Establishment, Maintenance, Differentiation, Genetic Manipulation, and Transplantation of Mouse and Human Lacrimal Gland Organoids
In This Article
Summary
This protocol describes how to establish, maintain, genetically modify, differentiate, functionally characterize, and transplant lacrimal gland organoids derived from primary mouse and human tissue.
Abstract
The lacrimal gland is an essential organ for ocular surface homeostasis. By producing the aqueous part of the tear film, it protects the eye from desiccation stress and external insults. Little is known about lacrimal gland (patho)physiology because of the lack of adequate in vitro models. Organoid technology has proven itself as a useful experimental platform for multiple organs. Here, we share a protocol to establish and maintain mouse and human lacrimal gland organoids starting from lacrimal gland biopsies. By modifying the culture conditions, we enhance lacrimal gland organoid functionality. Organoid functionality can be probed through a "crying" assay, which involves exposing the lacrimal gland organoids to selected neurotransmitters to trigger tear release in their lumen. We explain how to image and quantify this phenomenon. To investigate the role of genes of interest in lacrimal gland homeostasis, these can be genetically modified. We thoroughly describe how to genetically modify lacrimal gland organoids using base editors-from guide RNA design to organoid clone genotyping. Lastly, we show how to probe the regenerative potential of human lacrimal gland organoids by orthotopic implantation in the mouse. Together, this comprehensive toolset provides resources to use mouse and human lacrimal gland organoids to study lacrimal gland (patho)physiology.
Introduction
The lacrimal gland is the glandular epithelium responsible for producing most of the aqueous layer of the tear film1. The aqueous layer of the tear film not only contains water to lubricate the ocular surface but also a large repertoire of antimicrobial components that protect the ocular surface from infections2. When the lacrimal gland is damaged or inflamed, dry eye disease occurs, which results in discomfort for patients and can eventually lead to loss of vision3. Over the years, model systems to study the lacrimal gland, in particular the human gland, have been limited4,5,6. This has contributed to a knowledge gap regarding lacrimal gland function under physiological and pathological conditions.
Recently, in vitro models have been developed to study the lacrimal gland in a dish7,8,9. These lacrimal gland organoids are derived from adult stem cells grown as three-dimensional structures in an extracellular matrix supplemented with a cocktail of growth factors that sustains their regenerative capacities in vitro7. The advantage of adult stem cell (ASC)-derived organoids is that they can be maintained for a very long time while recapitulating healthy tissue features. This type of organoid solely consists of epithelial cells, unlike induced pluripotent stem cell (iPSC)-derived organoids, which may also contain stromal cells, for instance. Unlike pluripotent stem cell (PSC)-derived organoids, ASC organoids are established directly from adult tissue and do not require any genetic modifications to be expanded. ASC organoids express adult characteristics10.
This protocol contains a toolbox to derive lacrimal gland organoids from mouse and human primary tissue. The protocol describes how to further enhance the organoids' functionality by simple growth factor withdrawal and how to provoke the organoids to secrete tear fluid by performing a swelling assay. This protocol additionally includes an electroporation-based transfection method to genetically engineer mouse organoids using CRISPR-derived base editors. Unlike conventional Cas9, the use of base editors allows the modification of single bases in the genome without generating a double-stranded break11,12. Lastly, the orthotopic transplantation of human lacrimal gland organoids into immunodeficient mice and the subsequent histological assessment of the engraftment is described. This lacrimal gland organoid toolkit can be used in research on lacrimal gland regeneration and function and for genetic and inflammatory disease modeling.
Protocol
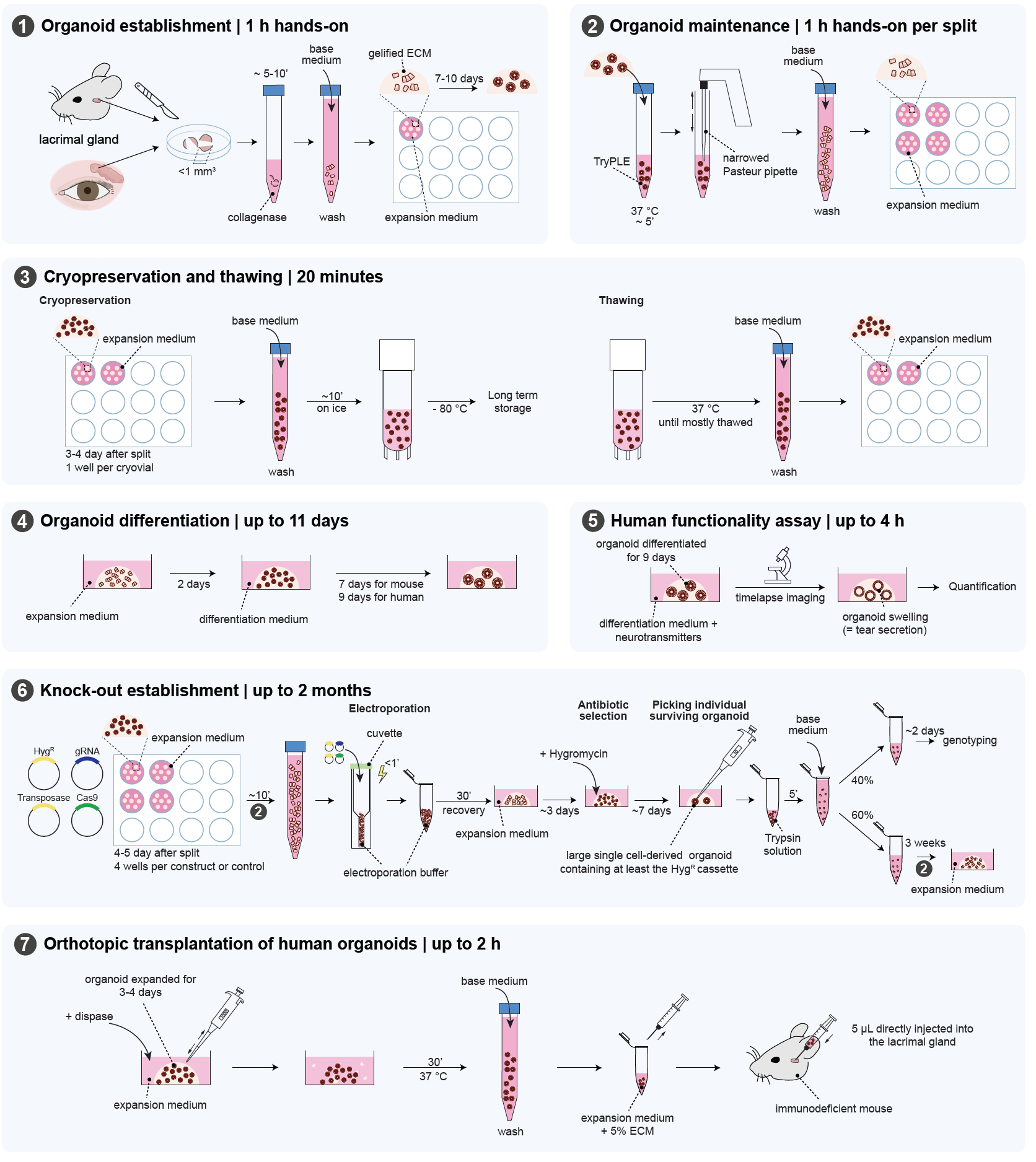
The mouse experiments were approved by the Animal Ethics Committee of the Royal Netherlands Academy of Arts and Sciences (KNAW) under project license AVD8010020151. The organoids were derived from mouse surplus material. The human lacrimal gland biopsies were collected from the waste material of patients undergoing surgery at the University Medical Centre Utrecht (UMCU) after approval by the medical ethical committee under protocol number 18-740. The protocol contains several sections that are outlined in Figure 1.

Figure 1: Overview of the protocol. This figure highlights the different steps of the protocol. Please click here to view a larger version of this figure.
{kind=link}
NOTE: All medium and buffer compositions are described in Supplementary Table 1.
1. Establishment of organoids from mouse and human lacrimal glands
- Dissecting out the mouse lacrimal gland
- Prepare the dissection tools, including scissors, forceps, and dissection pads. Euthanize a mouse by O2/CO2 inhalation13.
- Place the euthanized mouse on its abdomen on the dissection pad, and pin its limbs. Wet the hair located between the mouse ears and on the forehead using 70% ethanol.
- Using dissection scissors, make an opening behind the skull between the ears. Extend this opening over the forehead up to the nose. Still using scissors, cut the skin behind the ears to generate two flaps.
- Pull the flaps toward the nose firmly until the lacrimal glands are exposed, as shown in Figure 2A. Pin the flaps to the dissection pad. Using scissors, make a small incision in the membrane located above the lacrimal gland to completely expose the lacrimal gland.
- Using the forceps, pull the lacrimal gland. There should be some resistance until the main lacrimal duct is torn. If preferred, cut the main lacrimal duct directly with scissors.
- Place the mouse lacrimal gland into tissue culture-grade PBS until further processing. Proceed with processing the lacrimal gland within 2-4 h to limit cell death. If this takes longer, place the mouse lacrimal gland into the biopsy collection medium (Supplementary Table 1).
NOTE: Several lacrimal glands can be pooled if large numbers of organoids are needed immediately.
- Collecting human lacrimal gland biopsies
- Before the surgery, prepare 20 mL aliquots of biopsy collection medium (Supplementary Table 1) in a 50 mL tube, and give this to the surgeon ahead of the surgery. This medium can be kept at 4 °C for several months.
- On the day of surgery, let the surgeon sample a piece of the lacrimal gland (usually <1 mm3), and store it in the biopsy collection medium (Supplementary Table 1) at 4 °C.
- Collect the biopsy to process it as soon as possible. Ideally, this should be the same day within 2 h of biopsy sampling.
- Organoid derivation
- Prepare the following in advance: thawed extracellular matrix (ECM), room temperature (RT) mouse or human expansion medium (Supplementary Table 1), thawed collagenase, base medium (Supplementary Table 1), two scalpels, one 10 cm Petri dish, a 70 µm strainer fitted on a 15 mL tube, and 12-well suspension plates prewarmed at 37 °C for 30 min.
- Prepare the digestion medium by combining all the components shown in Supplementary Table 1. Pre-wet the scalpels in this medium to avoid tissue pieces sticking to them.
- Retrieve the mouse or human lacrimal gland tissue from the medium, and place it in the Petri dish.
- Using the pre-wetted scalpels, mince the tissue. Once the tissue pieces are very small (i.e., <0.5 mm3), place them in the digestion medium by scraping them off the Petri dish with the scalpel. If the human biopsy is already very small, place it directly in the digestion medium without mincing to avoid tissue loss.
- Incubate the tissue pieces for up to 15 min in a 37 °C water bath. Invert the 15 mL tube regularly to resuspend the pieces. Monitor the cell dissociation under a benchtop microscope so as not to over-digest the tissue.
- In the meantime, narrow a Pasteur pipette by placing its tip in a flame while turning, and pre-wet it in the base medium. To efficiently dissociate the tissue, ensure that the hole is slightly smaller than the largest pieces of tissue left. To facilitate the dissociation process, pipet the mixture up and down every 5 min with the pre-wetted narrowed Pasteur pipette.
- When many single cells and small clumps are visible under the microscope, stop the dissociation by adding 10 mL of the base medium. Spin down at 400 x g for 5 min to pellet the cells.
- Remove the supernatant, and resuspend the pellet in 10 mL of the base medium to repeat the wash. Filter it through a 70 µm strainer to remove the large undigested tissue pieces and remaining collagen fibers, which would prevent adequate polymerization of the ECM. Spin the eluate at 400 x g for 5 min.
NOTE: A red cell pellet indicates the presence of red blood cells, which normally does not hamper organoid derivation. However, for some applications, such as flow cytometry, the red blood cells should be lysed. For that, incubate the cells in 5 mL of fresh red blood cell lysis buffer at RT for 5 min, and pellet the cells at 400 x g for 5 min. - Remove the supernatant, and resuspend the cell pellet in 100 µL of cold ECM for a single mouse lacrimal gland and in 50 µL of cold ECM for a human biopsy. When resuspending, be cautious not to make bubbles, as these would also affect the ECM polymerization and stability.
- Seed up to 100 µL of cells per well of a 12-well suspension plate. Use a P200 to make ~20 µL droplets in the well. The smaller the droplets, the better the diffusion of the growth factors and nutrients through the matrix.
- Place the plate upside-down in a humidified incubator at 37 °C for 20-30 min to allow the ECM to solidify.
- Once the ECM is solidified, add ~1 mL of RT mouse or human expansion medium per well of a 12 well-plate. Refresh the medium every 2-3 days until the organoids reach a size of 300 µm. For that, aspirate the medium in the well, and gently add expansion medium on the side of the well without touching the ECM droplets so as not to disrupt them.
NOTE: Primocin (100 mg/mL; 1:1,000 from commercial stock) can be added upon isolation if bacterial contamination occurs. However, because it slows down organoid growth too, it is preferable not to add it or to remove it after one or two passages.
2. Expanding the mouse and human lacrimal gland organoids
- After ~7 days for mouse organoids and ~10 days for human organoids, when the organoids reach a size of ~300 µm, remove the culture medium.
- Resuspend the ECM droplets containing the organoids in 1 mL of trypsin solution by vigorously pipetting up and down with a P1,000 until the droplets fall apart. Transfer this mixture to a 15 mL tube, and incubate it briefly in a 37 °C water bath.
- After 2-3 min, use a narrowed and pre-wetted Pasteur pipette to pipet the organoid suspension up and down 10x-15x. Check the dissociation status of the organoids under a benchtop microscope; small clumps of ~20 cells should be obtained. Incubate longer in the 37 °C water bath, and repeat the pipetting step until the dissociation is satisfactory.
NOTE: This step may take longer for human organoids as they are multi-layered. Single cells will be generated; this does not affect organoid outgrowth as long as most of the organoid suspension consists of small clumps. - Stop the dissociation by adding 10 mL of the base medium. Then, pellet the cells at 400 x g for 5 min.
- After removing the supernatant and the ECM that may lay on top of the organoids (transparent, jelly-like layer without organoids), resuspend the cells in an appropriate volume of cold ECM before plating in a suspension plate, as indicated in step 1.3.10.
NOTE: Generally, mouse lacrimal organoids are split at a 1:5 ratio and human organoids at a 1:3 ratio. This means that, when starting with organoids contained in 100 µL of ECM, they need to be resuspended in 500 µL and 300 µL, respectively, after the split. - Incubate the plate upside-down in a 37 °C incubator until the solidification of the ECM for 30 min, and cover the organoids with the mouse or human expansion medium at RT.
3. Cryopreserving the mouse and human lacrimal gland organoids
- To cryopreserve the organoids, first split the organoids in the expansion medium according to the instructions given in section 2.
- About 3-4 days later, when the organoids are in a growth phase, remove the medium, and resuspend 100 µL of the ECM containing organoids in 10 mL of cold base medium. Incubate on ice for 10 min to help dissociate the ECM. Flip the tube regularly to facilitate this process.
- Pellet the organoids at 500 x g for 5 min. Remove the supernatant, and resuspend the organoid pellet in 1 mL of cryopreservation medium (leftover ECM does not impair cryopreservation). Transfer the organoid suspension to a cryovial and immediately to a −80 °C freezer.
NOTE: When using the cryopreservation medium described in the Table of Materials, the cryovials can be kept indefinitely in a −80 °C freezer. If another cryopreservation medium is used, transfer the cryovial to a liquid nitrogen tank after 24 h to ensure optimal preservation. - To thaw the organoids, retrieve the cryovial from the freezer, and transport it on dry ice to a 37 °C water bath. Hold the cryovial in the water until most of its contents are thawed. Transfer the contents to a 15 mL tube containing 10 mL of base medium, and spin down the cells at 400 x g for 5 min.
- Plate the organoids in 100 µL of ECM with the expansion medium, as described in step 2.5 and step 2.6.
4. Differentiating lacrimal gland organoids and assessing their functionality
- Organoid differentiation
- To differentiate lacrimal gland organoids, first split the organoids in the expansion medium according to the instructions in section 2.
- After 2 days, replace the expansion medium with mouse or human differentiation medium as described in step 1.3.12. Refresh the medium every 2-3 days to maintain the mouse organoids for 5 days in that medium and the human organoids for 9 days.
- To assess organoid differentiation after 5 days or 9 days in the differentiation medium, collect 100 µL of ECM containing organoids to extract the RNA. To do so, resuspend the ECM droplets in 1 mL of medium contained in the well using a P1,000, and transfer the organoid suspension in 3 mL of ice-cold base medium. Pellet the organoids at 500 x g for 5 min.
- Discard the supernatant, and resuspend the pellet in RNA extraction buffer. Perform the downstream RNA extraction according to the instructions of the RNA extraction kit. Use the obtained RNA, for instance, for RT-qPCR analysis of the expression of stem cells (TP63, KRT5, KRT14) and differentiated cell markers (LCN2, WFDC2, AQP5, LTF, ACTA2…)14.
NOTE: To obtain the relevant expression analysis, measure the expression of the chosen markers in one or several tissue samples. Organoids cultured under differentiation conditions tend to contain less RNA.
- Functional swelling assay
NOTE: For this part of the protocol, use human lacrimal gland organoids that have been differentiated for at least 7 days. The minimum time required is 7 days to ensure sufficient marker expression for functional tearing. Each well of a 12-well plate constitutes one condition. The minimum number of conditions is three: a positive control, a negative control, and a test condition.- Freshly prepare 1 mL of human differentiation medium containing individual components that induce secretion by the lacrimal gland organoids. For instance, add 100 µM noradrenaline and 1 µM forskolin, and mix thoroughly.
NOTE: Forskolin serves as a positive control that usually induces maximum swelling. - At an automated brightfield time-lapse microscope, set up the positions to be imaged in the plate, the time interval (5 min), and the duration (4 h). Ensure that an entire ECM droplet is visible at each position.
- Right before starting to image and without moving the plate from the microscope, remove the culture medium from the wells that will be imaged, and replace it with the well-resuspended medium prepared in step 4.2.1. Include as a negative control a well with differentiation medium replaced by differentiation medium only, as medium refreshment may trigger some organoid swelling.
- After up to 4 h, the swelling assay is over. Analyze the results.
NOTE: These steps are performed with an EVOS M7000 microscope that allows for automated brightfield time-lapse imaging. For that microscope, refer to the detailed manufacturer's manual to set up the time-lapse imaging. Of note, any other microscope with similar characteristics can be used. - To quantify organoid swelling, measure the diameter of each individual organoid at 0 h and 4 h. Open the images of a single organoid droplet at 0 h and 4 h in ImageJ.
- Click on the Straight line icon in the toolbar, and first draw the diameter of an organoid at 0 h. Then, use the Measure tool in ImageJ (Analyze > Measure) to measure the length of this line and, hence, the organoid diameter before swelling. Repeat the process on the same organoid at 4 h to obtain the organoid diameter after swelling.
- Measure the organoid diameter of ~20 organoids per condition before and after the swelling assay.
- Freshly prepare 1 mL of human differentiation medium containing individual components that induce secretion by the lacrimal gland organoids. For instance, add 100 µM noradrenaline and 1 µM forskolin, and mix thoroughly.
5. Constructing a plasmid to knock out Pax6
- gRNA design to knockout Pax6 using C > T base editors
NOTE: There are plenty of software programs available for gRNA design. Here, Benchling was used as this allows for integrated gRNA design, annotation, and the alignment of Sanger traces. All subsequent steps can, thus, also be performed using alternative software programs.- Start the gRNA design process by visualizing the target gene in Benchling by clicking New (+) > DNA Sequence > Import DNA Sequences.
- In the tab Import from Database, type the gene of interest, Pax6, and press Search.
- Select the newest build of the mouse reference genome GRCm38 (mm10, Mus musculus), and press Import.
- Select the exon where the knock-out gRNA can be designed by adhering to the following rules:
- Avoid putting a gRNA in the first coding exon, because alternative start sites in subsequent exons may be used by the cell to circumvent the early induced stop codon.
- Design gRNAs in exons that are present in all the alternative transcripts that can occur by splicing of the Pax6 mRNA. To ensure this, visualize Pax6 in the Ensembl genome browser, and select the exon that is used in all transcripts.
- To further circumvent alternative splicing, target an exon that has an incomplete codon (one or two remaining bases of a triplet). This is of lesser importance but gives more certainty about the effects of induced indels. For Pax6, exon 4 to exon 11 are a good target. In addition, choose an exon that contains tryptophan (W), glutamine (Q), or arginine (R) residues.
NOTE: Standard C > T base editors that use SpCas9 have an editing window that spans from nucleotide 4 to nucleotide 8 from the start of the gRNA (furthest away from the PAM). C > T base editors can introduce stop codons on all tryptophan (W) residues (TGG to TGA, TAG or TAA) with a gRNA on the reverse strand, on glutamine (Q) residues (CAG to TAG or CAA to TAA), and lastly, on arginine (R) residues (CGA to TGA) on the forward strand.
- Select the exon + 20 bases upstream and downstream. Click on the right side of the screen on the target sign CRISPR, and click on Design and Analyze guides.
- In the newly opened menu, under the tab Design Type, tick the Guides for "base editing" (Komor et al., 2016). Keep the Guide Length at 20 nucleotides, and under Genome for mouse, select GRCm38 (mm10, Mus musculus).
- After clicking the green + sign, the software program will automatically detect all the protospacer adjacent motif (PAM) sequences and, on the right side of the screen, create a list of all the potential gRNAs in the area around the exon of interest.
- Scroll through the list until the stop codon sign (*) in a red box, which signals for a gRNA that can potentially turn an amino acid into a stop codon and, thus, result in a knock-out.
- In the first column on the right of the gRNA sequence, for each target "C" around the editing window, the in silico predicted editing efficiencies are calculated. Ensure that the C > T edit that results in the stop codon has an editing efficiency of at least ~10.
- Click on the value in the Off-Target column to check which off-target loci the gRNA could bind to. Avoid choosing a gRNA that binds to any additional genes to increase specificity. For instance, a good gRNA to edit Pax6 with a C > T base editor targets exon 7 and is 5'-AAGCAACAGATGGGCGCAGA-3'.
- Create an annotation in the Benchling file by clicking on the Annotations icon in the top right of the screen, followed by New Annotation. Name the annotation, and ensure that the right gRNA orientation is selected in the Strand drop-down menu.
- gRNA plasmid generation
NOTE: There are various vectors available for gRNA delivery into organoids. The following plasmid was used as a base for gRNA construction: pFYF1320 (Addgene #47511, a kind gift from Keith Joung).- To clone the gRNAs in pFYF1320, implement an inverse PCR strategy utilizing the standard forward primer: "/5phos/ GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC. To design the reverse primer, paste the reverse complement of the gRNA spacer sequence (double check the gRNA orientation [+ or - strand]) in front of the following universal reverse primer part: CGGTGTTTCGTCCTTTCCACAAG. To target exon 7 of mouse Pax6 using the C > T base editor, the reverse primer is as follows: TCTGCGCCCATCTGTTGCTTCGGTGTTTCGTCCTTTCCACAAG. Order both primers.
- Run an inverse PCR reaction using 1 ng of pFYF1320 as a template using a high-fidelity DNA polymerase for 35 cycles at an annealing temperature of 61 °C.
- Run the PCR reaction on a 1% agarose gel in TAE buffer at 100 V for ~45 min to visualize the expected fragment of 2,281 base pairs (bp). Perform a gel clean-up using the kit of choice.
- Set up the following ligation reaction: 100 ng of cleaned-up PCR product, Dpn1 enzyme to remove the initial pFYF1320 template DNA, and T4 ligase. Incubate the mix for 15 min at 20 °C, 30 min at 37 °C, and 20 min at 80 °C.
- Transform the reaction mix into chemically competent DH5α bacteria, and spread the bacteria on agar plates containing ampicillin15.
- The next day, pick and expand three individual colonies in 3 mL of lysogeny broth (LB) medium supplemented with 50 µg/mL ampicillin.
- The day after, perform a miniprep on 1 mL of each of the three mini-cultures using a miniprep kit, and store the rest at 4 °C. Subject the obtained plasmid to Sanger sequencing with the U6_Forward primer to ensure the gRNA is correctly inserted into the vector16.
- After identifying a single bacterial colony with the correct plasmid, inoculate 500 µL of the corresponding mini-culture in 50 mL of LB supplemented with ampicillin to amplify the plasmid DNA overnight. Perform a midi-prep the day after using a midiprep kit. This plasmid will be used for the electroporation in section 6.
6. Generation of Pax6 KO clones
- Organoid electroporation
- Start with mouse organoids that were split at most 5 days before so they are in a proliferative state. Use ~300-400 µL of organoid droplets per knock-out. Include an additional condition for selecting a negative control that will be electroporated without any plasmid.
- Perform steps 2.1-2.4 to dissociate the organoids, but dissociate the organoids longer so that they are single cells. Discard the supernatant.
- Resuspend the cells in 80 µL of the electroporation buffer.
- In a 1.5 mL tube, prepare the following plasmids for a maximum of 20 µL: 2.5 µg of pFYF1320-gRNA plasmid, 2.8 µg of transposase-containing plasmid, 7.2 µg of hygromycin resistance transposon-containing plasmid, and 7.5 µg of pCMV_ABEmax_P2A_GFP.
NOTE: The pFYF1320-gRNA plasmid encodes the previously designed gRNA, which guides the base editor to the target locus. The pCMV_ABEmax_P2A_GFP plasmid encodes the base editor, as well as a GFP reporter, which can be used to monitor the cells that express the base editor following electroporation. The transposase-containing plasmid encodes the transposase, which pastes the hygromycin-resistance cassette provided by the hygromycin resistance transposon-containing plasmid somewhere randomly into the genome. Cells that have incorporated this cassette into their genome become resistant to hygromycin and can be positively selected by the addition of hygromycin. As the co-incorporation of several plasmids is very likely, hygromycin resistance constitutes a functional selection to enrich for cells that have been edited for Pax6. - Add the plasmids to the cells, and mix well by pipetting up and down.
- Set up the electroporator with the following parameters for poring pulse (voltage: 175 V; pulse length: 5 ms; pulse interval: 50 ms; number of pulses: 2; decay rate: 10%; polarity: +) and for transfer pulse (voltage: 20 V; pulse length: 50 ms; pulse interval: 50 ms; number of pulses: 5; decay rate: 40%; polarity: +/-).
- Place the cells with the plasmids in an electroporation cuvette. Immediately measure the resistance (Ω), which should be between 0.30 A and 0.55 A, and electroporate right away. Performing this step quickly is required to avoid the cells settling at the bottom of the cuvette and reducing the electroporation efficiency.
- Transfer the cells to a 1.5 mL tube, and add 400 µL of electroporation buffer supplemented with a Rho-kinase inhibitor. Allow the cells to recover at RT for 30 min.
- Pellet the cells at 500 x g for 5 min, discard the supernatant, and plate the cells in 200 µL of ECM. After solidification, add the mouse expansion medium.
- Cystic organoids grow out after 2-3 days. Monitor the GFP signal, which reveals the presence of C > T base editor, daily.
- Organoid selection
- After ~3 days, when the organoids have recovered, add 100 µg/mL hygromycin to the mouse expansion medium to select for organoids that have integrated the hygromycin resistance cassette. There is a high likelihood that cells that take up one plasmid will take up several plasmids. By selecting for hygromycin-resistant organoids, edited organoids on the Pax6 locus are enriched.
- When all the cells in the negative control are dead and surviving organoids are ~300 µm, pick the hygromycin-resistant organoids.
- Organoid picking and genotyping
- Prepare sterile P20 tips next to the microscope and 1.5 mL tubes containing 100 µL of trypsin solution each on ice.
- Bend a P20 tip with forceps. Observe the plate containing the surviving organoids on a benchtop microscope. When a surviving organoid is in focus, remove the lid of the plate. Using the bent P20 tip and still under the microscope, aspirate each surviving organoid individually, and place each of these in a separate tube containing trypsin solution.
- Pick up to 20 clones. The number of clones to be picked depends on the number of clones required for each experimental design and on the editing efficiency, which varies depending on each gRNA and locus.
- When all the clones have been picked, place the 1.5 mL tubes in a 37 °C water bath for up to 5 min. Vortex each tube regularly to increase the dissociation speed, and check the organoids under the benchtop microscope. When the organoids are dissociated into small clumps and/or single cells, stop the digestion by adding 1 mL of base medium.
- For each clone picked, keep ~400 µL to genotype in a separate 1.5 mL tube. Spin the ~600 µL left at 500 x g for 5 min, remove the supernatant, resuspend the cells in 20 µL of ECM, plate them as a single droplet in a well of a 24-well plate, and add mouse expansion medium without hygromycin.
- To genotype the clones, spin down the tubes containing ~400 µL of cell suspension at 500 x g for 5 min, remove the supernatant, and resuspend the cells in 50 µL of DNA extraction buffer to extract the DNA.
- Immediately incubate the cells as follows: 6 min at 60 °C, vortex, and 4 min at 95 °C. This DNA can be kept for up to 7 days at −20 °C, but performing the downstream PCR immediately is most optimal.
- To amplify the locus targeted with the nuclease, perform a PCR on 2 µL of the extracted DNA using adequate genotyping primers ordered beforehand and a low-fidelity polymerase. The genotyping primers are AGACTGTTCCAGGATGGCTG (Pax6_C>T_F) and TCTCCTAGGTACTGGAAGCC (Pax6_C>T_R). The amplicon should start about 100 bp from the expected edited locus to ensure it is sequenced efficiently.
- Run the PCR product on an agarose gel to assess the PCR efficiency, as mentioned in step 5.2.3. When a band of the correct size is detected, cut it out of the gel to perform a DNA gel extraction using a kit.
- Sequence the extracted DNA from each organoid clone previously picked with the genotyping primers.
- Align the sequences obtained to the Pax6 gene in Benchling. Open the imported gene sequence from step 4.1. Click on Alignments on the right side, and then on Create new alignment. Upload the .ab1 files obtained from the sequencing provider, select DNA as a nucleotide type, and press Next.
- In the following window, select Multisequence and the alignment program Auto (MAFFT) before clicking on Create alignment.
- Identify the genotypes that have a homozygous premature stop codon (as obtained with base editors) on both alleles. Keep the corresponding organoid clones to expand, and cryopreserve these for further analysis. Ideally, three different organoid clones should be analyzed side by side to circumvent potential nuclease off-target effects.
7. Orthotopic transplantation of human lacrimal gland organoids in NSG mice
- Organoid preparation
- Human lacrimal gland organoids must be split ~3 days before the transplantation day, as described in section 2. On the day of transplantation, ensure that the organoids are in the proliferative phase to increase the chances of engraftment.
- To extract the organoids from the ECM, add dispase to the culture medium to reach a final concentration of 0.125 U/mL. Using a P1,000, thoroughly resuspend the ECM droplets to disrupt them, and place the plate back in the 37 °C incubator for 30 min. A volume of 100 µL of ECM is sufficient to inject ~10 lacrimal glands.
- Resuspend the organoids in 10 mL of base medium to wash out the enzyme. Pellet the cells at 400 x g for 5 min.
- Resuspend the cells (~1,500,000) in 50 µL of cold human expansion medium supplemented with 5% ECM. Place the organoid suspension on ice, and proceed immediately to the transplantation.
- Orthotopic transplantation in mice
- In the animal facility, have the organoid suspension and insulin needles on ice. Aspirate the organoid suspension in a cold insulin needle.
- Sedate a NOD scid gamma (NSG) immunodeficient mouse with 3% isoflurane to initiate anesthesia. When the mouse is asleep, quickly place it on its side with the main lacrimal gland (located halfway between the eye and the ear) accessible.
- Inject 5 µL of the organoid suspension directly through the skin into the lacrimal gland. The maximum volume a mouse lacrimal gland can contain is 5 µL.
- Allow the mouse to recover, and monitor the mouse every day to assess the presence of any discomfort related to the transplantation, especially on the eye.
- Assess engraftment
- Sacrifice the mouse with O2/CO2 inhalation after up to 90 days depending on whether short-term or long-term engraftment should be assessed13.
- Dissect the lacrimal gland as described in section 1. Fix it in 4% formalin for 24 h at RT.
- Place the lacrimal gland in a cassette. Dehydrate the lacrimal gland by incubating it as follows: 2 h in 70% ethanol, 2 h in 96% ethanol, 1 h in 100% ethanol (2x), 2 h in xylene, and overnight in liquid paraffin at 58 °C in the oven.
- The next day, orientate the lacrimal gland as preferred in a metallic mold, top it up with liquid paraffin, and allow the block to solidify on a cold surface. This process is best performed with an embedding machine.
- When the paraffin block is solid, cut the entire block into 4-5 µm sections using a microtome. Keep all the sections (in the shape of ribbons) in a dry, draft-free environment. The sections can be kept indefinitely at room temperature.
- Sample one out of every 10 sections spanning the entire block.
- Mount the sections on a slide as follows: put a droplet of water on the slide, place the section on top, let it rest for ~2 min to allow it to stretch on a 42 °C hot plate, and remove the water gently with paper.
- Place the slides to dry overnight in a 58 °C oven. The slides can be kept indefinitely at RT past this stage.
- To distinguish human cells from mouse cells, perform human nuclear antigen staining on these sections. Rehydrate the sections by performing the following washes: 3 min in xylene (2x), 1 min in 100% ethanol (2x), 1 min each in 96% ethanol, 90% ethanol, 80% ethanol, 70% ethanol, 60% ethanol, and 25% ethanol, and finally, 1 min in demi water (2x).
- Incubate the slides for 15 min in PO buffer (Supplementary Table 1). Wash the slides 3x in ultrapure water.
- Perform a citrate-based antigen retrieval in the autoclave:
- Place the slides in an autoclave-proof slide holder in a basket filled with citrate buffer (Supplementary Table 1). Place the basket in the autoclave, and run an autoclave cycle (pressure: 10 PSI; temperature: 121 °C; time: 15 min).
- When the autoclave is depressurized, retrieve the basket containing the slides, and place it in an RT water bath to bring the slides to RT.
NOTE: The antigen retrieval strategy depends on the antibody used. Refer to the provider's datasheet to perform the most adequate antigen retrieval.
- Place the slides horizontally, section-side up, on an incubation tray. Add 500 µL of blocking buffer containing 1% bovine serum albumin (BSA) in PBS on top, and incubate for 1 h. Remove the blocking buffer.
- Prepare the antibody solution by adding 1 µL of human nucleolar antigen-antibody to 500 µL of blocking buffer per slide to be stained. Add the antibody solution to each slide. Incubate overnight in a humidified incubation tray at 4 °C.
- The next day, wash the slides 3x in PBS. Incubate the slides with undiluted HRP anti-mouse secondary antibody for 45 min. Wash the slides 3x in PBS.
CAUTION. In a chemical hood, prepare the DAB buffer (Supplementary Table 1). Incubate the slides with DAB buffer for 10 min. Discard any waste material in organic chemical liquid waste containers. - Wash the slides with water. Incubate the slides for 2 min in hematoxylin to counterstain the nuclei. Wash the slides in running tap water for 10 min.
- Dehydrate the slides by incubating them for 1 min each in 50% ethanol, 60% ethanol, 70% ethanol, 80% ethanol, and 90% ethanol, 1 min in 96% ethanol (2x), 1 min in 100% ethanol (2x), and 1 min in xylene (2x).
- Enclose the slides with mounting medium and a coverslip. After drying for about 20 min, observe the slides under a microscope.
Representative Results
Following the dissection of the mouse lacrimal gland (Figure 2A), the enzymatic and mechanical digestion generated small tissue fragments, among which the acini and ducts could be distinguished (Figure 2B). The remaining large pieces of tissue would destabilize the ECM, which would reduce initial organoid outgrowth. The mouse lacrimal gland organoid derivation was successful when cystic organoids of ~500 µm diameter were found after ~7 days, the stage at which the organoids were ready to be split (Figure 2C). Even if the overall organoid derivation is successful, some organoids may begin to grow before stopping eventually. Human lacrimal gland organoids grew out as cysts within 3-4 days and reached full-grown size in 10-14 days after tissue isolation (Figure 2D). For both mice and humans, organoid derivation sometimes failed, with no or few organoids growing out; this was generally caused by over-digestion of the tissue. The mouse lacrimal gland organoids could be passaged at least 40x and the human organoids at least 20x. Passaging was done every 7-10 days on average, depending on the organoid growth.

Figure 2: Establishment of mouse and human lacrimal gland organoids. (A) Photographs of the different stages of mouse lacrimal gland dissection. The arrow points at the lacrimal gland under its protective membrane. (B) Brightfield images of mouse lacrimal gland cells right after tissue digestion, with insets showing an acinus and a duct. (C) Brightfield images of a successful and an unsuccessful mouse lacrimal gland organoid derivation. (D) Brightfield images of human organoid outgrowth over the course of 14 days. Please click here to view a larger version of this figure.
{kind=link}
The lacrimal gland organoids contain a large proportion of stem cells when cultured in the expansion medium. To increase their differentiation level, we set up a mouse and a human differentiation medium with reduced growth factor content. After 5 days and 7 days, respectively, in the differentiation medium, the mouse and human lacrimal gland organoids became denser (Figure 3A-B). This morphological change correlated with increased functional properties. Applying the cyclic AMP activator forskolin or the neurotransmitter norepinephrine resulted in organoid swelling (i.e., apical water secretion) within less than 3 h (Figure 3C). When the swelling took longer than 3-4 h, this suggested that the organoids were not differentiated enough and/or did not express functional markers, such as receptors for neurotransmitters.

Figure 3: Differentiation of mouse and human lacrimal gland organoids and functional swelling assay in human organoids. (A) Brightfield images of mouse lacrimal gland organoids cultured in expansion medium for 7 days and in differentiation medium for 5 days after 2 days in expansion medium. (B) Brightfield images of human lacrimal gland organoids cultured in expansion medium for 11 days and in differentiation medium for 9 days after 2 days in expansion medium. (C) Brightfield images of differentiated human lacrimal gland organoids exposed to fresh differentiation medium (control), to 1 µM forskolin, and to 100 µM norepinephrine over the course of 3 h. Please click here to view a larger version of this figure.
{kind=link}
To knock out Pax6 in mouse lacrimal gland organoids, a plasmid containing the chosen Pax6-targeting gRNA was generated by PCR and ligation (Figure 4A). This gRNA-containing plasmid was electroporated along with the Piggy-Bac plasmids (hygromycin resistance transposon-containing and transposase-containing plasmids) and the C > T base editor Cas9 in mouse lacrimal gland organoids dissociated into single cells. After 3 days, when the organoids had recovered, they were exposed to hygromycin to select for clones that had incorporated the hygromycin resistance cassette. In successful electroporations, organoids resistant to hygromycin grew out (Figure 4B). The growing organoid clones that were larger than ~300 µm were picked, ideally before they started to spontaneously differentiate (Figure 4C). DNA was extracted from part of the organoid while keeping the remainder in culture. The PCR amplification of the Pax6 locus targeted by the gRNA yielded a 367 bp band for each of the clones picked (Figure 4D). After sequencing the amplified locus, clones that were homozygously C > T edited (n = 1) were kept. On the other hand, clones that were not edited (n = 4), heterozygously edited, or wrongly edited (n = 1) were discarded (Figure 4E). Overall, using this gRNA targeting Pax6, one homozygous knock-out mouse lacrimal gland clone out of six sequenced was obtained. Some clones grew out well, but some organoid clones were lost after picking or began to differentiate (Figure 4F). Out of the 10 organoid clones picked, 7 grew out well.

Figure 4: Base editing-mediated knock-out of Pax6 in mouse lacrimal gland organoids. (A) Sanger sequencing trace of pFYF1320 after correct integration of the gRNA targeting the Pax6 locus. (B) Brightfield images of mouse lacrimal gland organoids 5 days after exposure to hygromycin following electroporation. The organoids were cultured in mouse expansion medium. On the left is an example of a failed electroporation, with no clone resistant to hygromycin growing out. On the right is an example of a successful electroporation, with several hygromycin-resistant organoid clones surviving. (C) Brightfield images of clones that should be picked and clones that should not be picked. (D) Agarose gel showing the amplification of the Pax6 locus targeted with the gRNA. In green, the band of the expected size of 367 bp is highlighted. (E) Sanger sequencing trace of three organoid clones that were resistant to hygromycin. The top clone is unedited. The middle clone is a homozygous C > T edition and, hence, a homozygous knock-out. The bottom clone presents two heterozygous point mutations and is either a heterozygous knock-out or a mixed clone and has been wrongly edited. (F) Brightfield images of the picked organoid clones with various levels of outgrowth. Please click here to view a larger version of this figure.
{kind=link}
Lastly, to perform human lacrimal gland organoid orthotopic transplantation in mice, organoids that were split 3 days in advance (< 100 µm in diameter) were used. Organoid engraftment was confirmed 1 month after injecting the organoids into the mouse lacrimal gland by staining for a human-specific marker, the human nucleolar antigen (Figure 5). The absence of punctate staining from all the sections of the mouse lacrimal gland signified a lack of human organoid engraftment.

Figure 5: Transplantation of human lacrimal gland organoids into the mouse lacrimal gland. Staining of transplanted mouse lacrimal glands for the human nucleolar marker to monitor engraftment 1 month after transplantation. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1: Composition of the media and buffers. Please click here to download this Table.
Discussion
This protocol describes the establishment and use of lacrimal gland organoids for functional assays, mutation modeling, and transplantation. When establishing mouse and human lacrimal gland organoids, tissue dissociation is crucial. If the tissue is not sufficiently digested, the organoid yield will be low. If the tissue is over-digested, the cells will die and not grow out as organoids. Each tissue should be digested with a specific enzyme for a specific time to ensure optimal organoid outgrowth14,17,18. The lacrimal gland is a rather soft tissue for which a collagenase digestion of 5-10 min combined with pipetting-based mechanical dissociation is sufficient to isolate small pieces of tissue. If single cells need to be obtained for applications such as single-cell RNA sequencing, the dissociation can be carried out for longer until the single-cell stage is reached, but dissociation should be stopped as soon as single cells are obtained to limit any decrease in viability. As tissue dissociation is so crucial, over-digestion is the most likely reason for the failure of lacrimal gland organoid establishment.
The appropriate maintenance of lacrimal gland organoids is important to their use. Long-term maintenance is a hallmark of adult stem cell-derived lacrimal gland organoids, compared to induced pluripotent stem cell-derived organoids7,17. To achieve long-term maintenance, regular organoid splitting and medium changes should be performed. Without this, organoids begin to differentiate and develop decreased stem cell potential, which hampers their long-term maintenance7. At this step again, over-digestion can kill the stem cells and impair organoid maintenance. For regular maintenance, organoid dissociation into single cells is not required. However, to generate clonal knock-out organoid lines, it is critical to begin with single cells. If not, the organoids will be constituted of a mosaic of cells with different genetic backgrounds, making analyzing the effect of a single, defined mutation impossible. Here, we describe the use of a C > T base editor to generate stop codons. This genome editor relies on the presence of arginine, glutamine, or tryptophane codons within 12-18 bases from an NGG PAM. When these conditions are not met in designing a gRNA, conventional Cas9 or C >T base editors with alternative PAMs can be used7,18. However, conventional Cas9 introduces double-stranded breaks that result in indels upon repair11. As both alleles may harbor different indels, clone genotyping requires additional caution. Deconvolution of the modifications introduced should be performed to ensure both alleles contain out-of-frame indels and, hence, that the organoid clones are knocked out for the gene targeted19. The advantage of C > T base editors lies in the fact that they can be used to model point mutations that do not necessarily result in stop codons. For instance, they can be used to model specific Pax6 mutations found in patients with aniridia to study their impact on lacrimal gland physiology20.
The lacrimal gland secretes the aqueous part of the tear film1. Tear secretion can be recapitulated in human organoids after differentiation mediated by growth factor withdrawal and NOTCH inhibition. Under these conditions, organoids undergo terminal differentiation and cannot be further maintained. Yet, differentiated lacrimal gland organoids can guide the development of tearing-inducing drugs in the context of dry eye disease, potentially in high-throughput screens. The tearing assay presented in this protocol is the one that currently gives the biggest change in organoid size in a short amount of time, which makes it easier to quantify in the context of a drug screen7,9,17.
Stem cell therapy holds great promise for lacrimal gland regeneration in dry eye disease21. Adult stem cell-derived lacrimal gland organoids could be a source material for such applications. The protocol presented here results in human lacrimal gland organoid engraftment, mostly as cysts. Low organoid engraftment can arise due to organoids being injected in the wrong site. Training the injection procedure with a dye allows the tracking of the injection site and, ultimately, improves the injection accuracy. Alternatively, the mouse epidermis can be incised to have direct access to the lacrimal gland, as done in rats before22. This method takes longer but may be more accurate. On the other hand, with the present protocol, the organoids did not functionally integrate into the mouse lacrimal gland. Similar results have been observed for iPSC-derived lacrimal gland engraftment22. The transplantation method could be further improved by wounding the lacrimal gland in advance, using a dry eye mouse model, and/or injecting the organoids as single cells or small clumps. Nevertheless, adult stem cell-derived lacrimal gland organoids and the related toolkit can be the basis of future applications in lacrimal gland research and regenerative medicine.
Disclosures
Hans Clevers is the head of Pharma Research and Early Development at Roche, Basel, and holds several patents related to organoid technology.
Acknowledgements
We thank Yorick Post for the initial development of the protocol. This work was in part supported by an award from the Cancer Research UK Grand Challenge (C6307/A29058) and the Mark Foundation for Cancer Research to the SPECIFICANCER team.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL safe-lock centrifuge tubes | Eppendorf | EP0030 120.094 | |
| 3,3′-Diaminobenzidine tetrahydrochloride hydrate (DAB) | Sigma-Aldrich | D5637 | CAS: 868272-85-9 , CAUTION, 6 g/L solution can be stored aliquotted at -20 °C |
| 5x green GoTaq Flexi buffer | Promega | M891A | Store at -20 °C |
| A83-01 | Tocris | 2939 | Store at -20 °C, stock at 30 mM, 10000x |
| Advanced DMEM/F12 | Invitrogen | 12634-010 | store at 4 °C |
| Agar plates containing Ampicillin | Hubrecht Institute | ||
| Ampicillin sodium salt | Sigma-Aldrich | A9518 | |
| Autoclave VAPOUR-Line lite | VWR chemicals | ||
| B27 supplement | Invitrogen | 17504-044 | Store at -20 °C, 50x |
| BD Micro-Fine insulin needle 1 mL | BD Bioscience | 324825 | |
| Benchtop microscope DMI1 | Leica | ||
| Bovine serum albumine (BSA) | MP biomedicals | 160069 | Store at 4 °C |
| BTXpress | BTX | MDS450805 | |
| C57BL/6 mice | Hubrecht Institute | ||
| Cassettes | Klinipath | 410-02S | |
| CellBanker 1 | amsbio | 11910 | Cryopreservation medium, adhere to instructions |
| Centrifuge | Eppendorf | ||
| Citric acid monohydrate | J.T. Baker | 0088 | CAS: 5949-29-1 |
| Collagenase I | Sigma Aldrich | C9407 | Aliquots at 20 mg/mL, 20x, store at -20 °C |
| Conical tubes 15 mL | Greiner Bio-One | 5618-8271 | |
| Conical tubes 50 mL | Corning | CLS430828-500EA | |
| Coverslips 24 mm x 50 mm | Menzel-Gläzer | BB024050S1 | |
| Cultrex Basement Membrane Extract (BME), Growth Factor Reduced, Type 2 - extracellular matrix | R&D Systems, Bio-Techne | 3533-001-02 | Store at -20 °C, keep at 4 °C for up to 1 month |
| DAPT | Sigma Aldrich | D5942 | Store at -20 °C, stock at 10 mM, 1000x |
| Disodium hydrogen phosphate anhydrous | VWR chemicals | 28026.292 | CAS: 7558-79-4 |
| Di-sodiumhydrogenphosphate dihydrate | Sigma-Aldrich | 71643 | CAS:10028-24-7 |
| Dispase | ThermoFisher Scientific | 17105-041 | Aliquots at 50 U/mL, store at -20 °C until use, 400x |
| Disposable Scalpel Sterile N° 10 | Swann Morton | 3033838 | |
| DM4000 microscope | Leica | ||
| dNTPs 25 mM | Promega | U1420 | Mix all 4 nucleotides together, Store at -20 °C |
| Dpn1 | New England Biolabs | R0176 | |
| Dulbecco's Phosphate-bufferd Saline (DPBS) | Gibco | 14190144 | 1x |
| Easy strainers 70 µm | Greiner | 542170 | |
| Electroporation cuvette | Nepagene | EC002S | |
| EnVision+/HRP mouse | Agilent | K400111-2 | |
| Ethanol 100% | BOOM | 84045206;5000 | CAUTION, Use to prepare other Ethanol dilutions |
| Ethanol 70% | BOOM | 84010059.5000 | CAUTION |
| Ethanol 96% | BOOM | 84050065.5000 | CAUTION |
| EVOS FL Auto 2 Cell Imaging System | ThermoFisher Scientific | Live-imaging brightfield microscrope | |
| FGF10 | Peprotech | 100-26 | Store at -20 °C, stock at 100 mg/mL in base medium, 100x |
| Fiji | NIH, Fiji developers | ||
| Formaldehyde solution 4% | Sigma-Aldrich | 1.00496 | CAS: 50-00-0, CAUTION |
| Forskolin | Tocris | 1099 | Store at -20 °C, stock at 10 mM, 10000x |
| Glutamax | Gibco | 35050-061 | 100x |
| GoTaq G2 Flexi DNA Polymerase | Promega | M7805 | Store at -20 °C |
| Haematoxylin | VWR chemicals | 10047105 | Store at room temperature |
| HEPES | Gibco | 15630-080 | Store at 4 °C, 100x |
| Histocore H and C, Tissue embedding machine | Leica | ||
| Hot plate | Meidax | ||
| Human nucleolar antigen antibody | Abcam | ab-190710 | |
| Hydrochloric acid 5 N | ThermoFisher Scientific | 10605882 | CAS: 7647-01-0, CAUTION |
| Hydrogen peroxyde 30% | Chem-lab | CL00.2308.1000 | CAS: 7722-84-1, CAUTION |
| Hygromycin B-gold | InvivoGen | ant-hg | Stock at 100 mg/µL, 1000x |
| Hygromycin resistance cassette-containing plasmid | Andersson-Rolf et al, Nature Methods, 2017. doi: 10.1038/nmeth.4156 | ||
| IsoFlo 100% | Mecan | 5960501 | |
| LB medium | Hubrecht Institute | ||
| MgCl2 25 mM | Promega | A351H | Store at -20 °C |
| Microtome RM2235 | Leica | ||
| Midiprep DNA isolation kit | ThermoFisher Scientific | K210005 | |
| Miniprep DNA isolation kit | ThermoFisher scientific | K210003 | |
| N-acetylcysteine | Sigma Aldrich | A9165 | Store at -20 °C, stock at 500 mM, 400x |
| NEPA21 electroporator | Nepagene | ||
| Nicotinamide | Sigma Aldrich | N0636 | Store at -20 °C, stock at 1M, 100x |
| NOD Scid Gamma (NSG) mice | Hubrecht Institute colony | ||
| Noggin conditioned medium | U-Protein Express | Custom order | Store at -20 °C |
| Noradrenaline | Sigma Aldrich | A7257 | Store at -20 °C, stock at 100 mM |
| Oven | Memmert | Set at 58 °C | |
| P20, P200 and P1000 pipettes | Gilson | ||
| Paraffin | VWR chemicals | 10048502 | |
| Pasteur pipettes, glass plugged | ThermoFisher Scientific | 1150-6973 | |
| Pax6_C>T_F: AGACTGTTCCAGGATGGCTG | IDT | ||
| Pax6_C>T_R: TCTCCTAGGTACTGGAAGCC | IDT | ||
| pCMV_ABEmax_P2A_GFP | Addgene | 112101 | |
| Penicillin/Streptomycin | Invitrogen | 15140-122 | Store at -20 °C |
| Pertex | Klinipath | AM-08010 | |
| pFYF1320 | Addgene | 47511 | |
| Primocin | InvivoGen | ant-pm-1 | 1000X, store at -20 °C |
| Prostaglandin E2 (PGE2) | Tocris | 2296 | Store at -20 °C, stock at 10 mM, 10000x |
| Petri dish, 10 cm | Greiner | 633102 | |
| Q5 buffer | New England Biolabs | B9027S | |
| Q5 high-fidelity DNA polymerase | New England Biolabs | M0491S | |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | |
| QuickExtract DNA Extraction Solution | Lucigen | QE09050 | Store aliquots at -20 °C |
| R-spondin 3 conditioned medium | U-Protein Express | Custom order | Store at -20 °C |
| sgRNA Reverse Primer: TCTGCGCCCATCTGTTGCTT CGGTGTTTCGTCCTTTCCACAAG | IDT | ||
| Slides | StarFrost | MBB-0302-55A | Adhesive, ground |
| Sodium azide | Merck | 8.22335.1000 | CAS: 26628-22-8, CAUTION |
| Sodium cytrate dihydrate | J.T. Baker | 0280 | CAS: 6132-04-3 |
| Standard Forward Primer: “/5phos/ GTTTTAGAGCTAGAAATAGCAAG TTAAAATAAGGC | IDT | ||
| Subcloning efficiency competent cells DH5alpha | Invitrogen | 18265-017 | |
| Suspension cell culture plates (24-well) | Greiner Bio-One | 662102 | 24-well |
| Suspension cell culture plates (12-well) | Greiner Bio-One | 665102 | 12-well |
| T4 DNA ligase | New England Biolabs | M0202 | |
| TAE buffer | ThermoFisher Scientific | B49 | Stock at 50x, dilute to 1x with ultrapure water |
| Transposase-containing plasmid | Andersson-Rolf et al, Nature Methods, 2017. doi: 10.1038/nmeth.4156 | ||
| TrypLE Express Enzyme | Invitrogen | 12605-028 | store at 4 °C |
| U6_Forward primer: GGGCAGGAAGAGGGCCTAT | IDT | ||
| UltraPure Agarose 1000 | Invitrogen | 16550 | |
| Water bath | Tulabo | ||
| Xylene | Klinipath | 4055-9005 | CAS: 1330-20-7, CAUTION |
| Y-27632 | Abmole Bioscience | Y-27632 dihydrochloride | Store at -20 °C, stock at 10 mM, 1000x |
References
- Garg, A., Zhang, X. Lacrimal gland development: From signaling interactions to regenerative medicine. Developmental Dynamics. 246 (12), 970-980 (2017).
- Selinger, D. S., Selinger, R. C., Reed, W. P. Resistance to infection of the external eye: The role of tears. Survey of Ophthalmology. 24 (1), 33-38 (1979).
- Messmer, E. M. The pathophysiology, diagnosis, and treatment of dry eye disease. Deutsches Arzteblatt International. 112 (5), 71-81 (2015).
- Massie, I., et al. Development of lacrimal gland spheroids for lacrimal gland tissue regeneration. Journal of Tissue Engineering and Regenerative Medicine. 12 (4), 2001-2009 (2018).
- Tiwari, S., et al. Establishing human lacrimal gland cultures with secretory function. PloS One. 7 (1), 29458 (2012).
- Nguyen, D. H., Beuerman, R. W., Halbert, C. L., Ma, Q., Sun, G. Characterization of immortalized rabbit lacrimal gland epithelial cells. In Vitro Cellular & Developmental Biology. Animal. 35 (4), 198-204 (1999).
- Bannier-Hélaouët, M., et al. Exploring the human lacrimal gland using organoids and single-cell sequencing. Cell Stem Cell. 28 (7), 1221-1232 (2021).
- Hayashi, R., et al. Generation of 3D lacrimal gland organoids from human pluripotent stem cells. Nature. 605 (7908), 126-131 (2022).
- Jeong, S. Y., et al. Establishment of functional epithelial organoids from human lacrimal glands. Stem Cell Research & Therapy. 12 (1), 247 (2021).
- Hofer, M., Lutolf, M. P. Engineering organoids. Nature Reviews Materials. 6 (5), 402-420 (2021).
- Meyenberg, M., Ferreira da Silva, J., Loizou, J. I. Tissue specific DNA repair outcomes shape the landscape of genome editing. Frontiers in Genetics. 12, 728520 (2021).
- Geurts, M. H., et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell. 26 (4), 503-510 (2020).
- Overmyer, K. A., Thonusin, C., Qi, N. R., Burant, C. F., Evans, C. R. Impact of anesthesia and euthanasia on metabolomics of mammalian tissues: Studies in a C57BL/6J mouse model. PLoS One. 10 (2), 0117232 (2015).
- Driehuis, E., et al. Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer Discovery. 9 (7), 852-871 (2019).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. Journal of Visualized Experiments. (6), e253 (2007).
- Ran, F. A., et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Lian, J., Meng, X., Zhang, X., Hu, H. Establishment and genetic manipulation of murine hepatocyte organoids. Journal of Visualized Experiments. (180), e62438 (2022).
- Lõhmussaar, K., et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell. 28 (8), 1380-1396 (2021).
- Bloh, K., et al. Deconvolution of complex DNA repair (DECODR): Establishing a novel deconvolution algorithm for comprehensive analysis of CRISPR-edited Sanger sequencing data. The CRISPR Journal. 4 (1), 120-131 (2021).
- Latta, L., et al. Pathophysiology of aniridia-associated keratopathy: Developmental aspects and unanswered questions. The Ocular Surface. 22, 245-266 (2021).
- Veernala, I., et al. Lacrimal gland regeneration: The unmet challenges and promise for dry eye therapy. The Ocular Surface. 25, 129-141 (2022).
- Hayashi, R., et al. Generation of 3D lacrimal gland organoids from human pluripotent stem cells. Nature. 605 (7908), 126-131 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved