the embedding is unsuccessful, the EB can be recovered and re-encapsulated. Re-encapsulation in the fresh matrix can be done at any point to provide the organoid with additional support.
Similar to matrix embedding for scaffolding, PLGA fibers provide additional support for three-dimensional growth. Originally incorporated to produce elongated organoids and increase surface area38, the incorporation of PLGA fibers has progressively been identified as an additional tool to improve organoid differentiation and maturation39. As more labs look to reduce the use of the matrix or abolish it altogether, the self-organizing properties of organoids supported by the incorporated fibers provide enough scaffolding for three-dimensional tissue creation and differentiation39. Here, both methods were combined to increase the chances of long-term culture38,39. The incorporation of fibers during the initial aggregation is critical, as these may not be introduced at a later stage. After centrifugation, checking that a couple of fibers are among the aggregated cells will ensure that a fiber is incorporated in the EB. If not successful, a gentle aspiration of the well and re-centrifugation should ensure mixing.
Another critical step in organoid maintenance is the introduction of an orbital shaker for better media perfusion. In early iterations of organoid protocols, a spinning bioreactor was used to create agitation11. An orbital shaker at 90 rpm provides sufficient agitation without destroying the matrix droplet or damaging organoid morphology. Some groups refrain from using the matrix scaffold but retain shaker agitation to provide a suitable environment34. As with all protocols, the speed of the rotations must be adjusted depending on the shaker to reduce the amount of shear stress. If a dome embedding was chosen, a tilted shaker could be employed to reduce the amount of shear stress11,34.
Taken together, the integration of several selected techniques provides a robust method of iPSC-derived organoid formation. There are several ways of creating and maintaining organoids, but many of them focus on early differentiation trajectories. In this work, multiple different techniques were combined to culture organoids for extended periods of time, past the differentiation phase, and into a maturation period where aging phenotypes can start to develop. Incorporating these techniques allows for prolonged maturation without the need for exogenous biological factors to maintain the cultures, retaining the self-organization and natural progression of aging.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Netherlands Organ-on-Chip Initiative, an NWO Gravitation project (024.003.001) funded by the Ministry of Education, Culture and Science of the government of the Netherlands. D.C.B. thankfully acknowledges the financial support of Consejo Nacional de Ciencia y Tecnología (CONACyT) in the form of a doctoral scholarship.
Materials
| Name | Company | Catalog Number | Comments |
| 2-β-Mercaptoethanol | Thermo Fisher Scientific | 31350010 | |
| 4′,6-Diamidino-2-phenylindoledihydrochloride (DAPI) | Invitrogen | D1306 | 1:4000 |
| 6-well Clear Flat Bottom CELLSTAR Cell Culture Multiwell Plate | Greiner Bio-One | 657185 | |
| 6-well Clear Flat Bottom Ultra-Low Attachment Well Plate | Corning | 3471 | |
| 96-well Clear Round Bottom Ultra-Low Attachment Microplate | Corning | 7007 | |
| 96-Well, Nunclon Delta-Treated, Flat-Bottom Microplate | Thermo Fisher Scientific | 136101 | |
| Accutase | Sigma-Aldrich | 46964 | cell detachment solution |
| Antibiotic-Antimycotic (100x) | Gibco | 15240062 | |
| B27 Suppement (with Vitamin A) (50x) | Gibco | 17504044 | |
| B27 Supplement (minus Vitamin A) (50x) | Gibco | 12587010 | |
| BD Vacutainer™ Glass Mononuclear Cell Preparation (CPT) Tubes | Thermo Fisher Scientific | 02-685-125 | |
| Bovine Serum Albumin | Sigma-Aldrich | A9418 | |
| Centrifuge | Eppendorf | 5810 R | With plate holders |
| CHIR99021 | Selleck Chemicals | S2924 | |
| CytoTune Sendai Reprogramming Vector | Thermo Fisher Scientific | A1378001 | |
| ddPCR primers | human | MAPT | Bio-Rad | dHsaCPE192234 | |
| ddPCR primers | human | RBFOX3 (NeuN) | Bio-Rad | dHsaCPE5052108 | |
| DMEM/F12 | Thermo Fisher Scientific | 11320074 | |
| Doublecortin (DCX) | Santa Cruz Biotechnology | SC-8066 | 1:500 |
| Dulbecco’s phosphate buffered saline (DPBS) | Thermo Fisher Scientific | 14190144 | no calcium, no magnesium |
| Eppendorf cups, 1.5 mL | Eppendorf | 0030 125.215 | |
| Essential 6 | Gibco | A1516401 | |
| Essential 8 | Gibco | A1517001 | |
| Ethylenediaminetetraacetic acid (EDTA) | Invitrogen | 15575020 | |
| Falcon tubes, 15 mL, conical | Greiner Bio-One | 188271-N | |
| Formaldehyde | Sigma-Aldrich | 252549 | 37% Stock solution, diluted to 4% in PBS |
| Geltrex LDEV-Free Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher Scientific | A1413202 | |
| GlutaMax (100x) | Gibco | 35050038 | |
| Hemacytometer cell counter | Hausser scientific | 1490 | |
| HEPES Buffer | Thermo Fisher Scientific | 15-630-080 | |
| Insulin | Sigma-Aldrich | I9278 | |
| LDN 193189 | StemCell Technologies | 72147 | |
| MAP2 | Abcam | ab32454 | 1:200 |
| Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix, LDEV-free | Corning | 356230 | basement membrane matrix |
| MEM-Non Essential Amino Acid Solution (MEM-NEAA; 100x) | Thermo Fisher Scientific | 11140050 | |
| Multilabel Counter Victor 3 Plate Reader | Perkin Elmer | 1420 | luminometer |
| MycoAlert Mycoplasma Detection Kit | Lonza | LT07-318 | |
| N-2 Supplement (100x) | Thermo Fisher Scientific | 17502-048 | |
| NeuN | Millipore | MAB377 | 1:500 |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | |
| Oct-3/4 Antibody (C-10) Alexa Fluor 647 | Santa Cruz Biotechnology | sc-5279 AF647 | 1:100 |
| Parafilm | Bemis | PM-996 | thermoplastic film sheet |
| PAX6 | Thermo Fisher Scientific | 42-6600 | 1:200 |
| Penicillin/Streptomycin | Gibco | 15070063 | |
| Poly(lactic-co-glycolic acid) (PLGA) microfilaments | Ethicon | J463 | |
| QX200 Droplet digital PCR system | Bio-Rad | 1864001 | |
| ROCK inhibitor (Y27632) | Selleck Chemicals | S1049 | |
| SB431542 | R&D Systems | 1614/50 | |
| SOX2 Monoclonal Antibody (Btjce), Alexa Fluor 488, eBioscience | Invitrogen | 53-9811-80 | 1:100 |
| Synapsin I (SYN) | Calbiochem | 574777 | 1:200 |
| Triton-X 100 | Sigma-Aldrich | T8787 | |
| TUJ1 | Santa Cruz Biotechnology | sc-80005 | Beta-3-tubulin; 1:500 |
| XAV939 | Tocris Bioscience | 3748 |
References
- Liu, G. H., et al. Aging Atlas: a multi-omics database for aging biology. Nucleic Acids Research. 49, 825-830 (2021).
- Ferrucci, L., et al. Measuring biological aging in humans: A quest. Cell. 19 (2), 13080 (2020).
- Mertens, J., Reid, D., Lau, S., Kim, Y., Gage, F. H. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu Rev Genet. 52, 271-293 (2018).
- Mertens, J., et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer's patients. Cell Stem Cell. 28 (9), 1533-1548 (2021).
- Lin, Y. T., et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron. 98 (6), 1141-1154 (2018).
- Fang, E. F., et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nature Neuroscience. 22 (3), 401-412 (2019).
- Hu, J. L., Todhunter, M. E., LaBarge, M. A., Gartner, Z. J. Opportunities for organoids as new models of aging. Journal of Cell Biology. 217 (1), 39-50 (2018).
- Choi, S. H., et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 515 (7526), 274-278 (2014).
- Gonzalez, C., et al. Modeling amyloid beta and tau pathology in human cerebral organoids. Molecular Psychiatry. 23 (12), 2363-2374 (2018).
- Yoon, S. -. J., et al. Reliability of human cortical organoid generation. Nature Methods. 16 (1), 75-78 (2019).
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Muratore, C. R., Srikanth, P., Callahan, D. G., Young-Pearse, T. L. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS ONE. 9 (8), 105807 (2014).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2020).
- Goto-Silva, L., et al. Computational fluid dynamic analysis of physical forces playing a role in brain organoid cultures in two different multiplex platforms. BMC Developmental Biology. 19 (1), 1-10 (2019).
- Sommer, A. G., et al. Generation of human induced pluripotent stem cells from peripheral blood using the STEMCCA lentiviral vector. Journal of Visualized Experiments. (68), e4327 (2012).
- Beers, J., et al. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Scientific Reports. 5 (1), 1-9 (2015).
- Wakao, S., et al. Morphologic and gene expression criteria for identifying human induced pluripotent stem cells. PLoS ONE. 7 (12), e48677 (2012).
- Schlaeger, T. M., et al. A comparison of non-integrating reprogramming methods. Nature Biotechnology. 33 (1), 58-63 (2015).
- JoVE. Basic Methods in Cellular and Molecular Biology. Using a Hemacytometer to Count Cells. JoVE Science Education Database. , (2022).
- Choy Buentello, D., Koch, L. S., Trujillo-De Santiago, G., Alvarez, M. M., Broersen, K. Use of standard U-bottom and V-bottom well plates to generate neuroepithelial embryoid bodies. PLOS ONE. 17 (5), 0262062 (2022).
- Ellis, P., et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Developmental Neuroscience. 26 (2-4), 148-165 (2004).
- Lee, S., et al. TuJ1 (class III β-tubulin) expression suggests dynamic redistribution of follicular dendritic cells in lymphoid tissue. European Journal of Cell Biology. 84 (2-3), 453-459 (2005).
- Suzuki, S., Namiki, J., Shibata, S., Mastuzaki, Y., Okano, H. The neural stem/progenitor cell marker nestin is expressed in proliferative endothelial cells, but not in mature vasculature. Journal of Histochemistry and Cytochemistry. 58 (8), 721-730 (2010).
- Soltani, M. H., et al. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. The American Journal of Pathology. 166 (6), 1841-1850 (2005).
- Wilson, P. G., Stice, S. S. Development and differentiation of neural rosettes derived from human embryonic stem cells. Stem Cell Reviews. 2 (1), 67-77 (2006).
- Gusel'nikova, V. V., Korzhevskiy, D. E. NeuN as a neuronal nuclear antigen and neuron differentiation marker. Acta Naturae. 7 (25), 42-47 (2015).
- Rao, M. S., Hattiangady, B., Shetty, A. K. The window and mechanisms of major age-related decline in the production of new neurons within the dentate gyrus of the hippocampus. Aging Cell. 5 (6), 545-558 (2006).
- Meng, L., et al. A synapsin I cleavage fragment contributes to synaptic dysfunction in Alzheimer's disease. Aging Cell. 21 (5), 13619 (2022).
- Moon, S. H., et al. Optimizing human embryonic stem cells differentiation efficiency by screening size-tunable homogenous embryoid bodies. Biomaterials. 35 (23), 5987-5997 (2014).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Lee, S. -. W., et al. Optimization of Matrigel-based culture for expansion of neural stem cells. Animal Cells and Systems. 19 (3), 175-180 (2015).
- Zhang, J., et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: The matrix sandwich method. Circulation Research. 111 (9), 1125-1136 (2012).
- Hocevar, S. E., Liu, L., Duncan, R. K. Matrigel is required for efficient differentiation of isolated, stem cell-derived otic vesicles into inner ear organoids. Stem Cell Research. 53, 102295 (2021).
- Birey, F., et al. Assembly of functionally integrated human forebrain spheroids. Nature. 545 (7652), 54-59 (2017).
- Kozlowski, M. T., Crook, C. J., Ku, H. T. Towards organoid culture without Matrigel. Communications Biology. 4 (1), 1-15 (2021).
- Kaiser, A., Kale, A., Novozhilova, E., Olivius, P. The effects of Matrigel® on the survival and differentiation of a human neural progenitor dissociated sphere culture. The Anatomical Record. 303 (3), 441-450 (2020).
- Kakni, P., et al. Intestinal organoid culture in polymer film-based microwell arrays. Advanced Biosystems. 4 (10), 2000126 (2020).
- Lancaster, M. A., et al. Guided self-organization and cortical plate formation in human brain organoids. Nature Biotechnology. 35 (7), 659-666 (2017).
- Tejchman, A., Znój, A., Chlebanowska, P., Frączek-Szczypta, A., Majka, M. Carbon fibers as a new type of scaffold for midbrain organoid development. International Journal of Molecular Sciences. 21 (17), 5959 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved