
Summary
Abstract
Introduction
Protocol
Representative Results
Discussion
Acknowledgements
Materials
References
Immunology and Infection
Evaluación simultánea del parentesco, el número de división y el fenotipo mediante citometría de flujo para células madre y progenitoras hematopoyéticas
Aquí se presenta una técnica basada en citometría de flujo que permite medir simultáneamente el número de divisiones celulares, el fenotipo celular superficial y el parentesco celular. Esas propiedades se pueden probar estadísticamente utilizando un marco basado en permutaciones.
Pocas técnicas pueden evaluar el fenotipo y el destino de la misma célula simultáneamente. La mayoría de los protocolos actuales utilizados para caracterizar el fenotipo, aunque capaces de generar grandes conjuntos de datos, requieren la destrucción de la célula de interés, lo que hace imposible evaluar su destino funcional. Por lo tanto, los sistemas de diferenciación biológica heterogéneos como la hematopoyesis son difíciles de describir. Sobre la base de los tintes de seguimiento de la división celular, desarrollamos un protocolo para determinar simultáneamente el parentesco, el número de división y el estado de diferenciación para muchos progenitores hematopoyéticos individuales. Este protocolo permite evaluar el potencial de diferenciación ex vivo de progenitores hematopoyéticos murinos y humanos, aislados de diversas fuentes biológicas. Además, como se basa en citometría de flujo y un número limitado de reactivos, puede generar rápidamente una gran cantidad de datos, a nivel de una sola célula, de una manera relativamente económica. También proporcionamos la tubería analítica para el análisis de una sola célula, combinada con un marco estadístico robusto. Como este protocolo permite la vinculación de la división y diferenciación celular a nivel de una sola célula, se puede utilizar para evaluar cuantitativamente el compromiso de destino simétrico y asimétrico, el equilibrio entre la autorrenovación y la diferenciación, y el número de divisiones para un destino de compromiso dado. En conjunto, este protocolo se puede utilizar en diseños experimentales con el objetivo de desentrañar las diferencias biológicas entre los progenitores hematopoyéticos, desde una perspectiva unicelular.
La última década estuvo marcada por la difusión mundial de enfoques unicelulares para la biología celular y molecular. Siguiendo los pasos de la genómica unicelular1,2, hoy en día es posible estudiar muchos componentes de una sola célula (por ejemplo, ADN, ARN, proteínas), con nuevas técnicas de una sola célula que florecen cada año. Estas técnicas han arrojado luz sobre viejas y nuevas preguntas para los campos de la inmunología, la neurobiología, la oncología y otros, tanto utilizando células humanas como de organismos modelo3. Al resaltar las diferencias entre células individuales, las ómicas unicelulares impulsaron la definición de un nuevo modelo de hematopoyesis, centrado en la heterogeneidad de las células madre y progenitoras hematopoyéticas (HSPC) y alejándose del modelo clásico de poblaciones homogéneas discretas 4,5.
Uno de los pocos inconvenientes de todas las técnicas -ómicas es la destrucción de la célula de interés, lo que impide la posibilidad de evaluar su funcionalidad. Por el contrario, otros métodos unicelulares, como el ensayo de trasplante de células individuales y las tecnologías de rastreo de linaje, proporcionan una lectura de la funcionalidad de la célula ancestra al evaluar el destino de células individuales in vivo 6,7. Las tecnologías de rastreo de linaje implican etiquetar la célula de interés con un7 genético hereditario o una etiqueta fluorescente8,9, lo que permite seguir el destino de múltiples células individuales al mismo tiempo. Sin embargo, la caracterización de las células iniciales se limita típicamente a un número restringido de parámetros, como la expresión de unas pocas proteínas de superficie evaluadas por citometría de flujo10. Además, las tecnologías de rastreo de linaje de una sola célula requieren una detección laboriosa de la etiqueta celular, generalmente a través de secuenciación de ADN / ARN o imágenes. Este último punto en particular limita el número de condiciones que se pueden probar en un solo experimento.
Otra clase de métodos que se utilizan para estudiar la funcionalidad de células individuales son los sistemas de cultivo celular ex vivo de HSPC individuales. Fáciles de realizar, estos ensayos estándar de oro implican la clasificación de células individuales en recipientes de cultivo celular de 96 pocillos y, después del cultivo, caracterizando el fenotipo de la progenie celular, generalmente por citometría de flujo o análisis morfológico. Estos ensayos se han utilizado principalmente para caracterizar la diferenciación a largo plazo de las CPHS en células maduras, típicamente después de 2-3 semanas de cultivo11,12. Alternativamente, se han utilizado para tratar de mantener y expandir las HSPC ex vivo 13,14,15,16,17,18, con la promesa de un beneficio médico para el trasplante de células madre humanas 19. Por último, se han utilizado para estudiar el compromiso temprano de las HSPCs mediante cultivo a corto plazo20, siendo el bajo número de células generadas en este cultivo el principal factor limitante. Un inconveniente de estos diferentes tipos de ensayos ex vivo es que solo reflejan parcialmente la complejidad in vivo; aún así, son una de las raras formas de estudiar la diferenciación de HSPC humana.
Una pieza faltante de información de los métodos unicelulares existentes (ómicas de células individuales, rastreo de linaje y cultivo ex vivo) es la detección precisa de divisiones celulares, un parámetro esencial a considerar cuando se estudia la dinámica de HSPC21. Una forma sencilla de evaluar el número de divisiones a través de la citometría de flujo es el uso de "colorantes proteicos" solubles, como el éster succinimidílico de diacetato de 5-(y 6)-carboxifluoresceína (CFSE)22. Estos colorantes de división se difunden dentro del citoplasma de las células teñidas, y se diluyen a la mitad y pasan a las dos células hijas en cada división celular, lo que permite enumerar hasta 10 divisiones. Combinando varios tintes de división, es posible sembrar múltiples progenitores individuales en el mismo pozo, ya que cada tinte individual permite la separación de los diferentes descendientes. Este es el principio detrás del uso de colorantes celulares para el seguimiento clonal y de división múltiple que se introdujo por primera vez para los linfocitos murinos23,24.
Aquí, presentamos el desarrollo del ensayo MultiGen para su uso con HSPC murinos y humanos. Permite probar muchas células individuales simultáneamente por sus propiedades de diferenciación, división y parentesco ex vivo. Este ensayo de alto rendimiento, fácil de realizar y económico permite medir el fenotipo celular, el número de divisiones realizadas y el parentesco celular y la relación clonal con las otras células en el pozo, todo al mismo tiempo. Se puede utilizar para evaluar cuantitativamente el compromiso de destino simétrico y asimétrico, el equilibrio entre la autorrenovación y la diferenciación, y el número de divisiones necesarias para un destino de compromiso dado. El protocolo requiere un clasificador de células activadas por fluorescencia (FACS) y un citómetro de flujo con un lector de placas, además del equipo necesario para realizar el cultivo celular. Además del protocolo técnico para la ejecución del ensayo en HSPC humanas, también proporcionamos el marco de análisis detallado, incluyendo las pruebas estadísticas necesarias para evaluar las propiedades celulares relacionadas con el concepto de familia celular25. Este protocolo ya ha sido utilizado con éxito para describir el compartimento murinoHSPC 26,27.
El siguiente protocolo utiliza células CD34+ enriquecidas magnéticamente como material de partida28. De esta manera, es posible teñir y aislar eficientemente las HSPC humanas de diferentes fuentes de sangre (por ejemplo, sangre del cordón umbilical, médula ósea, sangre periférica). Es importante no descartar la fracción CD34- , ya que se utilizará como parte del protocolo para establecer diferentes tipos de controles experimentales. Las cantidades y volúmenes de celdas mencionados se pueden ampliar o reducir, de acuerdo con el flujo de trabajo y las necesidades experimentales. Del mismo modo, el protocolo se puede adaptar al estudio de diferentes tipos de progenitores, simplemente modificando los anticuerpos utilizados para los pasos de clasificación celular y citometría de flujo.
Para el siguiente protocolo, se utilizó sangre de cordón umbilical no identificada como fuente de HSPC y se recolectó de acuerdo con las pautas definidas por el biobanco de sangre del cordón umbilical del Hospital Saint-Louis (autorización AC-2016-2759) y con la Declaración de Helsinki.
NOTA: Antes de comenzar, asegúrese de que todos los reactivos y equipos necesarios para este protocolo estén disponibles, como se indica en la Tabla de materiales y se menciona en el protocolo. Prepare los reactivos relevantes frescos y no los almacene, a menos que se mencione explícitamente.
1. Tinción de colorante celular
NOTA: Esta sección describe la tinción con cuatro combinaciones de colorantes de división celular CFSE y colorante violeta (CTV). Procese todos los tubos simultáneamente, incluso si no se agrega solución de colorante celular. Todos los pasos se realizan en condiciones estériles para permitir el siguiente paso de cultivo celular. Tiempo requerido: aproximadamente 100 min.
- Procesar la unidad de sangre del cordón umbilical de acuerdo con un protocolo de clasificación magnética29. Asegúrese de que haya dos fracciones disponibles: una fracción CD34- grande y una fracción CD34+ más pequeña. Gire ambos tubos durante 5 minutos a 300 x g. Aspirar el sobrenadante sin alterar la bolita.
- Para la fracción CD34+ , resuspenderla en 1 ml del medio Eagle modificado de Dulbecco (DMEM) sin suero fetal bovino (FBS). Contar las células usando un hemocitómetro; la densidad celular no debe ser superior a 3 x 106 células/ml. Si este es el caso, adapte el volumen en consecuencia. Para la fracción CD34- , resuspender en DMEM sin FBS y ajustar el volumen a un máximo de 6 x 106 células/ml.
- Alícuota 250 μL de la fracción CD34+ en cuatro tubos de polipropileno de 15 ml. Etiquete los tubos de la siguiente manera: CD34+/CF (CFSE_only), CD34+/CV (CFSE_high CTV_low), CD34+/VC (CFSE_low CTV_high) y CD34+/VI (CTV_high). Alícuota 250 μL de la fracción CD34- en otros cuatro tubos de polipropileno de 15 ml. Etiquete los tubos de la siguiente manera: CD34-/CF (CFSE_only), CD34-/CV (CFSE_high CTV_low), CD34-/VC (CFSE_low CTV_high) y CD34-/VI (CTV_high). Las células restantes de la fracción CD34- pueden ser descartadas.
- Prepare dos soluciones CFSE, denominadas CFSE_high y CFSE_low. Para CFSE_high (10 μM), mezclar 1,1 ml de DMEM sin FBS con 2,2 μL de solución de CFSE (5 mM). Para CFSE_low (5 μM), mezclar 550 μL de DMEM sin FBS y 0,55 μL de solución de CFSE (5 mM).
- Añadir 250 μL de la solución CFSE_high a los tubos CF y CV, 250 μL de la solución CFSE_low a los tubos VC y 250 μL de DMEM sin FBS al tubo VI. Para garantizar una mezcla eficiente de suspensión celular y tinte celular, incline el tubo casi 90 grados y deposite las soluciones CFSE en la pared del tubo. Luego, sostenga el tubo verticalmente para mezclar las dos soluciones y pipete tres o cuatro veces para garantizar una mezcla rápida de las soluciones CFSE con las células resuspendidas. Incubar a 37 °C durante exactamente 8 min.
- Después de la incubación, agregue 5 mL de DMEM + 10% FBS. Mantener los tubos a 37 °C durante 5 min.
- Gire los tubos durante 5 minutos a 300 x g. Retire el sobrenadante por aspiración sin alterar el pellet y lave el pellet con 5 ml de solución salina tamponada con fosfato 1x/ácido etilendiaminotetraacético (PBS 1x/EDTA). Girar de nuevo durante 5 minutos a 300 x g. Desechar el sobrenadante sin alterar el pellet y resuspender el pellet celular en 250 μL de 1x PBS /EDTA.
- Prepare dos soluciones CTV, denominadas CTV_high y CTV_low. Para CTV_high (10 μM), mezclar 1,1 ml de PBS 1x/EDTA y 2,2 μL de CTV (5 mM). Para CTV_low (5 μM), mezclar 550 μL de PBS 1x/EDTA con 0,55 μL de material CTV (5 mM).
- Agregue 250 μL de la solución CTV_high a los tubos VC y VI, 250 μL de la solución CTV_low al tubo CV y 250 μL de 1x PBS / EDTA al tubo CF. Utilice la misma técnica que se describe en el paso 1.5. Incubar a 37 °C durante exactamente 8 min.
- Después de la incubación, agregue 5 mL de DMEM + 10% FBS. Conservar a 37 °C durante 5 min.
- Gire los tubos durante 5 min a 300 x g, deseche el sobrenadante sin alterar el pellet y luego lave el pellet con 5 ml de 1x PBS / EDTA. Girar de nuevo durante 5 minutos a 300 x g.
- Desechar el sobrenadante sin alterar el pellet y resuspender las fracciones CD34- en 1x PBS/EDTA para una concentración final de 1,5 x 106 células/ml. Resuspender las fracciones CD34+ en 40 μL de tampón de tinción y transferir las células a tubos de 1,5 ml.
2. Tinción de anticuerpos
NOTA: La tinción de anticuerpos se puede personalizar de acuerdo con las necesidades experimentales. Solo las fracciones CD34+ se someten a tinción de anticuerpos; las fracciones CD34- se utilizan como un único control de tinción para las combinaciones de colorantes de división celular (fracciones CV, VC, CF y VI). El siguiente panel está diseñado para la detección de cuatro tipos de CPH: células madre hematopoyéticas (HSC), progenitores multipotentes (MPP), progenitores multipotentes preparados para linfoides (LMPP) y células progenitoras hematopoyéticas (HPC)12. Sin embargo, se presenta la identificación de HSC y MPP. Tiempo requerido: 75 min.
- Prepare la tinción única para la tinción superficial, utilizando perlas de compensación. Mezcle las perlas negativas y las perlas de inmunoglobulina G (IgG) en una proporción de 1:1, para obtener un volumen total equivalente a 20 μL x el número de marcadores de superficie (por ejemplo, 120 μL si el panel de tinción contiene seis anticuerpos).
- Enviar 20 μL de perlas en tubos individuales de 1,5 ml para cada marcador. Añadir el volumen correspondiente al factor de dilución para cada anticuerpo en el tubo correspondiente (por ejemplo, si el factor de dilución es 1:20, añadir 1 μL).
- Para teñir las células CD34+ , prepare una mezcla maestra de anticuerpos12, basada en la Tabla 1. Mezcle los anticuerpos en un solo tubo de 0,5 ml. Agregue 7 μL de la mezcla maestra de anticuerpos a cada una de las cuatro condiciones CD34+.
- Incubar las perlas de compensación y las muestras CD34+ a 4° C durante al menos 30 min.
NOTA: El tiempo de incubación debe adaptarse a los detalles técnicos de los anticuerpos utilizados para la tinción. - Durante la incubación, preparar la placa de fondo redondo de 96 pocillos que se utilizará para la clasificación, añadiendo 100 μL de medio de cultivo celular a cada pocillo utilizando una pipeta multicanal.
NOTA: Deje vacíos los pozos H8-H12. - Etiquete los tubos de polipropileno de 5 ml para los controles de tinción superficial (5, usando perlas), los controles de colorantes de división celular (4, usando las fracciones CD34- ) y las muestras CD34+ (4).
- Al final de la incubación, lave las células y las perlas con 1 ml de tampón de tinción. Transfiera el volumen total a los tubos de polipropileno de 5 ml. Centrifugar los tubos durante 5 minutos a 300 x g, luego aspirar el sobrenadante sin alterar el pellet.
- Resuspender las células en tampón de tinción, utilizando aproximadamente 500 μL cada una para las perlas y las células CD34+ , y 1 ml para los tubos CD34- .
Tabla 1: Plantilla para preparar la mezcla maestra de anticuerpos para un experimento de clasificación celular. Haga clic aquí para descargar esta tabla.
3. Clasificación celular
NOTA: Los números de celda ordenados pueden variar según la cantidad total de celdas disponibles. En el protocolo, se proporciona un número de celda mínimo para cada control. Tiempo requerido (para una sola placa): 100 min.
- Abra el experimento de plantilla o establezca un nuevo experimento. Cree una sola muestra y varios tubos, uno para cada condición.
- Establezca la estrategia de acceso detallada en la Figura 1, creando seis diagramas de diagramas de puntos. Primero, visualice las celdas en un diagrama de puntos FSC-A / SSC-A y haga doble clic en la herramienta de activación de polígonos para seleccionar una población con dispersión lateral baja (Figura 1A). En el siguiente diagrama de puntos (FSC-A / FSC-H), haga clic derecho en el gráfico y seleccione la puerta "Celdas" en el menú desplegable haciendo clic en ella. Utilice la misma herramienta de compuerta para seleccionar una población estrecha en la diagonal entre los dos ejes (Figura 1B).
- En el tercer diagrama de puntos (APC vs. FSH-H), muestre la población "Single Cells" y compuerta las celdas negativas para la expresión de APC Lineage (Lin) (Figura 1C). En la cuarta gráfica (CFSE vs. CTV), muestre la población "Lin-" y cree cuatro puertas separadas, una para cada combinación de tinte (Figura 1D).
NOTA: Estas puertas deben ser herméticas, para seleccionar solo una pequeña fracción de celdas teñidas homogéneamente. - Utilice la quinta y sexta gráfica (APC-Cy7 vs. BV650 y PE-Cy7 vs. PE) para identificar los progenitores de interés. Abra generosamente la población CD34+CD38- y la CD34+CD38+ en la quinta gráfica (Figura 1E). Luego, seleccione la población CD34+CD38- en la sexta gráfica y dibuje tres puertas, de acuerdo con la Figura 1F.
- Ejecute los tubos de tinción individuales que contienen las perlas de compensación, haciendo clic en el botón Adquirir . Ajuste los voltajes del tubo fotomultiplicador (PMT) desde el menú desplegable Parámetros , particularmente para los colorantes de división celular (entre 104 y 105 en una escala biexponencial).
- Refine la matriz de compensación de acuerdo con el panel utilizado para la clasificación, utilizando la pestaña Compensación . Registre al menos 5,000 eventos en la puerta de cuentas, haciendo clic en el botón Grabar .
- Ejecute las fracciones CD34- y vuelva a comprobar la matriz de compensación. Registre al menos 10,000 eventos en la puerta de celda única.
- Ejecute las fracciones CD34+ , registrando al menos 5.000 eventos en la puerta de celda única. Ajuste la puerta para cada combinación de tinte, estableciendo una puerta apretada para seleccionar una población homogénea (Figura 1D). Del mismo modo, ajuste la puerta para seleccionar HSC y MPP.
- Una vez que se complete el análisis y se registren todos los tubos, inserte la placa en el soporte apropiado, después de realizar la calibración estándar de Aria para clasificar en placas de 96 pocillos. Se recomienda enfriar la placa.
- Prepare la plantilla de clasificación de placas de acuerdo con el esquema presentado en la Tabla 2, utilizando el diseño de clasificación del experimento. Los pozos llamados "CD34-" contienen 5.000-10.000 células, clasificadas en la puerta CF/CV/VC/VI. Los pocillos "a granel" contienen al menos 500 células, clasificadas en la puerta CD34+CD38-. Finalmente, los pocillos unicelulares contienen solo un evento por combinación de colorante de división celular por pozo, por lo que cuatro eventos por pozo en total.
NOTA: Las poblaciones "a granel" se pueden adaptar al subconjunto específico de progenitores; No clasifique menos de 500 celdas. - Para la clasificación, proceda en orden, completando cada combinación de colorante de división celular antes de pasar a la siguiente. Por ejemplo, comience con la clasificación del CD34-CF, en modo de pureza de rendimiento. Haga clic en el botón de adquirir, luego en el botón de ordenación.
- Al final de la clasificación CD34- , inserte el tubo CF CD34+ . Adquiera, luego haga clic en el botón de ordenar, asegurándose de haber marcado 0/ 16/0 como grado de pureza. Finalmente, ordene las celdas de interés, una celda por pocillo, en pureza de celda única , asegurándose de marcar la opción Clasificación de índice .
- Pase a la siguiente combinación de colorante de división celular, repitiendo el mismo orden. Como referencia, la Tabla 2 proporciona un ejemplo de una placa clasificada.
NOTA: La función de ordenación de índices genera archivos individuales para cada condición ordenada. - Al final de la ordenación, exporte los archivos como archivos .fcs 3.0. Poner las células en una incubadora de 37 °C, 5% CO2 . Las células se cultivan durante varios días, de acuerdo con el diseño experimental, durante al menos 24 h26.
Tabla 2: Plantilla para una placa de 96 pocillos de clasificación celular, basada en los requisitos específicos para el análisis sucesivo de citometría de flujo. Haga clic aquí para descargar esta tabla.
4. Análisis de datos de clasificación celular
NOTA: Para validar la calidad de la clasificación celular, es necesario el análisis de los datos del sistema de control de los bienes sobre el terreno antes de continuar. El resultado principal de este paso es la generación de una hoja de cálculo que contiene las intensidades de marcador de cada celda individual ordenada.
- Cargue los archivos .fcs 3.0 en el software de análisis.
- Verifique la configuración de compensación utilizada durante la clasificación de celdas, utilizando los archivos de tinción individuales registrados antes de la clasificación real.
- Establezca la estrategia de acceso revisada utilizando los archivos correspondientes a los diferentes bultos. Copie y pegue esas puertas en los archivos de clasificación de índice.
- Compruebe que las celdas ordenadas del índice cayeron en la puerta establecida. Si hay algunas celdas ordenadas que se cerraron incorrectamente, se pueden identificar exportando las coordenadas de la placa registradas durante la clasificación del índice y eliminarlas más adelante en el análisis.
- Exporte el evento desde los archivos de clasificación de índice como parámetros compensados. Expórtelos como archivos .csv, marcando las opciones "valores de escala" y "parámetros compensados". Estos archivos deben exportarse en una carpeta llamada "Archivos exportados".
- Combine todos los archivos en un único archivo .csv, utilizando el script del archivo complementario 1. Establezca la ruta correcta con la función "setwd". El resultado de este script es una hoja de cálculo que contiene todos los eventos cerrados de forma diferente y las intensidades relativas para todos los parámetros.
5. Tinción de anticuerpos después del cultivo
NOTA: Realice esta parte del protocolo en condiciones estériles; Varios reactivos se comparten con los pasos anteriores, y necesitan permanecer estériles. Para el análisis de citometría de flujo, utilice un citómetro de flujo con un lector de placas. Esto permite realizar la tinción directamente en la placa de cultivo de tejidos, reduciendo al mínimo la pérdida celular al limitar la cantidad de pipeteo y centrifugado. Prepare la tinción monocolor del marcador de superficie utilizando las perlas de compensación, excepto los pocillos A1-A4, que representan la tinción única para los colores CF/CV/VC/VI y ya están presentes en la placa de 96 pocillos. Las poblaciones a granel ordenadas según el colorante celular ayudan a establecer la estrategia de compuerta para el número de divisiones y el gating general. Tiempo requerido: 120 min.
- Antes de comenzar el protocolo, marque los pocillos que contienen al menos una celda revisando la placa bajo un microscopio invertido. Este paso permite optimizar la cantidad de anticuerpos utilizados para la tinción y acelera el procedimiento.
- Preparar la mezcla maestra de anticuerpos, de acuerdo con la Tabla 3. Como hay una cantidad significativa de pipeteo, la tabla considera el error técnico debido al pipeteo, incluido un volumen adicional del 5%. Los anticuerpos descritos en la tabla permiten caracterizar una gama de CPHS a partir de muestras de sangre del cordón umbilical humano12.
- Centrifugar la placa durante 5 min a 300 x g. Invierta rápidamente la placa debajo de la capucha y sobre una toalla de papel, para quitar el sobrenadante.
- Agregue 8 μL de tampón de tinción a los pocillos A1-A4. Agregue 8 μL de la mezcla a los otros pocillos.
- Mezcle las perlas negativas y las perlas de compensación de IgG en una proporción de 1:1, para obtener un volumen total equivalente a 120 μL. Envíe 20 μL en un tubo de 1,5 ml por cada marcador. Añadir el volumen de anticuerpos correspondiente al factor de dilución (por ejemplo, si el factor de dilución es 1:20, añadir 1 μL).
NOTA: Adapte el volumen total al número de marcadores utilizados para la tinción (por ejemplo, 100 μL si el panel de tinción contiene cinco anticuerpos). - Incubar la placa y los controles de compensación de tinción simple a +4 °C, durante al menos 30 min.
NOTA: El tiempo de incubación debe adaptarse a los detalles técnicos de los anticuerpos utilizados para la tinción. - Lave las perlas con 1 ml de tampón de tinción. Transfiera el volumen total a los tubos de polipropileno de 5 ml previamente etiquetados. Centrifugar los tubos durante 5 minutos a 300 x g, luego retirar el sobrenadante por aspiración.
- Lave las células de la placa añadiendo 100 μL de tampón de tinción por pocillo con una pipeta multicanal. Centrifugar la placa a 300 x g durante 5 minutos, luego invertir rápidamente la placa debajo de la capucha y sobre una toalla de papel, para eliminar el sobrenadante.
- Vuelva a suspender las células en 85 μL de tampón de tinción, utilizando una pipeta multicanal.
- Inicie el análisis en el citómetro de flujo (modo de adquisición), utilizando la plantilla dedicada y haciendo clic en personalizado. Esta plantilla personalizada tiene en cuenta las características técnicas de la placa de fondo redondo de 96 pocillos, en particular las dimensiones de cada pozo (diámetro, profundidad y espesor). La sonda debe llegar al fondo del pozo, así que colóquela en el centro exacto de los pozos A1 y H12.
- Después de seleccionar los fluoróforos de interés de la lista propuesta por el software, establezca la configuración de la placa siguiendo la plantilla de placa de la Tabla 2, corregida por el número de pocillos que contienen al menos una celda.
- Seleccione 100 μL como límite de volumen de adquisición. Marque la opción de agitación . Establezca la tasa de adquisición en 1 μL / s como máximo, ya que una velocidad más baja mejora el volumen total analizado por pozo.
- Agregue las soluciones de limpieza y lavado adecuadas a los pozos H8-H12. La plantilla en la Tabla 2 deja específicamente los pozos H8-H12 vacíos, ya que el citómetro de flujo necesita ejecutar una variedad de condiciones de lavado al final del análisis.
NOTA: Este paso se adapta a las características específicas del citómetro de flujo utilizado. - En la sección de gráficos y puertas, primero establezca la puerta de celda única, utilizando el diagrama de dispersión FSC-A/SSC-A y luego el diagrama de dispersión FSC-H/FSC-A. Cree un histograma para cada marcador de interés.
- Una vez confirmada la configuración, proceda a la sección Análisis . Analice primero la tinción única, registrando no menos de 5,000 eventos (rango óptimo: 5,000-15,000 eventos), tanto para las perlas de compensación como para las fracciones teñidas con CD34. Ajuste los voltajes si es necesario.
- Una vez que se registran todas las tinciones individuales, es posible iniciar la adquisición real, haciendo clic en la función Adquisición .
Tabla 3: Mezcla maestra de anticuerpos para un experimento de citometría de flujo, específicamente para la identificación de HSPC a partir de sangre del cordón umbilical humano. Haga clic aquí para descargar esta tabla.
6. Análisis de datos de citometría de flujo post-cultivo
NOTA: El análisis de datos descrito es específico para el software mencionado en la Tabla de materiales. El resultado principal es la generación de una hoja de cálculo que contiene información sobre la intensidad del marcador de superficie, el número de divisiones y el parentesco por cada celda analizada. En esta parte del protocolo se incluye un script escrito en R, necesario para este flujo de trabajo para generar la hoja de cálculo de análisis final.
- Exporte archivos desde el citómetro de flujo como archivos .fcs. Subirlos al software de análisis, agrupándolos como "tinción única", "a granel" y "celda única".
- Prepare una matriz de compensación utilizando los archivos de tinción individuales y aplíquela a los otros dos grupos arrastrando y soltando.
NOTA: Si se utiliza una herramienta de compensación automática, compruebe la calidad a mano antes de continuar. - Para tener un gating representativo, concatenar los diferentes pozos a granel en un solo archivo. Este paso resalta rápidamente si dos colores se superponen (típicamente CV y VC) u otras anomalías y, por lo tanto, deben excluirse. Después de hacer clic en la opción concatenar poblaciones , seleccione todos los parámetros no compensados en el menú "parámetros", luego haga clic en concatenar.
- Cargue el archivo concatenado en el espacio de trabajo y, a continuación, aplique la matriz de compensación mediante arrastrar y soltar.
- Prepare la estrategia de acceso definida en la figura 2 utilizando el archivo concatenado. En la puerta de celda única, muestre los eventos en un diagrama de dispersión con CFSE y CTV. Cree una primera puerta llamada Labeled, que incluya los cuatro colores y excluya la posible autofluorescencia (Figura 2C). Luego, puerta cada color individualmente.
- Las celdas etiquetadas con CV y VC necesitan un valor transformado, considerando que el color es el resultado de las señales CFSE y CTV. Por lo tanto, las dos señales coordinadas se giran en una escala logarítmica de 45 °, para permitir que la dilución por división proceda en paralelo al eje x. Este valor transformado se deriva manualmente, haciendo clic en Herramientas y luego en Derivar parámetro. Pegue la siguiente fórmula en el cuadro de fórmula :

NOTA: La ecuación26 asume que CFSE y CTV son los parámetros 03 y 17. - Para visualizar correctamente este nuevo parámetro denominado Parámetro derivado, establezca un eje lineal que oscile entre ~3-7, haga clic en la opción Parámetro Eje y seleccione Personalizar eje.
- Aplique el gatillo a cada color individualmente como un diagrama de histograma: para CF y VI, establezca CFSE-A y CTV-A en el eje x, respectivamente. Para CV y VC, establezca el parámetro recién derivado en el eje x. Establezca las puertas correspondientes a cada pico, como se muestra en la Figura 3.
- Aplique el gating a cada pozo individual de una sola célula. Asegúrese de agregar el parámetro derivado a cada pozo analizado. Verifique manualmente cada puerta de color para cada pozo, para detectar eventos que se asignan incorrectamente a un pico determinado. En la figura 4 se muestran ejemplos de compuertas.
- Una vez completado el análisis y verificados todos los pozos, seleccione todas las compuertas CF/CV/VC/VI que contengan al menos una celda. Expórtelos como archivos .csv, marcando las opciones "valores de escala" y "parámetros compensados". Estos archivos se exportan en una carpeta denominada "Archivos exportados".
- Combine todos los archivos en un solo .csv, utilizando el script R en Archivo complementario 1. Recuerde establecer la ruta correcta con la función "setwd". El resultado de este script es una hoja de cálculo que contiene todos los diferentes eventos cerrados y las intensidades relativas para todos los parámetros.
- Abra la hoja de cálculo y cambie el nombre de las columnas para cada parámetro, por ejemplo, utilizando los siguientes nombres: CFSE, CTV, CD90, CD123, CD45RA, CD34, CD38. Estos nombres se utilizarán para identificar el umbral de acceso para asignar correctamente a cada celda su identidad.
- Agregue seis columnas llamadas "Bueno", "Condición", "Color", "Generación", "Original_cell" y "Culture_time". Estas variables son las definidas experimentalmente y se infieren de cada fila:
export_A10 CD34 + PBS_CV_Peak 1.csv.1 = A10 (pozo), CD34+ (Original_cell), PBS (condición), CV (color), Peak_1 (generación). - Exporte los pozos a granel para identificar los valores umbral para el gating: exporte la población compensada de interés (por ejemplo, CD34 + CD38-) como archivos .csv, marcando las opciones "valores de escala" y "parámetros compensados". Exporte estos archivos en una carpeta llamada "Archivos exportados".
- Para encontrar el umbral para CD38, identifique el mayor valor numérico para este parámetro. Por el contrario, para encontrar el umbral para CD34, identifique el valor numérico más pequeño para este parámetro. Repita este proceso para todos los parámetros de interés.
NOTA: Para el análisis presentado en el protocolo, el marcador CD45RA se utiliza tanto para identificar LMPPs en la puerta CD34+CD38- como CMP/GMP en la puerta CD34+CD38+. Esto significa que es necesario extraer dos valores umbral diferentes para este marcador. - Copie y pegue los valores de umbral en un archivo de Excel llamado "gating_matrix". Este archivo está organizado de acuerdo con la Tabla 4, y permite el análisis de múltiples experimentos independientes. Es muy importante nombrar cada columna exactamente con este esquema: XXYYMMDD_xxh, donde XX representa las dos iniciales del operador, YY los dos últimos números para el año, MM para el mes, DD para el día y xx para el punto de tiempo de análisis.
Tabla 4: Matriz de acceso para la asignación del destino celular, antes del análisis estadístico. El CD45h se refiere a la intensidad de CD45RA para el subconjunto HPC gating, mientras que CD45l se refiere a la intensidad de CD45RA para los subconjuntos CD34+CD38- . Haga clic aquí para descargar esta tabla.
7. Análisis estadístico
NOTA: La prueba estadística de los datos generados implica una canalización de análisis personalizada, codificada con el lenguaje de programación Python (Archivo complementario 2, Archivo complementario 3 y Archivo complementario 4). El script está organizado en tres bloques: el primero para procesar la hoja de cálculo, el segundo bloque para generar el mapa de calor para la visualización de datos y el último bloque para generar múltiples histogramas para analizar y probar las propiedades de diferenciación y división.
- A partir del bloque "0_process_data" (Archivo complementario 2), asegúrese de que las rutas de gating_matrix y de hoja de cálculo de datos estén correctamente definidas en el script.
- Defina el diccionario "cell_cols", para asignar los destinos de celda relevantes a cada celda. En el caso específico, los destinos son HSC, MPP, LMPP, progenitores mieloides comunes (CMP), progenitores granulomonocíticos (GMP), progenitores eritroides megacariocíticos (MEP) y CD34-.
- Utilizando los valores umbral definidos a partir de los pozos a granel (paso 6.16), defina la función "cell_class_exp_time". Es esencial ser coherente en la nomenclatura de las columnas, para definir correctamente estos umbrales, utilizando el mismo nombre utilizado para definir cada columna en el paso 6.12.
- Los fenotipos celulares se definen en el guión utilizando una serie de declaraciones "if-else", basadas en los umbrales detectados durante el análisis de citometría de flujo.
NOTA: Se pueden mostrar diferentes fenotipos modificando estas instrucciones para acomodar otras combinaciones de marcadores. - Especifique las condiciones específicas experimentales, utilizando la función "cond_rule" (por ejemplo, diferentes tratamientos experimentales). Para el conjunto de datos proporcionado, las condiciones se denominan "GT" y "Diff". Describir los dos medios de cultivo celular diferentes utilizados para cultivar las células. Esta información será utilizada por el bloque "1_dot_plot" (Archivo Suplementario 3) para trazar el mapa de calor.
- En el bloque "2_bar_plot" (Archivo suplementario 4), defina el diccionario "class_dct", incluidos los destinos de celdas discretas de interés. Para el conjunto de datos proporcionado, los destinos de celda de interés son los mismos descritos para el diccionario "cell_cols".
- Defina "conds" (condiciones), "or_cells" (celda original), "sym_labs" (etiquetas de simetría) y "times" (el punto de tiempo experimental). Estos son filtros de reiteración necesarios para trazar. "conds" toma las condiciones definidas en "cond_rule" nuevamente, "or_cells" son HSC y MPP, y "sym_labs" describe el tipo de divisiones.
- En el bloque "2_bar_plot", es posible trazar celdas que han progresado hasta la división 6.
NOTA: El conjunto de datos proporcionado sólo incluye celdas hasta la división 4, por lo que aparece un mensaje de error, pero esto no impide que el script funcione. - Las figuras generadas por el script se pueden recuperar en la carpeta llamada "figuras" como archivos pdf. Los archivos denominados "Test" representan las diferentes pruebas estadísticas realizadas para el histograma correspondiente.
Clasificación FACS
Las estrategias de clasificación de compuerta presentadas en este protocolo se basan en estrategias ampliamente aceptadas 12,30,31. Para la estrategia de compuerta presentada en la Figura 1, el material de partida son progenitores de sangre del cordón umbilical previamente purificados a través del enriquecimiento magnético CD34+, lo que explica el porcentaje insignificante de células positivas para el linaje. Es esencial utilizar puertas estrechas para las cuatro combinaciones de colorantes intracelulares (por ejemplo, el CTV en la figura), para mejorar la resolución de los picos durante el siguiente análisis y para bloquear la población celular correcta (Figura 1D). En el caso que se muestra en la figura, las puertas seleccionan para la población más grande y mejor definida. La presencia de poblaciones múltiples y cercanas para cada combinación de colorante de división celular no es, en nuestra experiencia, representativa de las diferencias biológicas. En cambio, podría indicar a) un procedimiento de tinción no óptimo, o b) una gran heterogeneidad (especialmente en tamaño) en el grupo inicial de células. Esto no es inesperado cuando se parte de la sangre del cordón umbilical u otras fuentes biológicas complejas (por ejemplo, aspirados de médula ósea, sangre periférica). Si la compuerta no está bien definida, la dilución progresiva de las diferentes combinaciones de colorantes puede conducir a la fusión de los picos posteriores, específicamente para las condiciones CV y VC (Figura 2D). Otra consecuencia negativa de una compuerta subóptima es la incapacidad de distinguir eficientemente diferentes picos después del cultivo celular, ya que una población inicial heterogénea puede conducir a picos poco profundos.
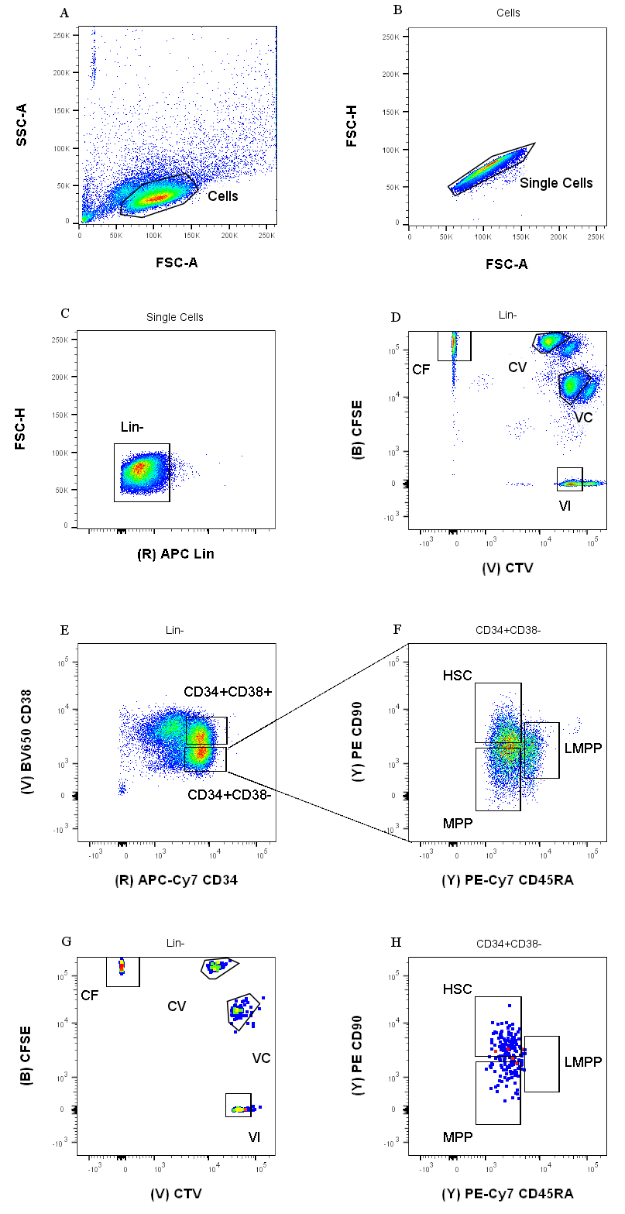
Figura 1: Estrategia de compuerta para la clasificación celular. (A) FSC-A versus SSC-A, para excluir desechos y células contaminantes. (B) FSC-A versus FSC-H, para excluir dobletes y grupos celulares. (C) Lin versus FSC-H, para excluir las células que son Lin+. (D) CTV versus CFSE, para identificar unívocamente las células teñidas con las combinaciones de colorantes CF, CV, VC y VI. Las puertas deben ser lo suficientemente estrictas como para incluir una población homogénea. (E) CD34 versus CD38, para separar los progenitores restringidos CD34+CD38+ (también llamados HPCs) del compartimento multipotente CD34+CD38-. (F) CD45RA versus CD90, de la población CD34+CD38-, para separar entre los progenitores más inmaduros enriquecidos en el HSC (CD90+CD45RA-), el LMPP (CD90midCD45RA+), y el MPP más comprometido (CD90-CD45RA-). (G) Índice de eventos ordenados, representados aquí para su tinción combinada de colorante celular y (H) la expresión de marcadores de superficie CD90 y CD45RA. Haga clic aquí para ver una versión más grande de esta figura.
Análisis de citometría de flujo después del cultivo celular
Los datos de la Figura 2 son representativos de las HSC de la sangre del cordón umbilical humano, mantenidas en cultivo durante 72 h, en presencia de múltiples citoquinas capaces de soportar una gama de progenitores y precursores mieloides. Los paneles 2A a 2D representan la puerta necesaria para establecer el parentesco de cada una de las células individuales, mientras que los paneles 2E a 2G permiten el fenotipado celular. La presencia reducida de eurodiputados en la figura es probablemente la consecuencia de las condiciones de cultivo utilizadas para este experimento representativo (Figura 2F). El uso de diferentes citoquinas y condiciones de cultivo altera el porcentaje relativo de cada subconjunto, de manera similar a la selección de diferentes células iniciales para el experimento.
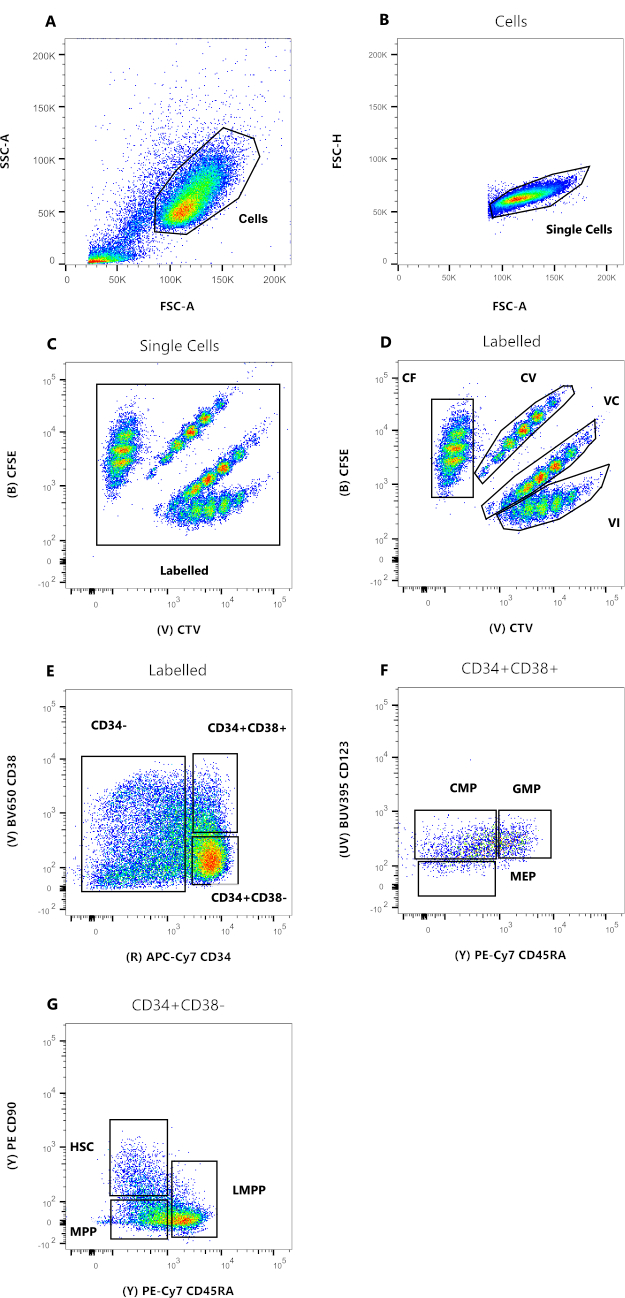
Figura 2: Estrategia de gating para el análisis de citometría de flujo. (A) FSC-A versus SSC-A, para excluir desechos y células contaminantes. (B) FSC-A versus FSC-H, para excluir dobletes y grupos celulares. (C) CTV versus CFSE, la puerta etiquetada permite excluir cualquier evento autofluorescente que pueda afectar la resolución de datos. (D) CTV versus CFSE. Es extremadamente importante vigilar estrictamente las cuatro poblaciones, basándose en las diluciones de colorante de división celular. (E) CD34 versus CD38, para distinguir entre precursores comprometidos (CD34-), progenitores restringidos (HPC) (CD34+CD38+) y progenitores inmaduros (CD34+CD38-). (F) CD45RA versus CD123, para distinguir tres tipos de progenitores restringidos: CMP (CD123+CD45RA-), MEP (CD123-CD45RA-) y GMP (CD123+CD45RA+). (G) CD45RA versus CD90, del CD34+CD38-, para identificar HSCs, LMPPs, y MPPs. Haga clic aquí para ver una versión más grande de esta figura.
La definición de picos y los pasos de asignación (Figura 3 y Figura 4) son aspectos cruciales del protocolo y requieren la definición de puertas estrictas. Para la definición del pico (Figura 3), se necesitan al menos 1.000 eventos para una identificación confiable. En este sentido, podría ser beneficioso aislar más células durante el paso de clasificación celular para los pozos "a granel". La Figura 4 describe cuatro ejemplos de pozos individuales que contienen múltiples familias. Esta figura aclara la importancia de la Figura 2D y la Figura 3 gating, especialmente para la identificación de cada familia y cada pico. La Figura 4A ilustra un ejemplo sencillo, ya que todas las celdas de la puerta CF están muy cerca unas de otras y se pueden asignar fácilmente a un solo pico. La Figura 4C muestra otro ejemplo de una familia distribuida unívocamente en dos picos bien separados, como se muestra claramente en el histograma de la Figura 4D. La Figura 4E,G revela la importancia de la puerta cerrada estricta basada en un gran número de eventos; Ambos muestran pocos eventos que están cerca, pero fuera de las puertas de combinación de tinte. Esos eventos podrían incluirse incorrectamente en las puertas VI y CF, basándose exclusivamente en el análisis de un solo pozo. Finalmente, la Figura 4F,H muestra dos ejemplos diferentes de familias distribuidas en múltiples picos, con un ejemplo de dos picos de intensidad similares (Figura 4F) y uno con dos picos de intensidad desiguales (Figura 4H).
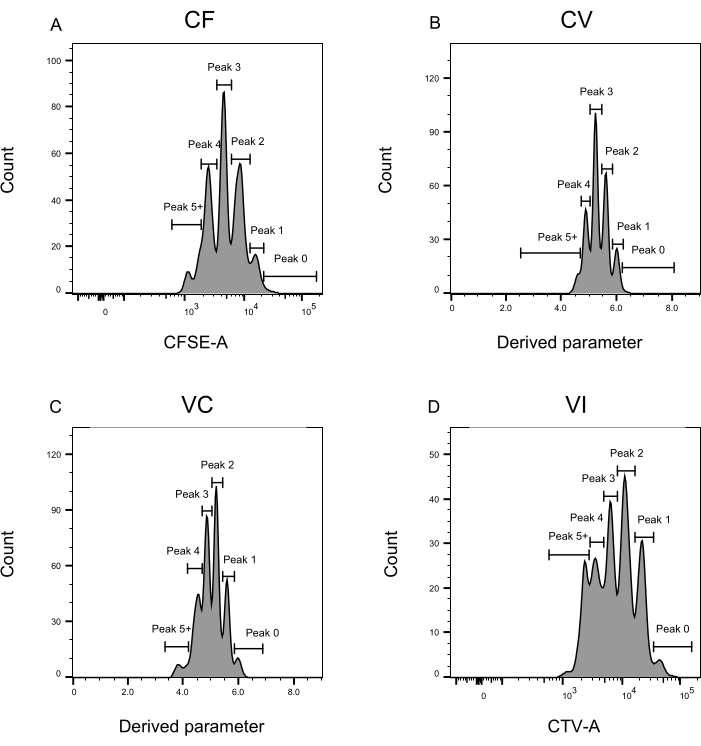
Figura 3: Definición de picos para el análisis de citometría de flujo. (A-D) Los picos deben definirse registrando al menos 500 eventos, para asegurar una buena representación para cada pico individual. (A) Histograma para la intensidad CFSE-A. Se pueden identificar varios picos, cada uno correspondiente a una población diferente de células en división. (B,C) Histogramas para la intensidad del parámetro derivado, que representa la mezcla CFSE-CTV, CV (B) y VC (C), respectivamente. (D) Histograma para la intensidad CTV-A. Haga clic aquí para ver una versión más grande de esta figura.
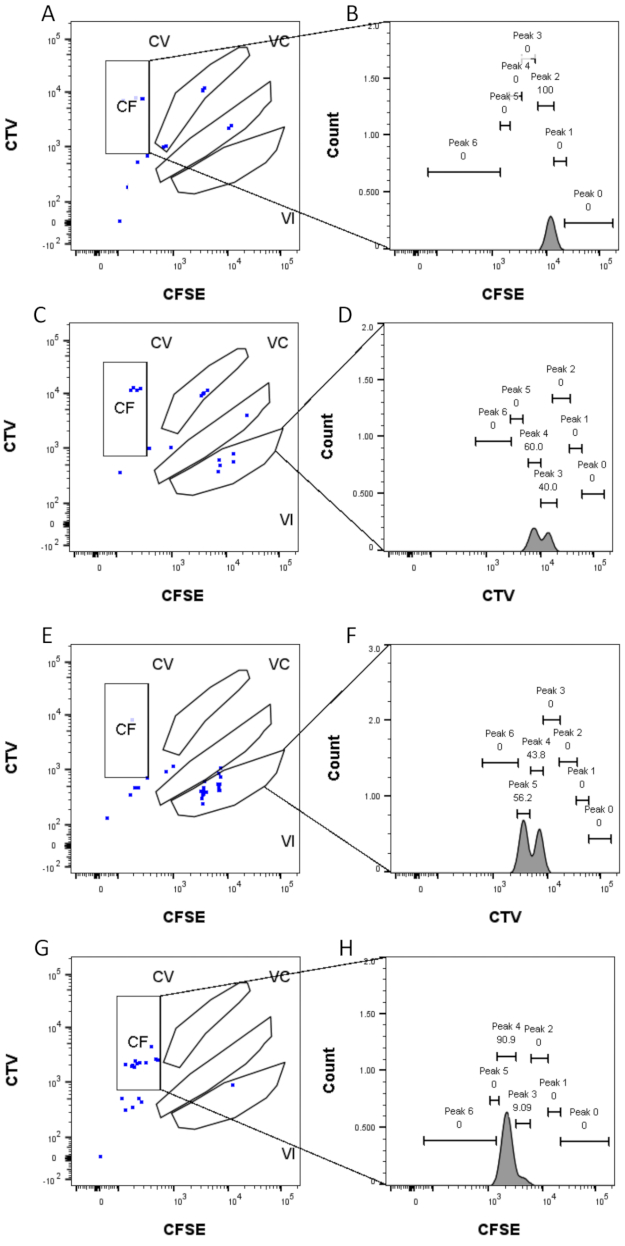
Figura 4: Asignación de picos . (A,B) Solo se puede detectar un pico para este pozo, en la puerta CF. (C,D) Se pueden detectar dos picos de intensidad casi igual en este pozo, en la puerta VI. Los picos están bien resueltos. (E,F) Se pueden detectar dos picos de intensidad comparable en este pozo, en la puerta VI. Solo se han considerado los eventos en la puerta, según la estrategia establecida utilizando los pozos a granel. (G-H) Se pueden detectar dos picos de intensidad desigual en este pozo, en la puerta CF. Haga clic aquí para ver una versión más grande de esta figura.
Representación de datos y pruebas estadísticas
La Figura 5 muestra diferentes tipos de representación de datos de dos experimentos separados, ambos realizados después de 72 h de cultivo celular. Las HSC y MPP se han cultivado en dos medios de cultivo celular diferentes, que se supone que alteran las propiedades de división y diferenciación celular. Estos medios se denominan "Diff" (Diferenciación)32 y "GT"33; el primero promueve la diferenciación mieloide y eritroide, ya que contiene eritropoyetina (EPO) y factor estimulante de colonias granulomonocítico (GM-CSF), mientras que el segundo se ha desarrollado en el contexto de ensayos clínicos de terapia génica, con el objetivo de mantener y amplificar un alto porcentaje de HSPC. La figura 5A es un mapa de calor representativo para la condición "Diff", que representa una variedad de familias de células, tanto en destinos celulares como en divisiones. En este mapa de calor, cada fila representa una familia individual, cada cuadrado una celda individual y las columnas agrupan todas las celdas que están en la misma generación (por ejemplo, las celdas de la generación 2 divididas al menos dos veces). Es posible distinguir familias altamente homogéneas, compuestas por un solo tipo de célula y que muestran el mismo número de divisiones (por ejemplo, familia #63), y familias heterogéneas, incluyendo tres tipos de células en dos generaciones (por ejemplo, familia #84). Debido a que la tasa de recuperación celular para este análisis es de aproximadamente el 70%, las familias completas, que se definen por tener todas sus células recuperadas en generaciones posiblemente diferentes (por ejemplo, una familia de una célula en la generación 1 y dos células en la generación 2), rara vez se observan (mostrando un hashtag junto a su número de identificación en la Figura 5A). Existen múltiples explicaciones que explican la detección incompleta, que podrían ser técnicas (problema de tinción, pérdida celular debido al protocolo) o biológicas (muerte celular y / o apoptosis). Las limitaciones técnicas se pueden superar utilizando un analizador diseñado para reducir el volumen muerto asociado con la muestra individual, y realizando la tinción celular directamente en la placa de cultivo celular para reducir el pipeteo de volumen. Por el contrario, los métodos ortogonales para determinar la cantidad de muerte celular (por ejemplo, a través de experimentos de imágenes de células vivas) pueden ayudar a distinguir los factores técnicos y biológicos que resultan en una detección incompleta.
La Figura 5Bi muestra cómo visualizar el efecto de la condición de cultivo en la composición del tipo celular, como si se hubiera realizado un ensayo masivo. Aquí, la condición Diff promueve un mayor número de destinos y un mayor porcentaje de células CD34 + (definidas como todos los tipos de células excepto CD34-). Los intervalos de confianza se calculan en el script mediante arranque básico, con 250.000 conjuntos de datos bootstrap34. Vale la pena señalar que todos los demás histogramas de la Figura 5 muestran intervalos de confianza calculados de la misma manera. La Tabla 5 recapitula toda la información sobre el número de familias y el número de células en cada generación.
La figura 5Bii representa gráficamente la salida de las pruebas estadísticas realizadas en el script "2_bar_plot". Se dispone de una descripción formal del marco estadístico26. En resumen, este marco permite probar hipótesis estadísticas asumiendo que las células de la misma familia son dependientes (una suposición que es en sí misma comprobable), contrariamente a las estadísticas clásicas que requerirían independencia entre todas las células observadas. En el caso específico presentado en la figura, la prueba estadística desafía la hipótesis de que las opciones de destino celular de los MPP, medidas como las frecuencias de los diferentes tipos de células presentes en el cultivo, son independientes de las condiciones de cultivo celular utilizadas. Primero, el estadístico de la prueba G se usa para evaluar la discrepancia entre las frecuencias de tipo celular de diferentes medios celulares (para el ejemplo en Bii, esta estadística se representa con la barra roja). Luego, se realiza una aleatorización de los datos a través de la permutación, intercambiando familias enteras de células entre las dos condiciones de cultivo celular. Esto es para preservar la dependencia entre las células relacionadas con la familia, manteniendo el número de familias en cada conjunto consistente con los datos originales. El estadístico de la prueba G se calcula a partir del conjunto de datos aleatorios. Los valores azules representados en 5Bii son el estadístico de la prueba G para 250.000 permutaciones. Finalmente, el valor p se calcula para evaluar el grado en que el conjunto de datos original se desvía de la distribución de los permutados. En el ejemplo, la estadística original se desvía en gran medida de la distribución, lo que resulta en un valor de p pequeño y, por lo tanto, rechaza la hipótesis de que el destino celular de los MPP es independiente de las condiciones de cultivo.
La Figura 5C representa el porcentaje de familias celulares por generación máxima, para explorar cómo las diferentes condiciones cambian la división celular por familia celular. Esta gráfica de datos muestra que a las 72 h, las células cultivadas en la condición Diff completan un mayor número de divisiones que las células en la condición GT. Se representa el número máximo de generaciones por cada familia, por lo que una familia que muestra celdas en las generaciones 1 y 2 se considera como generación 2. El mismo marco estadístico utilizado para la Figura 5B se puede utilizar para probar estadísticamente la independencia entre la división celular y la condición de cultivo.
La Figura 5D explora el tipo de simetría/asimetría de la primera división para los diferentes tipos de ancestros (HSC o MPP). Para las familias celulares completas en la generación 1, la única generación donde es posible establecer definitivamente las dos células hijas como células hermanas, se pueden definir cuatro tipos diferentes de simetría/asimetría: la etiqueta "Sym Undiff" describe familias donde ambas hijas conservan el fenotipo de la célula de origen. "Sym Diff" significa que ambas hijas tienen el mismo fenotipo, y es diferente de la célula de origen. "Asym Undiff" significa que una hija sólo conserva el fenotipo de la célula de origen. Finalmente, "Asym Diff" describe familias donde ambas hijas tienen fenotipos diferentes, y ninguno de ellos es igual a la célula de origen. Para obtener poder estadístico en la evaluación de estos destinos simétricos / asimétricos, es deseable realizar el análisis MultiGen en puntos de tiempo tempranos, con el fin de observar más familias cuya descendencia se encuentra en la generación 1.
Finalmente, la Figura 5E representa los porcentajes de tipos de células en función del número de divisiones, para obtener información sobre la progresión del patrón de diferenciación entre divisiones. Por ejemplo, los datos mostrados en la figura sugieren que las células progresan al estado CD34- , con más del 50% de las células detectadas en esta clase después de solo tres divisiones. Además, es posible inferir que los MPP no favorecen la división de auto-renovación, ya que un pequeño porcentaje de células retienen el fenotipo original. Algunas de estas conclusiones pueden probarse utilizando el marco estadístico presentado en las figuras anteriores.
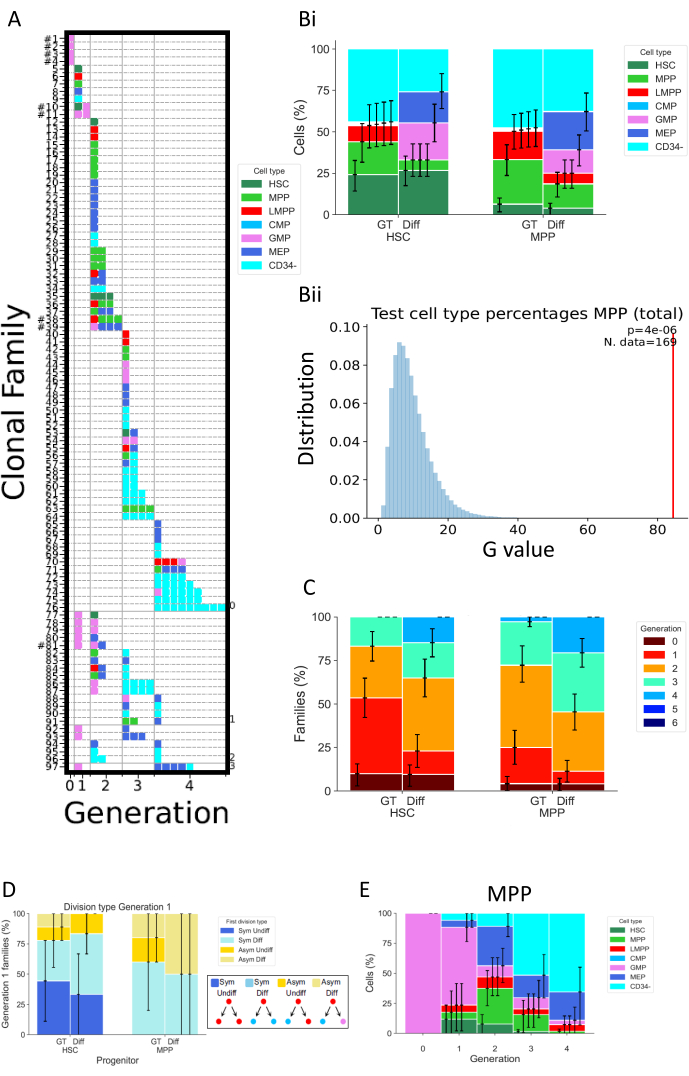
Figura 5: Ejemplo de representación de datos para un experimento de 72 h utilizando HSPC de sangre del cordón umbilical. (A) Mapas de calor para un conjunto de datos seleccionado (HSC, en medio "Diff", después de 72 h de cultivo). Los gráficos representan todas las celdas individuales (cuadrados) de acuerdo con su parentesco (filas), número de divisiones realizadas (columnas, llamada generación) y fenotipo (colores). (Bi) Histograma que compara proporciones de los tipos celulares de las progenies celulares de HSCs y MPPs, entre la condición GT y la condición Diff. (Bii) El gráfico representa las pruebas estadísticas realizadas en el script "2_bar_plot" para MPPs a las 72h de cultivo, comparando entre los cócteles de citoquinas "Diff" y "GT". El valor experimental se muestra en rojo, y los valores generados a través de 250.000 permutaciones en azul. El valor p. de la prueba G se indica en la esquina superior derecha con el número de familias utilizadas para la prueba. (C) Histograma que compara el porcentaje de familias (314 familias en total) en cada generación (codificado por colores), para HSC y MPP por condición de cultivo. Los intervalos de confianza se calculan con 250.000 conjuntos de datos bootstrad. (D) Histograma que representa el tipo de simetría/asimetría entre el destino de las células hijas para las familias con dos células en la generación 1: Sym Undiff (ambas hijas conservan el fenotipo de la célula de origen), Sym Diff (ambas hijas tienen el mismo fenotipo, y es diferente de la célula de origen), Asym Undiff (solo una hija conserva el fenotipo de la célula de origen), y Asym Diff (ambas hijas tienen fenotipos diferentes y ninguna de ellas se parece a la célula de origen). E) Histogramas de la contribución de los tipos celulares clasificados por generación para MPP cultivados con el cóctel "Diff"; n = 204 células y 97 familias. Haga clic aquí para ver una versión más grande de esta figura.
Tabla 5: Descripción del número de familias y células analizadas por cada condición experimental (célula de origen y medio de cultivo celular). Haga clic aquí para descargar esta tabla.
Archivo complementario 1: Haga clic aquí para descargar este archivo.
Archivo complementario 2: Haga clic aquí para descargar este archivo.
Archivo complementario 3: Haga clic aquí para descargar este archivo.
Archivo complementario 4: Haga clic aquí para descargar este archivo.
El ensayo MultiGen es un ensayo de alto rendimiento, fácil de realizar y poco costoso, que ha sido fundamental para estudiar los linfocitos 23,24,35 y las células hematopoyéticas murinas26,27. Aquí, presentamos un nuevo desarrollo del enfoque que permite descifrar ex vivo la fase temprana del compromiso de HSPC humano, a nivel unicelular utilizando cultivo a corto plazo (Figura 6). Los sistemas de cultivo ex vivo unicelulares se utilizan típicamente para evaluar el destino a largo plazo de las HSPC en células maduras, pero algunos destinos ocurren antes que otros36, lo que podría sesgar el análisis hacia menos destinos. Además, estos sistemas de cultivo generalmente pierden información sobre las divisiones durante el compromiso del destino. Se ha demostrado que los primeros pasos de compromiso ocurren tan pronto como al comienzo de la cultura, a veces sin división26,37, lo que hace que la cultura a corto plazo y la división de seguimiento sean esenciales para estudiar el compromiso de destino temprano. Al seguir simultáneamente el destino, la división y el parentesco, este ensayo permite comprender el papel de la primera división y la decisión del destino en las HSPC humanas. Usando el ensayo, es posible inferir después de cuántas divisiones ocurre el proceso de compromiso, el equilibrio entre la autorrenovación y la diferenciación para esos progenitores tempranos, y cómo esas propiedades se heredan a través de las generaciones. Hasta donde sabemos, este es el único ensayo que permite este tipo de mediciones para HSPC humanas, a resolución de una sola célula. Además, utilizando diferentes combinaciones de colorantes de división celular, aumentamos el rendimiento del análisis, haciendo de este ensayo una herramienta valiosa para generar grandes conjuntos de datos rápidamente. Las combinaciones de colorantes permiten seguir varias familias en los mismos pocillos, aumentando el número de células disponibles para su análisis en cultivo a corto plazo. El número de combinaciones podría aumentarse aún más, mediante la adición de otros colorantes (por ejemplo, colorante amarillo) o modificando la proporción de CFSE y CTV. Sin embargo, esto reduce el número de otros parámetros que se pueden analizar.
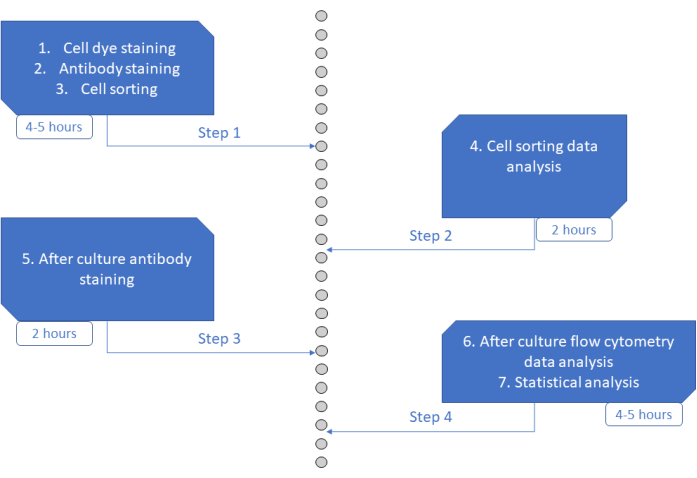
Figura 6: Representación esquemática del protocolo. Haga clic aquí para ver una versión más grande de esta figura.
Para realizar el análisis con éxito, debido a la gran cantidad de pocillos y al número reducido de células a analizar, es necesario ejecutar el análisis de citometría de flujo en un analizador equipado con un lector de placas. La nueva generación de analizadores de banco está particularmente adaptada a este ensayo, ya que la mayoría de ellos tienen un volumen muerto menor para reducir el porcentaje de pérdida celular. Esto a su vez garantiza una mayor eficiencia en la recuperación de la totalidad de cada pozo, lo que provoca una eficiencia estimada en el rango del 70%26. La estimación de la pérdida celular durante la adquisición de citometría de flujo es crucial para el análisis de cada familia individual. Por ejemplo, suponiendo que no hay muerte celular y contando el número de divisiones, es posible estimar el número de células por cada familia. Sin embargo, es deseable realizar algunos experimentos confirmatorios, particularmente en la estimación de la muerte celular en las condiciones de cultivo probadas y la medición de la tasa de recuperación experimentalmente utilizando un número definido de células.
Uno de los pasos cruciales de este protocolo es la asignación de picos. Como ya se mencionó, una distribución de picos de buena calidad depende en gran medida del aislamiento de picos muy estrechos en la clasificación celular. Sin embargo, todavía es difícil asignar el número correcto de divisiones basadas únicamente en la distribución. Como la clasificación celular y el análisis de citometría de flujo se realizan en dos máquinas diferentes, no es posible comparar directamente la intensidad de cada señal, por lo que podría ser difícil saber si el primer pico observado en el extremo derecho del histograma es el pico 0 o el pico 1. En este sentido, pocas soluciones son posibles; Una forma es realizar un experimento ortogonal para medir con precisión el número de divisiones realizadas por estas células (por ejemplo, imágenes de células vivas). Otra posibilidad es simplemente contar el número de células en el pozo bajo un microscopio de campo claro invertido, antes de ejecutar el análisis de citometría de flujo. Esto inferirá un número promedio de divisiones (suponiendo que no haya muerte celular). Finalmente, una solución post-hoc para la asignación de picos es la detección de un número inusual de "familias imposibles"; Estas familias están compuestas por un número mayor de lo posible de células por generación (por ejemplo, cinco células en la generación 2, o dos células en la generación 1 y una célula en la generación 2). La posibilidad de excluir familias imposibles se codifica en el paso de análisis estadístico y marca la familia imposible. Si la ocurrencia de estos errores es demasiado alta, es razonable suponer que la asignación de pico necesita ser revisada.
En este protocolo, incluimos algunos ejemplos de representación y análisis de datos para el ensayo, ya que se ha convertido en un paso esencial en la generación e interpretación de grandes conjuntos de datos38. El primer ejemplo es el mapa de calor que muestra la totalidad de todas las celdas analizadas, organizadas por familia. Esta es una herramienta eficiente para explorar las propiedades generales de los datos y las posibles conclusiones: ¿las familias están compuestas por múltiples tipos de células o tienden a ser homogéneas en composición? ¿Las familias están distribuidas a lo largo de varias generaciones, o en su mayoría se dividen el mismo número de veces? Este análisis exploratorio debe complementarse con gráficos más específicos y pruebas estadísticas. Se puede utilizar para evaluar cuantitativamente el compromiso de destino simétrico y asimétrico, la diferenciación sin división, el equilibrio entre la autorrenovación y la diferenciación, y el número de divisiones para un destino de compromiso dado. Es fundamental, durante la planificación experimental, establecer la duración del cultivo celular de acuerdo con el tipo de pregunta formulada; Por ejemplo, para las dos primeras preguntas (equilibrio simétrico/asimétrico y diferenciación sin división), la planificación de pasos de cultivos muy cortos permite el aislamiento de un gran número de familias que han realizado sólo una o ninguna divisiónen absoluto 26. Por el contrario, los experimentos más largos permiten la exploración del número de divisiones requeridas para un compromiso celular específico, ya que muestrean familias en diferentes etapas de diferenciación. Sin embargo, este método no está diseñado para cultivos a largo plazo (2-3 semanas), ya que la dilución del colorante celular no es capaz de rastrear con precisión más de siete u ocho divisiones22. Como consecuencia, esta herramienta está adaptada principalmente para estudiar el compromiso temprano de los progenitores hematopoyéticos, y no está diseñada para sacar conclusiones sólidas sobre las propiedades de diferenciación a largo plazo de estas células.
El marco estadístico fue desarrollado específicamente para el análisis de este tipo de datos y basado en el concepto de permutaciones26. Esto fue necesario debido a la observación de una dependencia familiar en la distribución del tipo celular y en el número de divisiones realizadas. En otras palabras, las células que forman parte de la misma familia también tienen más probabilidades de mostrar fenotipos similares y dividirse el mismo número de veces. Si bien un análisis en profundidad está más allá del alcance de este trabajo, el conjunto de pruebas estadísticas proporcionadas debería ser suficiente al evaluar diferentes condiciones.
En conclusión, este protocolo constituye una valiosa herramienta para evaluar la dinámica celular de las células madre y progenitoras hematopoyéticas ex vivo, de forma rápida y económica. Debido a su flexibilidad y versatilidad en cuanto al punto de tiempo, las condiciones de cultivo y el tipo de HSPC analizadas, permite probar una variedad de condiciones experimentales. Como ensayo basado en citometría de flujo, se puede implementar en la mayoría de los laboratorios y no requiere un amplio conocimiento previo, lo que lo convierte en un buen candidato para exámenes y experimentos piloto.
Los autores declaran no tener ningún conflicto de intereses relevante para este trabajo. Los financiadores no tuvieron ningún papel en el diseño del estudio, la recopilación e interpretación de datos, o la decisión de enviar el trabajo para su publicación.
Nos gustaría agradecer a los miembros del Institut Curie Flow Facility por su ayuda en la puesta en marcha de los experimentos de citometría de flujo. También queremos reconocer las contribuciones de los otros miembros del Equipo Perié, durante múltiples discusiones. Julia Marchingo y al Prof. Phil Hodgkin (Walter end Eliza Hall Institute of Medical Research) por compartir su protocolo de multiplexación de colorantes de división celular en linfocitos. Agradecemos al biobanco de sangre del cordón umbilical del hospital de Saint Louis por proporcionar los recursos biológicos necesarios para el desarrollo de este protocolo. El estudio fue apoyado por una subvención ATIP-Avenir del CNRS y la Fundación Bettencourt-Schueller (a L.P.), subvenciones del Labex CelTisPhyBio (ANR-10-LBX-0038) (a L.P. y A.D.), Idex Paris-Science-Lettres Program (ANR-10-IDEX-0001-02 PSL) (a L.P.), el Canceropole INCA Emergence (2021-1-EMERG-54b-ICR-1, a L.P.) y la subvención ITMO MIIC (21CM044, a L.P.). Además de la financiación del Consejo Europeo de Investigación (ERC) en el marco del programa de investigación e innovación Horizonte 2020 de la Unión Europea ERC StG 758170-Microbar (a L.P.), A.D. contó con el apoyo de una beca de la Fundación de Francia.
| Name | Company | Catalog Number | Comments |
| 1.5 mL polypropylene microcentrifuge tubes | vWR | 87003-294 | |
| 15-mL polypropylene tubes | vWR | 734-0451 | |
| 50-mL polypropylene tubes | vWR | 734-0448 | |
| 96-well U-bottom culture plate | Falcon | 353077 | |
| Anti-human Lin APC | Thermo Fisher | 22-7776-72 | Dilution 1/40 |
| ARIA III | BD | Can be replaced with any FACS sorter able to sort individual cells in 96-wells plate | |
| Carboxyfluorescein succinimidyl ester (CFSE) | Life Technologies | C34570 | |
| Cell Trace Violet (CTV) | Life Technologies | C34571 | |
| Compensation beads | BD | 552843 | |
| Dulbecco’s modified Eagle medium (DMEM) | Life Technologies | 11320033 | |
| Ethylenediaminetetraacetic acid (EDTA) | Thermo Scientific | J62948-36 | Prepare a solution 0.5 M, in sterilised water |
| FC block Fc1.3216 | BD | 564220 | Dilution 1/50 |
| Fetal Bovine Serum (FBS) | Dutscher | S1900-500C | Batch S00CH |
| FlowJo v10.8.1 | BD | ||
| Mouse anti-human CD10 PerCP-5.5, clone HI10a | Biolegend | 312216 | Dilution 1/20 |
| Mouse anti-human CD123 BUV395, clone 7G3 | BD | 564195 | Dilution 1/15 |
| Mouse anti-human CD34 APC-Cy7, clone 581 | Biolegend | 343513 | Dilution 1/40 |
| Mouse anti-human CD38 BV650, clone HB7 | Biolegend | 356620 | Dilution 1/40 |
| Mouse anti-human CD45RA AF700, clone HI100 | BD | 560673 | Dilution 1/20 |
| Mouse anti-human CD45RA PE-Cy7, clone HI100 | BD | 560675 | Dilution 1/20 |
| Mouse anti-human CD90 PE, clone 5E10 | Biolegend | 328110 | Dilution 1/20 |
| Phosphate Buffered Saline (PBS) 1X | Life Technologies | 10010001 | |
| Python | |||
| R | |||
| Sterile 12x75 mm conical polypropylene tubes | Falcon | ||
| ZE5 | Biorad | Can be replaced with any flow cytometry analyzer equipped with a plate reader | |
| Laboratory prepared | |||
| Cell culture media | Depends from the specific experiment. Prepare fresh daily and store at +4 °C until use | ||
| DMEM + 10% FBS | Can be stored in sterile conditions, at +4 °C up to 1 year. To prepare 500 mL, add 50 mL of FBS to 450 mL DMEM | ||
| PBS 1X + EDTA 0.1% | Can be stored in sterile conditions, at room temperature, up to 1 year. To prepare 500 mL, add 3.42 mL of EDTA 0.5 M to 500 mL PBS 1X | ||
| Staining buffer | Can be stored in sterile conditions, at +4 °C up to 1 year. To prepare 500 mL, add 2 mL of EDTA 0.5 M and 1 mL FBS to 500 mL PBS 1X |
- Ginhoux, F., Yalin, A., Dutertre, C. A., Amit, I. Single-cell immunology: Past, present, and future. Immunity. 55 (3), 393-404 (2022).
- Ke, M., Elshenawy, B., Sheldon, H., Arora, A., Buffa, F. M. Single cell RNA-sequencing: A powerful yet still challenging technology to study cellular heterogeneity. Bioessays. 44 (11), 2200084 (2022).
- Regev, A., et al. The human cell atlas. Elife. 6, 27041 (2017).
- Laurenti, E., Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 553 (7689), 418-426 (2018).
- Haas, S., Trumpp, A., Milsom, M. D. Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell. 22 (5), 627-638 (2018).
- Loughran, S. J., Haas, S., Wilkinson, A. C., Klein, A. M., Brand, M. Lineage commitment of hematopoietic stem cells and progenitors: insights from recent single cell and lineage tracing technologies. Experimental Hematology. 88, 1-6 (2020).
- Perié, L., Duffy, K. R. Retracing the in vivo haematopoietic tree using single-cell methods. FEBS Letters. 590 (22), 4068-4083 (2016).
- Yu, V. W. C., et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 167 (5), 1310-1322 (2016).
- Ganuza, M., et al. Lifelong haematopoiesis is established by hundreds of precursors throughout mammalian ontogeny. Nature Cell Biology. 19 (10), 1153-1163 (2017).
- Naik, S. H., Schumacher, T. N., Perié, L. Cellular barcoding: A technical appraisal. Experimental Hematology. 42 (8), 598-608 (2014).
- Quek, L., et al. Genetically distinct leukemic stem cells in human CD34 − acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. The Journal of Experimental Medicine. 213 (8), 1513-1535 (2016).
- Karamitros, D., et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nature Immunology. 19 (1), 85-97 (2018).
- Boitano, A. E., et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 329 (5997), 1345-1348 (2010).
- Delaney, C., et al. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nature Medicine. 16 (2), 232-236 (2010).
- Fares, I., et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 345 (6203), 1509-1512 (2014).
- Guo, B., Huang, X., Lee, M. R., Lee, S. A., Broxmeyer, H. E. Antagonism of PPAR-γ 3 signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nature Medicine. 24 (3), 360-367 (2018).
- Vannini, N., et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell. 24 (3), 405-418 (2019).
- Gupta, R., et al. Nov/CCN3 enhances cord blood engraftment by rapidly recruiting latent human stem cell activity. Cell Stem Cell. 26 (4), 527-541 (2020).
- Horwitz, M. E., et al. Omidubicel vs standard myeloablative umbilical cord blood transplantation: results of a phase 3 randomized study. Blood. 138 (16), 1429-1440 (2021).
- Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D., Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 367 (6479), 3381 (2020).
- Loeffler, D., Schroeder, T. Understanding cell fate control by continuous single-cell quantification. Blood. 133 (13), 1406-1414 (2019).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. Journal of Visualized Experiments. (70), e4287 (2012).
- Marchingo, J. M., et al. T-cell stimuli independently sum to regulate an inherited clonal division fate. Nature Communications. 7, 13540 (2016).
- Horton, M. B., et al. Multiplexed division tracking dyes for proliferation-based clonal lineage tracing. Journal of Immunology. 201 (3), 1097-1103 (2018).
- Lehmann, E. L., Romano, J. P., Casella, G. . Testing statistical hypotheses. , 784 (2005).
- Tak, T., et al. HSPCs display within-family homogeneity in differentiation and proliferation despite population heterogeneity. Elife. 10, 360624 (2021).
- Sommerkamp, P., et al. Mouse multipotent progenitor 5 cells are located at the interphase between hematopoietic stem and progenitor cells. Blood. 137 (23), 3218-3224 (2021).
- Kato, K., Radbruch, A. Isolation and characterization of CD34+ hematopoietic stem cells from human peripheral blood by high-gradient magnetic cell sorting. Cytometry. 14 (4), 384-392 (1993).
- Miltenyi, S., Müller, W., Weichel, W., Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry. 11 (2), 231-238 (1990).
- Doulatov, S., et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nature Immunology. 11 (7), 585-593 (2010).
- Goardon, N., et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 19 (1), 138-152 (2011).
- Laurenti, E., et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 16 (3), 302-313 (2015).
- Aiuti, A., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 341 (6148), 1233151 (2013).
- Davison, A. C., Hinkley, D. V. . Bootstrap Methods and their Application. , (1997).
- Horton, M. B., et al. Lineage tracing reveals B cell antibody class switching is stochastic, cell-autonomous, and tuneable. Immunity. 55 (10), 1843-1855 (2022).
- Notta, F., et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 351 (6269), 2116 (2016).
- Grinenko, T., et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nature Communications. 9 (1), 1898 (2018).
- Saeys, Y., Van Gassen, S., Lambrecht, B. N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nature Reviews Immunology. 16 (7), 449-462 (2016).
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved