An Optimized Quantitative Pull-Down Analysis of RNA-Binding Proteins Using Short Biotinylated RNA
In This Article
Summary
Here, we present an optimized in vitro method to uncover, quantify, and validate protein interactors of specific RNA sequences, using total protein extract from human cells, streptavidin beads coated with biotinylated RNA, and mass spectrometry analysis.
Abstract
Protein-RNA interactions regulate gene expression and cellular functions at transcriptional and post-transcriptional levels. For this reason, identifying the binding partners of an RNA of interest remains of high importance to unveil the mechanisms behind many cellular processes. However, RNA molecules might interact transiently and dynamically with some RNA-binding proteins (RBPs), especially with non-canonical ones. Hence, improved methods to isolate and identify such RBPs are greatly needed.
To identify the protein partners of a known RNA sequence efficiently and quantitatively, we developed a method based on the pull-down and characterization of all interacting proteins, starting from cellular total protein extract. We optimized the protein pull-down using biotinylated RNA pre-loaded on streptavidin-coated beads. As a proof of concept, we employed a short RNA sequence known to bind the neurodegeneration-associated protein TDP-43 and a negative control of a different nucleotide composition but the same length. After blocking the beads with yeast tRNA, we loaded the biotinylated RNA sequences on the streptavidin beads and incubated them with the total protein extract from HEK 293T cells. After incubation and several washing steps to remove nonspecific binders, we eluted the interacting proteins with a high-salt solution, compatible with the most commonly used protein quantification reagents and with sample preparation for mass spectrometry. We quantified the enrichment of TDP-43 in the pull-down performed with the known RNA binder compared to the negative control by mass spectrometry. We used the same technique to verify the selective interactions of other proteins computationally predicted to be unique binders of our RNA of interest or of the control. Finally, we validated the protocol by western blot via the detection of TDP-43 with an appropriate antibody.
This protocol will allow the study of the protein partners of an RNA of interest in near-to-physiological conditions, helping uncover unique and unpredicted protein-RNA interactions.
Introduction
RNA-binding proteins (RBPs) have emerged as crucial players in transcriptional and post-transcriptional gene regulation, since they are involved in processes such as mRNA splicing, RNA cellular localization, translation, modification, and degradation1,2,3. Such interactions between the two macromolecules are highly coordinated, precisely balanced, and essential for the formation of functional and processing hubs. Variations or dysregulations within these hubs have the potential of disrupting the finely regulated protein-RNA networks and are increasingly associated with a variety of human diseases, including cancer4,5 and neurodegenerative disorders6,7,8. The interactions between RNA molecules and their protein binding partners can be either stable and easy to validate experimentally, or highly dynamic, transient, and more difficult to characterize.
In recent years, intensive efforts have been undertaken to understand these interactions. Among the most established methods, protein pull-down assays (PDs) are probably the most appreciated and commonly used approaches to unravel the main players constituting ribonucleoprotein (RNP) complexes and other protein-RNA interaction networks3,9,10. PDs include a broad umbrella of informative techniques, such as the immunoprecipitation of either the RNA (RIP)11,12 or the protein (CLIP)13,14 of interest. Some of these RNA-PD protocols employ a known RNA as bait for proteins15, most frequently by taking advantage of high affinity tags such as biotin. In this instance, the interaction partners of a biotinylated RNA can be detected by anchoring the RNA on streptavidin-coated beads, enabling efficient isolation of the RNPs. The main limitations of these approaches are usually the design of the biotinylated probes and the testing of their ability to bind target proteins. For this purpose, it could be useful to rely on published CLIP data of the protein of interest, if available, since they reveal, with high precision, the short RNA regions that correspond to peaks of interactions with the target protein13,16. These same regions could be used to develop probes for PDs. An alternative method to design such RNA baits might be the systematic evolution of ligands by exponential enrichment (SELEX)17, which enable the design of aptamers through in vitro selection, starting from a comprehensive randomized library and via a series of PCR-driven optimization cycles. However, SELEX is complex and time consuming, and the final results are highly dependent on the initial library. To select the RNA bait to use in the protocol presented here, yet another approach was exploited, consisting of using an RNA bait designed de novo by means of the computational power of the algorithm catRAPID, which predicts the preferential binding of a given protein toward certain RNA sequences18,19,20.
The protocol introduced here is a version of an RNA-PD optimized to elute specific protein partners in near-to-physiological conditions, without the use of detergent, denaturing agents, or high temperatures. It relies on nano-superparamagnetic beads covalently coated with highly purified streptavidin and the use of a specific in silico designed biotinylated RNA as a bait. This protocol provides a rapid and efficient method to isolate the binding partners of biotinylated RNA molecules in native conditions, offering the potential for a wide range of downstream applications. To test this protocol, a 10-nucleotide single stranded RNA aptamer sequence, previously designed to bind the protein TAR DNA-binding protein 43 (TDP-43) with high affinity and specificity, was used20. Starting from HEK 293T cell lysates, the interactors of the biotinylated RNA aptamer were identified by means of mass-spectrometry analysis performed on samples detached from the RNA bait using a hypertonic buffer. This analysis confirmed the successful identification and quantification of TDP-43 as preferred binder.
This protocol enables the successful identification of protein interactors using only a short, in vitro synthesized RNA oligonucleotide. Moreover, the use of in silico designed RNA aptamers as PD probes21,22 guarantees specificity for the targets at significantly reduced costs.
Protocol
1. General methods and material
- Prepare the appropriate medium for the chosen mammalian cell culture and pre-warm it at 37 °C for 20 min before use.
- Prepare the required material in advance, as described in the Table of Materials. Autoclave glassware, plasticware, and buffer stocks.
- Prepare the buffers as described in Table 1. Adjust the pH of the stock solutions using concentrated HCl or NaOH before diluting the components to their final volumes.
2. Mammalian cell line preparation
- Grow HEK 293T cells in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 µg/mL penicillin/streptomycin solution. Incubate them at 37 °C in a humidified incubator supplied with 5% CO2. Routinely split the cells.
- Before detaching, rinse the cells with enough phosphate-buffered saline (PBS) solution to cover the growing surface.
- Remove the PBS and add an ultra-thin layer of trypsin-EDTA solution.
- Incubate the cells at 37 °C in a humidified incubator provided with 5% CO2 for 5 min, or until the cells are detached (they should look dispersed under microscopic observation).
- Dilute the trypsin-EDTA solution tenfold by adding complete DMEM to inactivate it and count the cells.
- Plate 1.5 x 105 cells/mL in 6-well plates, considering two wells/condition to test.
- Incubate the cells at 37 °C for 48 h in a humidified incubator with 5% CO2.
NOTE: Check the manufacturer instructions for the type of medium and supplement apt for the cell line. Also, the amount and the incubation time of trypsin-EDTA depend on the cell line. Some cell types grow faster/slower than what was reported in this protocol; thus, seeding concentration should be tested beforehand.
3. Total protein harvest
- Remove medium from the wells where cells are growing.
- Wash each well of the 6-well plates with 1 mL of PBS.
- Discard the PBS.
- Move the plates on ice and either proceed with step 3.5 or freeze the dry plates at -80 °C to facilitate lysis.
- Add 200 µL of lysis buffer to each well.
- Use a cell scraper to detach and break the cells.
- Transfer the cell extract deriving from two wells into the same 1.5 mL tube.
- Place the tube containing the protein extract on ice for 30 min.
- Centrifuge the cell lysates at 17,000 x g for 15 min at 4 °C.
- Transfer each supernatant into a pre-cooled tube.
NOTE: A total of 106-107 cells for each PD condition are recommended. Cell lysis and protein harvest should be performed with ice-cold buffers. Protease inhibitors should be added to the lysis buffer to prevent protein degradation.
4. Protein concentration determination
- Prepare Bradford reagent, as indicated by the producer, by diluting it fivefold in dH2O.
- Distribute 1 mL of reagent in a 1 cm cuvette, add 1 µL of the sample, and mix by inversion.
- Incubate in the dark at room temperature for 5-10 min.
- Read the absorbance at 595 nm.
- Calculate the volume of protein extract corresponding to 1.5 mg of proteins and bring all the samples to a final volume of 600 µL using lysis buffer.
- Keep the samples on ice until use.
NOTE: Any other protein concentration determination method can be used, following buffer compatibility recommendations. In any case, the lysis buffer should be used as a blank. Many reagents are not compatible with dithiothreitol (DTT). It is recommended to add DTT or other reducing agents only after protein quantification (DTT to a final concentration of 1 mM).
5. Bead preparation
- Mix the beads in their storage buffer by flicking the tube.
- Calculate 100 µL of slurry medium/sample and place the volume into a magnetic rack.
- Bead wash
- Remove the storage solution and wash the beads by adding 1 mL of lysis buffer/tube and invert it manually.
- Remove the buffer using the magnetic rack.
- Repeat the washing step.
- Add a volume of lysis buffer equal to the initial volume of slurry medium, mix by flicking the tube, and dispense the medium uniformly into as many 1.5 mL tubes as there are samples.
- Bead blocking
- Remove the buffer using the magnetic rack and add 600 µL of a 0.25 mg/mL solution of yeast tRNA prepared in lysis buffer.
- Incubate for 1 h at room temperature on a rotating wheel.
- Remove the tRNA solution using the magnetic rack.
- Add 600 µL of lysis buffer and wash by mixing manually.
- Repeat the washing step and discard the buffer.
6. Bead loading
- Prepare 200 µg of RNA oligonucleotide in 600 µL of lysis buffer for each tube containing the initial 100 µL slurry medium (now blocked beads).
- Add the oligo to the beads and incubate for 1 h at room temperature while rotating.
- Remove the solution, add 600 µL of lysis buffer, and wash the beads twice by rotating the tubes for 5 min at room temperature.
- Discard the buffer.
NOTE: Never vortex the beads, but flick instead. Limit the number of pipetting steps, unless necessary. When possible, use cut, 1 mL tips. The amount of bead slurry medium/sample depends on the binding capability of the beads and the starting amount of the total proteins. If the RNA oligo is predicted to have a significant amount of secondary structure, we recommend to first denature it at 80 °C for 10 min and then cool it down slowly at room temperature or refold it by incubating it at 30 °C for 1 h. It is suggested to recover the RNA oligo after bead loading and determine the concentration left, in order to optimize the amount required for loading and to evaluate the possibility of reusing the RNA.
7. Protein binding on beads
NOTE: From now on, when possible, perform the steps at 4 °C.
- Take a 5% volume from the 600 µL protein solution and keep it as INPUT (IN) for further analysis (1.5 mg of proteins are dissolved in 600 µL, so 5% corresponds to 30 µL and 75 µg of proteins).
- Add the remaining protein mix to each tube of loaded beads and leave slowly rotating overnight at 4 °C.
8. Washing of nonspecific binders
- Remove the unbound fraction using the magnetic rack. Save 5% of the volume and label it as FLOWTHROUGH (FT) (the unbound volume is ca. 600 µL, so again keep 30 µL for further analysis).
- Add 1 mL of wash buffer 1 to the beads and leave it rotating for 5 min at 4 °C.
- Discard the buffer.
- Repeat steps 8.2 and 8.3.
- Add 1 mL of wash buffer 2 to the beads and leave it rotating for 5 min at 4 °C.
- Discard the supernatant.
9. Elution of specific binders
- Add 100 µL of elution buffer 1 or elution buffer 2 to the beads.
- Mix manually by flicking and incubate for 5 min at room temperature.
- Place the tubes in a thermomixer and shake vigorously for 5 min at 95 °C.
- Place the tube into the magnetic rack and collect the eluted fraction into a clean tube.
- Quickly spin the beads with a bench centrifuge to maximize the recovery of the eluate.
- Save 5% of the total ELUATE (EL) volume for further analyses (total volume is 100 µL, so separate 5 µL into another tube).
- If needed, the protein concentration can be determined as in section 4, by using elution buffer as a blank.
NOTE: It is recommended to refrain from adding DTT to the elution buffer until protein concentration has been determined. If protein quantification is not required, or if reducing agent-compatible protein quantification kits are available, 1 mM DTT can be added to the elution buffer from the start. In this protocol, both elution buffer 1 (containing 1 M NaCl) and elution buffer 2 (with 2 M NaCl) were tested. No difference in the elution efficiency of the target protein was observed with increased ionic strength, but it is recommended to test both conditions before establishing the most appropriate buffer. If a high presence of salt in the elution buffer represents a limitation for further analysis, the very low amount of detergents in the elution buffer allows for buffer-exchange. As an alternative, the eluate can be diluted to reach the desired salt concentration.
10. Identification of protein binders by mass spectrometry
- Acetone precipitation
- Concentrate the eluted proteins by diluting it fourfold in cold (-20 °C) acetone.
- Vortex and incubate the tube at -20 °C overnight.
- Spin at 17,000 x g for 30 min at 4 °C.
- Gently remove the supernatant and allow the acetone to evaporate until the pellet has completely dried.
- In-solution protein digestion
- Dissolve the protein pellet by adding 50 µL of denaturation buffer.
- Add DTT to a final concentration of 5 mM, allowing protein reduction for 30 min at 55 °C.
- Cool down the samples at room temperature and proceed with the protein alkylation reaction, adding iodoacetamide (IAA) at a concentration of 10 mM for 15 min.
- Digest the proteins using a suitable enzyme (trypsin, LysC) and incubate the samples overnight at 37 °C.
- Stop the digestion by adding 1 µL of 10% trifluoroacetic acid (TFA).
- Clean up and concentrate the peptides on a custom reversed phase C18 micro-column, as previously described19.
- Elute peptides from the C18 tip with Buffer B.
- Remove the organic component using a vacuum centrifuge and resuspend the peptides in 5 µL of 0.1% formic acid for further analysis.
NOTE: Alternatively, protein digestion can be performed "on-beads" immediately after washing nonspecific binders (steps 8.1-8.6), thus making the protocol faster. However, it is advisable to test the efficiency of the enzyme working "on-beads" immobilized-proteins compared to standard in solution digestion, in order to guarantee the optimal experimental protein sequence coverage.
- Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
- Plug in the analytical column (C18-stationary phase) and keep it at 45 °C during the time of the run.
- Connect the column to the outlet of a six-port rotary valve of the LC pump through a capillary finger-tight fitting (20 µm x 550 mm) in a single-column configuration.
- Adjust the LC settings as follows:
- Load the peptides under controlled pressure (980 bar) in Buffer A.
- Apply a 5%-20% Buffer B gradient at 300 nL/min over 59 min, followed by a 20%-30% Buffer B gradient over 15 min and a 30%-65% Buffer B gradient over 5 min.
- Add a wash step by increasing the concentration of Buffer B up to 95% over 5 min plus a 5 min isocratic step at 95% Buffer B.
- Operate the mass spectrometer in data-dependent acquisition (DDA) mode to switch automatically between MS and MSMS events.
- Define a loop count equal to 15 using an automatic gain control (AGC) target value of 3 x 106 and 1 x 105 for the MS and MSMS events, respectively.
- Set the maximum allowed ion accumulation time to 20 ms for MS with a resolution of 60 K, and 100 ms for MSMS with a resolution of 15 K.
- Perform a high collision dissociation (HCD) fragmentation experiment using a normalized collision energy of 28%, with a dynamic exclusion time of 20 s.
- Operate the source parameters as follows:
Spray voltage: 1.7 kV
Capillary voltage: 275 °C
Neither a sheath nor auxiliary gas used
NOTE: In this protocol, ultra-high performance liquid chromatography (UHPLC) mass spectrometric (MS) analysis has been specifically performed using an LC single column setup, coupled to a hybrid triple quadrupole orbitrap instrument (Table of Materials). Other LCMS systems can be used, but an adaption of parameters is recommended.
- Data analysis
- Use the Load button to import the raw files.
- Define the experiment names by clicking on the Set Experiment button.
- Enter the group-specific parameters section to specify all theparameters related to identification:
Enzyme used for digestion: Trypsin/P
Missed cleavages: up to three
Fixed modification: Carbamidomethylation
Variable modification: N-acetyl (Protein), Oxidation (M) - Upload an updated FASTA file, available from public databases such as UniprotKB.
- Specify the correct parse rules according to the source of the chosen database.
- Define a parentage false discovery rate (FDR) value = 1 for both proteins and peptides.
- Add the label free quantification (LFQ) option in the Label Free Quantification tab.
- Keep the minimum LFQ ratio count at two.
NOTE: Here, we describe data analysis using MaxQuant24 and Perseus software25 to perform protein quantification and subsequent statistical analysis, respectively. However, data analysis can be performed with any other commercially available or free bioinformatic. FDR is estimated using a target-decoy database-based approach26. Peptide and protein FDR equal to 0.01 means that peptides and proteins identified are expected to contain 1% of false positives.
- Statistical analysis
- Load the proteingroups.txt file to perform statistical analysis at the protein level.
- Define the LFQ values as main columns.
- Remove "reverse" and "contaminants" by filtering rows based on the categorical column.
- Use the categorical annotation rows to group the different experimental conditions.
- Reduce the data matrix, selecting the number of valid values in each of the previously defined groups.
- Select the statistical test that better suits the experimental conditions (i.e., t-test, multiple sample test ANOVA).
- Determine a cut-off for significant hints with an FDR-based calculation. Typically, both 0.01 and 0.05 are accepted as thresholds for the adjusted p value.
- Visualize the results of differential analyses based on t-test statistics using a volcano plot representation.
- Export the final matrix in .txt format for further editing of the final result table.
NOTE: The configuration folder contains a FASTA file with proteins such as keratins that are considered common contaminants in global proteomic experiments, which are flagged with a + in the output table. In the present study, having two sample conditions, a t-test is used for the statistical analysis.
11. Results validation by western blot
- Sample preparation
- Add the appropriate volume of 4x sample loading buffer to each aliquot of IN, FT, and EL.
- Boil the samples for 5 min at 95°C.
- Fast-spin to recover evaporated sample from the top of the tubes.
- SDS-PAGE and gel transfer
- Load samples on a 4%-12% denaturing polyacrylamide gel.
- Run the gel with MES SDS running buffer for 1.5 h at 120 V.
- Transfer the gel on a nitrocellulose membrane with a semi-dry transfer cassette, following the manufacturer instructions. We recommend a 10 min transfer at 15 V.
- Immunodetection
- Block the membrane with 10% bovine serum albumin (BSA) for 1 h at room temperature while gently agitating.
- Add the primary antibody prepared in 5% BSA in TBST, according to the manufacturer's instructions. Leave overnight at 4 °C or for 1 h at room temperature under gentle agitation.
- Wash the membrane three times with TBST, each time for 5 min.
- Add the secondary antibody prepared in TBST for 1 h at room temperature under agitation.
- Wash the membrane three times with TBST, each time for 5 min.
- Visualize the results using a blot imager.
NOTE: For detecting TDP-43, a known binder of our RNA, recombinant rabbit monoclonal antibody was used and left with the membrane overnight at 4 °C. As a secondary antibody, the anti-rabbit IgG horseradish peroxidase (HRP) was used, but a fluorescent secondary antibody would work as well. To visualize the antibodies on the membrane, the membrane was incubated with the Clarity Western ECL substrate for 1 min, before imaging with the ChemiDoc imaging system.
Representative Results
To verify the validity of the proposed protocol, the PD experiments presented here were performed with a biotinylated RNA aptamer designed in silico to specifically bind TDP-4320. This RNA binds its protein target with high binding affinity (Kd = 90 nM)20. Here, this RNA, of sequence 5'-CGGUGUUGCU-3', is referred to with the name "+RNA". As a negative control, the reverse complementary sequence of +RNA, which is here called "-RNA", was used. Its sequence is 5'-AGCAACACCG-3'. -RNA shows a significantly lower binding affinity toward TDP-43 (Kd = 1.5 µM)19. For the purpose of the protocol described here, these RNA oligonucleotides have been purchased conjugated to a biotin molecule, to allow binding to the streptavidin beads. +RNA was purchased with a biotin-TEG at its 3' end, which includes a 15-atom triethylene glycol spacer between the biotin and the phosphate group of the nucleic acid; -RNA instead had a biotin at its 5' end, conjugated to the nucleic acid via an amino-C6 linker. However, if the design of the RNA bait is robust, and as long as there is no structural or chemical interference between the linker and the RNA, other positions for the biotin conjugation and other linker lengths could be employed.
Knowing the identity of the main protein to be found bound to the +RNA probe after the PD enabled the validation of the protocol by identification of TDP-43 in the eluate, using both mass spectrometry (MS) and western blot (WB) (Figure 1).
MS analysis was carried out on four PD replicates performed using either +RNA or -RNA (Figure 2). The identification of the interactomes of +RNA and -RNA is beyond the scope of this protocol, however some results that validate the accuracy of the protocol are reported. Of note, plotting the significantly enriched proteins in a volcano plot revealed that the total protein content and the enriched proteins eluted from +RNA was significantly higher that what was recovered from -RNA (Figure 2). This means that, despite having the same length and structural content (linear), +RNA can establish a higher number of specific interactions, which are retained up to the elution step with high salt. It is likely that -RNA instead establishes a higher number of nonspecific contacts that are disrupted during the washing steps. As expected, TDP-43 was identified as a unique interactor of +RNA20; the average label-free quantification (LFQ) for the four PD replicates performed with +RNA is 31.96 ± 0.56, while the protein is not identified among the interactors of -RNA. In addition, among all unique interactors of +RNA, TDP-43 was found to be the most abundantly enriched protein.
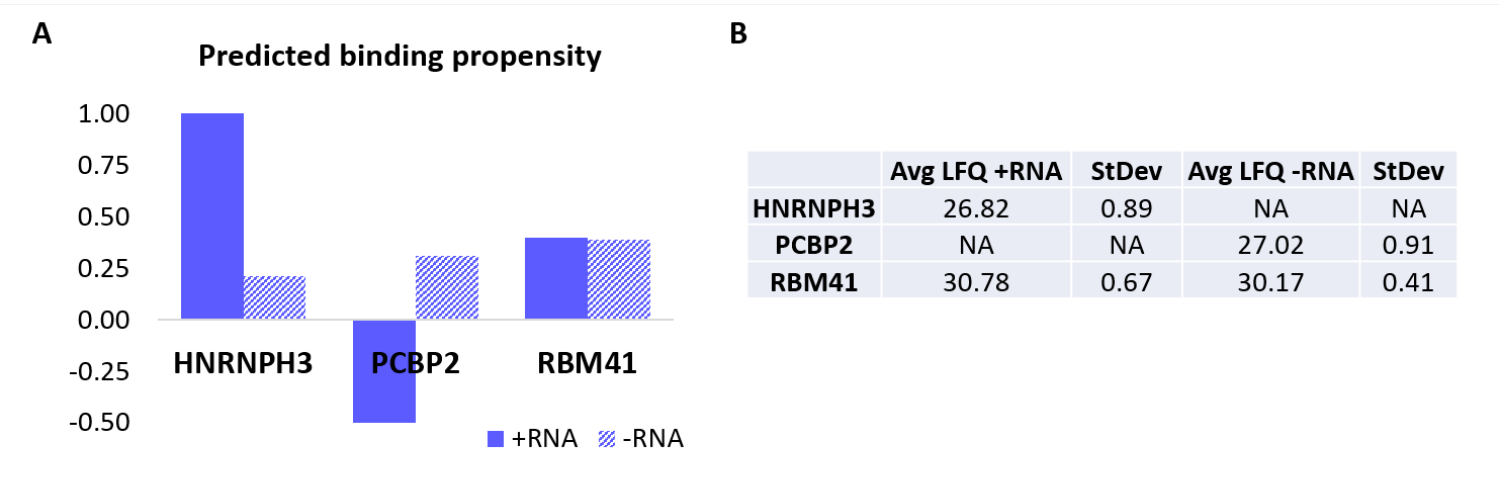
To further validate the protocol, the in-house algorithm catRAPID18,19 was used to computationally predict which other proteins would specifically bind either +RNA or -RNA. In particular, interaction scores for +RNA and -RNA with the proteins composing the human proteome were computed using the catRAPID 'interaction propensity' feature, as defined in our previous work27. Among the proteins scored with high confidence, HNRNPH3 was predicted to bind selectively +RNA (+RNA interaction score = 1.01; -RNA interaction score = 0.21) and PCBP2 to interact specifically with -RNA (+RNA interaction score = -0.5; -RNA interaction score = 0.31) (Figure 3A). In addition, the protein RBM41 was predicted to be promiscuous for both RNA oligonucleotides (+RNA interaction score = 0.4; -RNA interaction score = 0.39) (Figure 3A). The MS analysis indeed confirmed the presence of HNRNPH3 and PCBP2 in the PD of +RNA and -RNA respectively, while RBM41 was found interacting with both (Figure 3B).
WB was used to detect the presence of TDP-43 to further confirm the results and during protocol optimization (Figure 4). In the procedure described here, different samples were collected at different stages. The input sample (IN) consisted of the total proteins diluted in lysis buffer. The flowthrough (FT) was obtained after an overnight incubation of the total proteins with the streptavidin beads pre-coated with the biotinylated RNA, representing the fraction of proteins that did not bind the RNA. Finally, the eluate (EL) contained all the proteins that recognized specifically the RNA under investigation, since between the FT and the EL steps three washing steps with 150 mM salt and 0.1% triton-X should have removed the weakest interactions.
For each replicate, the same amount (5% v/v) of IN, FT, and EL was run in parallel on an SDS-PAGE and stained with an anti-TDP-43 antibody (Figure 4). In the case of +RNA, the band of TDP-43 was observed in IN and in EL, indicating that the protein, present from the start in the total protein extract, is retained by +RNA during the washing steps and is only eluted at the end with a high salt buffer. TDP-43 was also present in IN for -RNA, however the band corresponding to the protein is also visible in FT, indicating that this RNA does not bind TDP-43. The absence of the TDP-43 band in EL confirms this result.
During the optimization of the protocol, the elution of the proteins specifically bound to the RNA sequences was probed both with an elution buffer containing 1 M NaCl (EB1) and with an elution buffer complete with 2 M NaCl (EB2) (Figure 4). Eluates obtained with either EB were compared on an SDS-PAGE and blotted with the anti-TDP-43 antibody. The images obtained were then analyzed with ImageJ28 to quantify any difference in TDP-43 amount eluted with the two buffers. Overall, no significant difference was observed, and we concluded that, within these assays, 1 M salt is sufficient to disrupt even the strongest protein-RNA interactions.
Overall, the results reported here for MS and WBs demonstrate that this protocol is efficient in capturing the protein interactors of a given RNA in a specific manner, and that it enables the elution in buffers compatible with downstream analysis.

Figure 1: Sketch of the experimental pipeline used in the proposed protocol. (A) The biotinylated RNA oligonucleotide is prepared in lysis buffer at the appropriate concentration. (B) Magnetic streptavidin beads are washed, blocked with yeast tRNA, and loaded with the biotinylated RNA. (C) Total protein extract derived from cultured mammalian cell lines are added to the beads-RNA mixture. (D) Multiple washes are performed to remove nonspecific interactions. (E) The specific protein interactors are detached from the RNA with a hypertonic solution. (F) The identity of the interactors is revealed by mass spectrometry, and specific cases are validated by western blot. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Analytical strategy for label-free MS-based protein quantification. (A) Eluted proteins are precipitated in cold acetone overnight. Proteins are then denatured, and an in-solution digestion is performed. Proteolytic peptides are concentrated and desalted. (B) Peptides are analyzed via LC-MS/MS using a "shotgun approach". (C) Raw data processing and analysis is performed using MaxQuant and Perseus software, respectively. (D) Statistically significant enriched proteins are displayed in a volcano plot. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Correlation between predicted interaction propensities and experimentally determined interactions of +RNA and -RNA. (A) catRAPID interaction scores relative to HNRNPH3, PCBP2, and RBM41, indicating preferential binding of HNRNPH3 for +RNA and of PCBP2 for -RNA, while RBM41 is predicted to indiscriminately bind both RNA sequences. (B) Label-free quantification averages determined by mass-spectrometry analysis from the pull-downs performed with +RNA and -RNA. The analysis confirms that HNRNPH3 solely binds +RNA, PCBP2 solely binds -RNA, and RBM41 binds both equally. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Western blot validation of the presence/absence of TDP-43 among the interactors of chosen RNA sequences. The WB membrane has been treated with anti-TDP-43 antibody. IN = input; FT = flow-through; EL (EB1) = elution with elution buffer 1; EL (EB2) = elution with elution buffer 2; the sign "+" indicates samples derived from the pull-down performed with +RNA; the sign "-" indicates samples derived from the pull-down performed with -RNA; lane 1 contains a protein ladder. TDP-43 is indicated by an arrow. The WB indicates that TDP-43 is found among +RNA interactors but not among -RNA interactors. Please click here to view a larger version of this figure.
{kind=link}
| Buffer Name | Composition | |||||
| 10x Tranfer buffer | 250 mM tris, 1.92 M glycine, 1% SDS, 20% methanol. Dilute 10 folds prior use | PD | ||||
| 20X MES SDS running buffer | 1 M MES, 1 M tris, 2% SDS , 20 mM EDTA. Adjust pH to 7.3. Dilute 20 folds prior use | |||||
| 4x Sample loading buffer | 0.25 M Tris base, 0.28 M SDS, 40% glycerol, 20% 2-mercapto-ethanol, 4 mg/ml bromphenol blue | |||||
| Elution buffer 1 | 20 mM phosphate pH 7.5, 1 M NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT (to be added after quantification) | |||||
| Elution buffer 2 | 20 mM phosphate pH 7.5, 2 M NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT (to be added after quantification) | |||||
| Lysis buffer | 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT and protease inhibitors | |||||
| Tris-buffered saline with Tween-20 | 1 M Tris-HCl pH 7.4, 3 M NaCl, 2.0% Tween-20 | |||||
| Wash buffer 1 | 10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton TM X-100, 1 mM DTT and protease inhibitors | |||||
| Wash buffer 2 | 25 mM Hepes pH 8, 150 mM NaCl, 0.5 mM EDTA, 0.1 % Triton X-100, 1 mM DTT and protease inhibitors | |||||
| Buffer A | 0.1% formic acid | MS | ||||
| Buffer B | 60% acetonitrile, 0.1% formic acid | |||||
| Denaturation buffer | 8M urea, 50 mM Tris-HCl | |||||
Table 1: PD and MS buffers. Names and composition of the buffers used for either the pull-down experiments (PD) or for the mass-spectrometry analysis (MS).
Discussion
This work reports the optimization of a PD protocol performed with biotinylated RNA oligonucleotides to capture their protein interactors. The protocol described here is simple to perform, requires little material, and produces highly reliable results. Importantly, the most novel aspects of this protocol consist of the use of an RNA bait designed fully in silico and specific for the protein target, and the elution of all proteins bound to the RNA bait by directly disrupting their interactions with a high-salt solution, rather than by dissociating the streptavidin from the biotin with detergent and high temperature treatment.
This protocol takes advantage of the strength of the bond between biotin and streptavidin29,30. According to the chosen streptavidin beads, loading of the biotinylated RNA must be tested and quantified before proceeding. Also, the RNA tri-dimensional folding might affect the loading efficiency on the beads, since it might limit the exposure of the biotin to the streptavidin. Blocking the beads with non-biotinylated tRNA improves the cleanliness of the results by limiting nonspecific interactions with the beads. The loading buffer and the elution buffer must be chosen depending on the downstream applications. Here, very mild conditions, suitable to the majority of the applications and developed to preserve potential protein complexes, were proposed. This method is however highly adaptable; the user can choose any cell line and any RNA size, and could decide to repeat the protocol after folding/unfolding of the RNA to determine the effect of the structure on the binding properties.
Another original aspect of this protocol is the use of in silico prediction tools to ensure the correctness of the results20. Knowing in advance which proteins should be identified as interactors of the RNA of interest gives the unprecedented advantage of validating the technical aspects of the protocol. For example, using a simple WB analysis, it is possible to verify the presence of a known protein target in the samples derived from the different steps of the protocol before proceeding with the MS analysis, which requires specialized instrumentation and is more costly. In addition, a method to use catRAPID20, an in-house protein-RNA prediction algorithm, to design de novo RNA specific for a target protein was recently reported. Until recently, the only available pipeline to design DNA/RNA aptamers for a target protein was the SELEX (systematic evolution of ligands by exponential enrichment) approach31. The in silico method enables for a much faster and cost-effective design of RNA aptamers.
The main limitations of this method are associated with the need of working in nuclease-free buffers and tools. Moreover, if it is considered necessary to confirm in vitro the binding between a de novo-designed RNA and a target protein prior PD, the protein needs to be produced and purified and the binding determined with biophysical approaches. This is a limitation that is shared with the production of monoclonal antibodies.
Despite these minor issues, reliable methods to map RNA-protein interactions, such as the one presented here, can bring scientists closer to unveil macromolecular networks and complex main actors of many physiological and pathological mechanisms, such as the ones involved in cancer, cardiomyopathies, diabetes, microbial infections, and genetic and neurodegenerative disorders.
Acknowledgements
The authors would like to thank Prof. Tartaglia's and Dr. Cuomo's research group for the support offered. E.Z. received funding from the MINDED fellowship of the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 754490.
Materials
| Name | Company | Catalog Number | Comments |
| 6-well tissue culture plates | VWR | 10861-554 | CELLS |
| Cell scrapers | BIOSIGMA | 10153 | CELLS |
| Dulbecco′s Modified Eagle′s Medium (DMEM) | Thermo Fisher Scientfic | 11995065 | CELLS |
| Fetal Bovine Serum, qualified, heat inactivated, Brazil | Thermo Fisher Scientfic | 10500064 | CELLS |
| Phosphate Buffer Saline (PBS, Waltham, MA) | Thermo Fisher Scientfic | 14190169 | CELLS |
| Trypsin (0.25%), phenol red | Thermo Fisher Scientfic | 15050065 | CELLS |
| Anti-rabbit IgG horseradish peroxidase (HRP) | Cellsignal | 7070 | PD |
| Biotinylated RNA | Eurofins | Custom RNA oligonucleotides | PD |
| Bovine serum albumin | Sigma-Aldrich | A9418 | PD |
| Clarity Western ECL Substrate, 500 ml | Biorad | 1705061 | PD |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Merck - Sigma Aldrich | 5056489001 | PD |
| NuPAGE 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well | Invitrogen | NP0321BOX | PD |
| Recombinant anti-TDP43 antibody | Abcam | ab109535 | PD |
| Ribonucleic acid, transfer from baker's yeast (S. cerevisiae) | Merck - Sigma Aldrich | R5636-1ML | PD |
| Streptavidin Mag Sepharose | Merck - Sigma Aldrich | GE28-9857-99 | PD |
| Trans-Blot Turbo RTA Mini 0.2 µm PVDF Transfer Kit | Biorad | 1704272 | PD |
| Acetone | Thermo Fisher Scientfic | 022928.K2 | MS |
| C18 cartridge | Thermo Fisher Scientfic | 13-110-018 | MS |
| Dithiothreitol (DTT) | Thermo Fisher Scientfic | 20290 | MS |
| EASY-Spray HPLC Columns | Thermo Scientific | ES902 | MS |
| iodoacetamide (IAA) | Sigma Aldrich S.r.l. | I6125 | MS |
| Lys-C/Trypsin | Promega | V5073 | MS |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientfic | 28904 | MS |
| Urea | Thermo Fisher Scientfic | J75826.A7 | MS |
| Equipment | |||
| ChemiDoc imaging system | Bio-Rad | CELLS | |
| Dyna Mag -2 , Magnetic rack | Invitrogen | CELLS | |
| Forma Series 3 water jacketed C02 incubator | Thermo Scientific | PD | |
| PROTEAN II xi cell , power supply for PAGE applications | Bio-Rad | PD | |
| Rotating wheel, rotator SB3 | Stuart | PD | |
| Water bath set at 37 °C | VWR | PD | |
| XCell SureLock Mini-Cell electrophoresis system | ThermoFisher Scientific | MS | |
| Easy-nLC 1200 UHPLC | Thermo Scientific | MS | |
| Q exactive Mass Spectrometer | Thermo Scientific | MS | |
| Software | Version | ||
| MaxQuant | 2.0.3.0 | MS | |
| Perseus | 1.6.14.0 | MS |
References
- Gebauer, F., Schwarzl, T., Valcárcel, J., Hentze, M. W. RNA-binding proteins in human genetic disease. Nature Reviews Genetics. 22 (3), 185-198 (2021).
- Hentze, M. W., Castello, A., Schwarzl, T., Preiss, T. A brave new world of RNA-binding proteins. Nature Reviews Molecular Cell Biology. 19 (5), 327-341 (2018).
- Castello, A., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 149 (6), 1393-1406 (2012).
- Cooper, T. A., Wan, L., Dreyfuss, G. RNA and disease. Cell. 136 (4), 777-793 (2009).
- Qin, H., et al. RNA-binding proteins in tumor progression. Journal of Hematology & Oncology. 13 (1), 90 (2020).
- Nussbacher, J. K., Tabet, R., Yeo, G. W., Lagier-Tourenne, C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron. 102 (2), 294-320 (2019).
- Duan, R., Sharma, S., Xia, Q., Garber, K., Jin, P. Towards understanding RNA-mediated neurological disorders. Journal of Genetics and Genomics. 41 (9), 473-484 (2014).
- Maziuk, B., Ballance, H. I., Wolozin, B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Frontiers in Molecular Neuroscience. 10, 89 (2017).
- Zielinski, J., et al. In vivo identification of ribonucleoprotein-RNA interactions. Proceedings of the National Academy of Sciences. 103 (5), 1557-1562 (2006).
- Armaos, A., Zacco, E., Sanchez de Groot, N., Tartaglia, G. G. RNA-protein interactions: Central players in coordination of regulatory networks. BioEssays. 43 (2), 2000118 (2021).
- Weidmann, C. A., Mustoe, A. M., Jariwala, P. B., Calabrese, J. M., Weeks, K. M. Analysis of RNA-protein networks with RNP-MaP defines functional hubs on RNA. Nature Biotechnology. 39 (3), 347-356 (2021).
- Graindorge, A., et al. In-cell identification and measurement of RNA-protein interactions. Nature Communications. 10 (1), 5317 (2019).
- Ule, J., Hwang, H. W., Darnell, R. B. The future of cross-linking and immunoprecipitation (CLIP). Cold Spring Harbor Perspectives in Biology. 10 (8), 032243 (2018).
- Ascano, M., Hafner, M., Cekan, P., Gerstberger, S., Tuschl, T. Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdisciplinary Reviews. RNA. 3 (2), 159-177 (2012).
- McHugh, C. A., Russell, P., Guttman, M. Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biology. 15 (1), 203 (2014).
- Sugimoto, Y., et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biology. 13 (8), (2012).
- Bayat, P., et al. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie. 154, 132-155 (2018).
- Armaos, A., Colantoni, A., Proietti, G., Rupert, J., Tartaglia, G. G. CatRAPID omics v2.0: Going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Research. 49, 72-79 (2021).
- Agostini, F., et al. CatRAPID omics: A web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 29 (22), 2928-2930 (2013).
- Zacco, E., et al. Probing TDP-43 condensation using an in silico designed aptamer. Nature Communications. 13 (1), 3306 (2022).
- Leppek, K., Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Research. 42 (2), 13 (2014).
- Zhang, Y., Lai, B. S., Juhas, M. Recent advances in aptamer discovery and applications. Molecules. 24 (5), 941 (2019).
- UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research. 51, 523-531 (2023).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11 (12), 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Bellucci, M., Agostini, F., Masin, M., Tartaglia, G. G. Predicting protein associations with long noncoding RNAs. Nature Methods. 8 (6), 444-445 (2011).
- Gallo-Oller, G., Ordoñez, R., Dotor, J. A new background subtraction method for Western blot densitometry band quantification through image analysis software. Journal of Immunological Methods. 457, 1-5 (2018).
- Weissinger, R., Heinold, L., Akram, S., Jansen, R. P., Hermesh, O. RNA proximity labeling: A new detection tool for RNA-protein interactions. Molecules. 26 (8), 2270 (2021).
- Hirsch, J. D., et al. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: Uses for protein labeling, detection, and isolation. Analytical Biochemistry. 308 (2), 343-357 (2002).
- Sefah, K., Shangguan, D., Xiong, X., O'Donoghue, M. B., Tan, W. Development of DNA aptamers using cell-SELEX. Nature Protocols. 5 (6), 1169-1185 (2010).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved