Multiplex Immunohistochemical Analysis of the Spatial Immune Cell Landscape of the Tumor Microenvironment
* These authors contributed equally
In This Article
Summary
This protocol describes in detail how immune cell characterization of the tumor microenvironment using multiplex immunohistochemistry is carried out.
Abstract
The immune cell landscape of the tumor microenvironment potentially contains information for the discovery of prognostic and predictive biomarkers. Multiplex immunohistochemistry is a valuable tool to visualize and identify different types of immune cells in tumor tissues while retaining its spatial information. Here we provide detailed protocols to analyze lymphocyte, myeloid, and dendritic cell populations in tissue sections. Starting from cutting formalin-fixed paraffin-embedded sections, automatic multiplex staining procedures on an automated platform, scanning of the slides on a multispectral imaging microscope, to the analysis of images using an in-house-developed machine learning algorithm ImmuNet. These protocols can be applied to a variety of tumor specimens by simply switching tumor markers to analyze immune cells in different compartments of the sample (tumor versus invasive margin) and apply nearest-neighbor analysis. This analysis is not limited to tumor samples but can also be applied to other (non-)pathogenic tissues. Improvements to the equipment and workflow over the past few years have significantly shortened throughput times, which facilitates the future application of this procedure in the diagnostic setting.
Introduction
Immune cells play a crucial role in the protection against pathogens such as viruses and bacteria, but also against cancerous cells1. Therefore, the immune system within the tumor microenvironment (TME) holds a lot of promise for discovering prognostic and predictive biomarkers2. Immune cell infiltrates have been correlated to prognosis in various types of cancer, although this has not been implemented in clinical care yet3,4. In most tumor types, high numbers of cytotoxic T cells and T helper 1 cells and/or low numbers of regulatory T cells are linked to good prognoses. Efforts are ongoing to incorporate a so-called "Immunoscore" into the TNM staging of colorectal cancer, turning it into TNM-I staging5,6. The Immunoscore is derived from the total number of T cells (detected with CD3) and cytotoxic T cells (detected with CD8) in two different tumor regions: the tumor core versus the invasive margin (IM) of tumors. The Immunoscore has also been proposed to be of prognostic value in other cancer types, such as melanoma, lung cancer, and breast cancer6,7,8,9. Furthermore, immune cell infiltrates may also correlate to response to checkpoint blockade immunotherapy10. However, these predictive biomarkers must be validated in prospective studies before they can be routinely implemented in clinical practice. Moreover, it has also been proposed that a single biomarker will be insufficient for meaningful prediction11. Therefore, creating a complete map of a patient sample by combining different biomarkers has been proposed as a more comprehensive predictive biomarker in a so-called "cancer immunogram"12.
Among the methods for studying immune cells within the TME, the oldest and most well-known technique is immunohistochemistry (IHC), routinely used for diagnostic testing in several diseases, especially cancer13. This technique was limited to the use of one or only a few markers14 for a long time and therefore, was outcompeted in research settings by other techniques such as flow cytometry and gene expression profiling (GEP). However, the formalin-fixed and paraffin-embedded (FFPE) tumor tissues typically used in routine diagnostics and research are not (optimally) suitable for flow cytometry and GEP. Furthermore, although GEP and flow cytometry provide a lot of insight into cell phenotype and function, the lack of spatial information is a major disadvantage. Therefore, heterogeneity within a sample, such as differences in immune cell-infiltrated versus immune cell-excluded areas of a tumor, could go undetected15. Novel platforms have been developed for multiplex analysis of FFPE tissues, such as multiplex IHC, imaging mass cytometry, and CO-Detection by indEXing (CODEX) that can be used to detect multiple markers simultaneously within a tissue section16. Immune cells in the TME are being widely studied to find the best biomarkers for immunotherapy. However, multiplex techniques and automated image analysis pose hurdles of their own.
Our laboratory has extensive experience in multiplex IHC staining using the Opal/Tyramide signal amplification (TSA) method and has automated this on a IHC platform (see the Table of Materials)17,18,19,20,21,22,23,24,25,26,27,28,29,30,31. We have optimized immune cell panels for the detection of different subsets of lymphocytes, myeloid cells, and dendritic cells (DCs). Tissues that contain dense immune cell areas - for lymphocytes or complex cell morphologies (i.e., myeloid cells and DCs) - are particularly challenging to analyze, with a risk of over- or underestimating the number of immune cells present. To overcome this problem, ImmuNet analysis software was developed by our group32, and this machine-learning pipeline improved the quality of the detection of these different types of immune cells immensely. A detailed protocol from obtaining the FFPE material to the analysis of immune cell densities in different tissue compartments and distances between immune cell types is described here.
This protocol outlines how the multiplex IHC panels are performed at the Radboud University Medical Center since the implementation of the digital pathology imager in 2022. The described multiplex IHC panels can be used for different carcinomas (e.g., lung, prostate, colorectal, bladder, breast) with the use of a pan-cytokeratin antibody as a tumor marker or for melanoma with the use of melanocyte-associated antibodies as tumor markers. These multiplex IHC protocols have been carefully optimized in terms of primary antibody concentration, fluorophore combinations, and the sequence of the staining procedure. We and others have described multiplex IHC panel optimization earlier17,33,34,35. Multiplex IHC panels can be adapted, but the described analysis pipelines need to be evaluated and potentially adjusted or retrained accordingly. The described seven-color multiplex IHC protocols make use of the Opal fluorophores Opal480, Opal520, Opal570, Opal620, Opal690, Opal780, and 4',6-diamidino-2-phenylindole (DAPI), so that easy unmixing and fast scanning on the imager is enabled with "Multispectral One Touch ImmunoFluorescence" (MOTiF). Nine-color staining and scanning is not described in this protocol as this requires even more finetuning of the experimental setup and another mode of scanning on the imager that uses the liquid crystal tunable filter.
Protocol
Patient material that is shown for this protocol was part of a previously conducted study and was officially deemed exempt from medical ethical approval by the local Radboudumc Medical Ethical Committee concurrent with Dutch legislation (file number 2017-3164)30.
1. Collection of FFPE material, selection of blocks, and preparation of samples
- Retrieve FFPE block identifiers from patient files through treating physicians or pathologists. Check with local regulations whether ethical permission is required.
- Request FFPE blocks from the local pathology archive or external hospital(s).
NOTE: It is also possible that tumor material or a biopsy is acquired for a particular study. This can be the case for small clinical trials or animal studies. In these cases, processing of the tissue sample may be the responsibility of the researcher. - When multiple FFPE blocks are available, select the most representative FFPE block containing viable tumor tissue, preferentially with surrounding stromal tissue present by assessing the hematoxylin and eosin (HE)-stained slides (Figure 1).
NOTE: It is advised to obtain an expert opinion for this selection (e.g., a pathologist). It is possible that HEs are unavailable for assessment of the content of an FFPE block and new ones need to be made for the selection. Go to section 2 for a description. - Cut FFPE ribbons of 4 µm thickness on a microtome.
NOTE: The thickness can be between 1 µm and 6 µm without noticeable staining impact; however, 4 µm is the most standard. - Mount the samples on glass slides at a position that is favorable for the fluidics of the autostainer (Figure 2A-C) using one of the methods described below:
- Place the sections on the surface of distilled 40 °C water in a water bath to stretch out and pick them up with a glass slide.
OR
Place glass slides on a 40 °C heating plate, making sure to cover the spot where the section is to be mounted on the slide with a drop of distilled water. Place the section on top of this drop with forceps and allow it to stretch out. Absorb distilled water using a paper towel and remove excess water by tapping the slide.
NOTE: Placing tissue sections too close to the label of the slide will result in suboptimal staining (Figure 2D,E). We tend to mount 6-10 glass slides per sample to perform the different multiplex IHC panels and to have a backup.
- Place the sections on the surface of distilled 40 °C water in a water bath to stretch out and pick them up with a glass slide.
- Let the mounted glass slides dry at 56 °C for 1 h or overnight at 37 °C.
- Use the mounted glass slides for the experiment or store these in boxes at 4 °C.
NOTE: In our experience so far, these mounted slides can be stored for years before multiplex IHC staining is carried out.
2. Generating hematoxylin and eosin-stained slides
NOTE: All following steps of section 2 are to be carried out in a fume hood.
- Deparaffinize slides in xylene (2 x 5 min).
- Rehydrate in ethanol (99.6% 1 x 5 min; 95% 1 x 5 min; 70% 1 x 2 min). Alternatively, dip the slides 3x in 99.6% ethanol.
- Wash the slides in distilled water (2 min).
- Stain the nuclei with hematoxylin (10 min).
- Wash the slides with distilled H2O (5 min).
- Stain the slides with eosin (5 min).
- Dehydrate the slides by dipping 3x in 99.6% ethanol.
- Dip the slides 2x in xylene.
- Add a few drops of mounting medium and seal with a coverslip.
- Let the slides harden and take the slides out of the fume hood when all the chemicals have evaporated.
3. Performing monoplex and multiplex IHC in the autostainer
- Calculate how much reagent is needed depending on the number of samples to be stained.
NOTE: Per run, the autostainer has a capacity of 30 slides and takes ~18 h to complete the multiplex IHC protocol with six antibodies. When more slides need to be stained, multiple batches can be put in every night of the (work)week; 4 nights of 30 slides = 120 slides per week.- Prepare all necessary reagents at the start of the week. The autostainer system dispenses 150 µL of reagent per slide. Use the 6 mL titration containers for antibody and Opal reagents and the 30 mL containers for the blocking reagent and the secondary antibody-horseradish peroxidase.
NOTE: The 6 mL containers have convenient inserts that can easily be taken out and replaced when necessary. With reagent calculations, one has to consider the dead volume of 1.6 mL or 300 µL for the 30 mL container or 6 mL titration container, respectively. - Dilute all Opal fluorophores and digoxigenin (DIG) 1:100 in the provided diluent; dilute Opal780 1:25 in the antibody diluent. Dilute all the primary antibodies in antibody diluent, with dilutions specified in Supplemental File 1.
- Prepare all necessary reagents at the start of the week. The autostainer system dispenses 150 µL of reagent per slide. Use the 6 mL titration containers for antibody and Opal reagents and the 30 mL containers for the blocking reagent and the secondary antibody-horseradish peroxidase.
- To follow this protocol, run monoplex IHC (Supplemental File 2) on slides containing both tonsil control tissue and other (tumor) tissue types of interest before starting with the actual multiplex IHC experiment to make sure all reagents are prepared well.
NOTE: Monoplex IHC takes ~3.5 h and can be checked before the end of that day for signal patterns and intensity. If certain signals are too weak (Figure 3), adjustments to reagents can be made. - For autofluorescence correction, prepare a slide with (tumor) tissue containing autofluorescent structures, such as blood and collagen. Prepare this slide simultaneously with monoplex IHC slides, but with blocking reagent replacing the antibody and Opal reagents (Supplemental File 3).
NOTE: In principle, such a slide can be reused for multispectral imaging until autofluorescence correction is not optimal anymore. However, with highly autofluorescent tissues, such as the brain and liver, it is advisable to use that tissue for the autofluorescence correction. - With each multiplex IHC run, load 29 samples into the autostainer system with one control tissue slide to check the performance of each multiplex IHC run.
- Download multiplex IHC protocols from the website of the autostainer under the Downloads tab and adjust them to fit each customized multiplex IHC panel36. For multiplex IHC, see Supplemental File 4 for the protocol and for customized multiplex IHC panels, see Supplemental File 1.
- After completion of the staining protocol, take the slides out of the autostainer and put them in a container with wash buffer.
- To prevent contamination of the autostainer system with DAPI as samples are already stained at very low concentrations, apply DAPI manually before covering the slides with coverslips. Add two drops of DAPI per mL of wash buffer and incubate for 5 min at room temperature in the dark.
NOTE: For building spectral libraries, it is important to not have any DAPI stained in the samples. One drop of DAPI per mL of wash buffer and 10 min of incubation at RT is also possible. - Wash the slides 3x with wash buffer.
- Place the slides on paper towels and tap the excess wash buffer off the slides.
- Pipet a few drops of mounting medium on the tissue.
- Place a glass coverslip gently on top of the mounting medium to cover the slide at an angle to avoid air bubbles.
- Remove excess mounting medium and air bubbles by gently pushing on the glass coverslip with forceps or a clean pipet tip.
- Leave the slides undisturbed for ~24 h before the mounting medium solidifies, either horizontally on a microscopy slide board or load them directly into the microscope for imaging.
- After the mounting medium is solidified or after the slides are imaged, store the slides in microscopy boxes at 4 °C.
4. Imaging using the digital pathology imager and annotation of scan files
- Turn on the imager by pushing the power button on the right of the machine. After at least 20 s, start up the software.
NOTE: Wait for 20 s to allow the hardware to start up correctly. - Load the slides into the cassettes per four slides.
- Optional: Enter the slides into a .csv file for which a template can be downloaded (Supplemental File 5). To load the .csv file into the program, save it at C:\Users\Public\Akoya\VectraPolaris\States.
NOTE: A maximum of 20 cassettes or 80 slides can be loaded simultaneously.
- Optional: Enter the slides into a .csv file for which a template can be downloaded (Supplemental File 5). To load the .csv file into the program, save it at C:\Users\Public\Akoya\VectraPolaris\States.
- Reference settings
- Open Check Dashboard from the main menu.
NOTE: A cassette with reference slides is provided by the manufacturer and can optionally be kept permanently in slot 20. - Set the brightfield references on the provided slide once per week according to the manufacturer's instructions (takes a few minutes).
- Set the fluorescence references on the provided slide once per month according to the manufacturer's instructions (takes more than 1 h).
- Open Check Dashboard from the main menu.
- Making or adjusting the protocol
- Go back to the main menu and click Edit Protocol to make a protocol.
- Click New… and select Fluorescence as Imaging Mode, Multispectral Slide Scan, and Opal Polaris 5, 6, and 7 color under the Staining option.
- Give the protocol a name under Protocol Name and save it under a study by selecting a study from Available Studies or create a study under Create New Study | Study Name.
- Finish by selecting Create Protocol.
- For this type of scanning, use only the left window Multispectral Slide Scan Settings; ignore the window on the right Multispectral Field Settings.
- Scan the slides at different magnifications. To follow this protocol, scan at 20x magnification by leaving the Pixel Resolution at 0.50 µm (20x).
- Set the exposure times by selecting Scan Exposures.
- Load the cassette in which the slides are kept by selecting the correct slot under the Load Carrier option.
- To help navigate through the slides, select Take Overview to acquire an overview image of the carrier containing the slides after the carrier is loaded. To turn this on or off automatically, click the gear icon on the top right, go to Preferences…, and tick the option on or off under Navigation Overview Image to enable Automatically image carrier when loading for interactive tasks.
- Set exposure times per filter on the corresponding monoplex IHC stained slides by selecting Set Scan Exposures and finding different spots with a positive signal. Manually focus or use Auto Focus and select Autoexpose after switching to the compatible filter for that signal. Select the lowest exposure time to prevent overexposure and take snapshots of each slide for reference after all exposure times are set (Figure 3).
NOTE: Ignore the Set Field Exposures option for this type of scanning. - Set the exposure times on a multiplex stained slide by checking all the filters on a few locations with positive signal. Reduce the lowest autoexposure time by 10% to prevent overexposure and take a few snapshots after all exposure times are set.
- Take snapshots of the unstained slide for autofluorescence compensation by using the Sample AF filter to navigate (Figure 3H).
NOTE: Locations with erythrocytes and collagen structures are of interest. The exposure time of the Opal480 filter may need to be reduced for strong autofluorescent regions. If the Opal480 signal is strong enough, it should still be separated well (see section 6) from the autofluorescent structures because of the implementation of the proprietary Sample AF filter. - Assess the quality of the staining and imaging using the software (see sections 5 and 6; Figure 4, Supplemental File 6: Supplemental Figure S1, and Supplemental Figure S2).
- Select the Save… button to make sure that the protocol and its adjusted exposure times are saved into the protocol.
NOTE: When the protocol is already saved, no extra notification of unsaved adjustments is given by the software so far.
- Automatic scanning of slides
- Go back to the main menu and click Scan Slides to scan the slides.
- Manually enter slide names/IDs and corresponding tasks and protocol under Configure Tasks or automatically from the previously made .csv file with Load Setup.
- Click Scan to start the scanning.
- Wait for a window to pop up to save the scan setup. Click Save to use the default settings and start scanning.
NOTE: Scanning using this method takes ~10-20 min per slide. Depending on the number of slides, scanning can take up to a full day. - Check whether the scanning of the slides was successful for all slides by looking for any error messages. To know if scanning is successful, look for a saved Akoya whole slide scan file (.qptiff) of the scan and the complete tissue in the scan.
5. Annotation of data using the slide viewer
- Go back to the main menu and click Launch Phenochart to open the slide viewer.
- If the scan files are not directly visible, appoint their location by first clicking on the gear icon in the upper right corner, go to Change Browser Location… and randomly select one of the .qptiff files of the dataset of interest.
NOTE: Data are by default stored at D:\Data\VectraPolaris. - Load a slide by selecting it and clicking Load in the upper right corner or by double-clicking on it.
- Login by clicking on the Login button in the upper right corner.
NOTE: The username can be just the initials or name and is used to keep track of who made which annotations. - To perform unmixing, click on the Unmixing button on the top and select the Opal + AF option.
NOTE: This is useful to get rid of some of the autofluorescent signal near the Opal 480 channel, but not all. - To generate an algorithm for batch-processing the data, select representative images using the Stamp using the for inForm Projects 1 x 1 images (image size: 928 µm x 696 µm) option.
NOTE: A few representative stamps containing tumor, stroma, background, and different types of immune cells are selected throughout the dataset to end up with ~20-30 images. - Depending on what needs to be analyzed in the tissue, select a region of interest using the ROI option and select for inForm Batch. Manually delete images that do not need to be analyzed, such as images that are too far away from the tumor or in the background.
NOTE: We tend to draw an ROI around the whole tumor and select one extra image away from the tumor region to be able to analyze an IM of ~0.5 mm.
If the drawn ROI is relatively small, the ROI will consist of 2-9 merged 20x images. As this is not preferred by us, manually stamp the tissue of interest (selected for inForm Batch) to circumvent this. - When finished annotating, let the annotations be automatically saved and load the next slide.
- During the annotation process, check if the slides are correctly scanned.
- If a .qptiff file is missing or a slide is not successfully scanned, check if any tissue is present on the slide, clean the slide with 70% ethanol, and scan again.
- If the tissue is not fully scanned, thereby missing a potentially important (tumor) region, or if the scanning of the important region was out of focus, clean the slide with 70% ethanol and scan again.
NOTE: In both cases, it can also help to encircle the tissue with a marker on top of the coverslip to help the system to locate the tissue and try scanning again (Supplemental File 6: Supplemental Figure S3). In our hands, a thin red marker worked better than a thick black marker.
- Once scanning and annotation of all samples are completed, back up the data by storing them on a different computer or external disk.
6. Spectral unmixing
- Open the inForm Automated Image Analysis Software.
- Load the images into the software by File | Open Image; select .qptiff files. Let the stamps, marked as inForm Projects in step 5.6, be loaded into the project.
- Load the .qptiff files that are imaged for the autofluorescence compensation.
- To compensate for autofluorescence, use the select autofluorescence on the image tool to draw a line on the image from the unstained slide through different types of structures that are autofluorescent, such as erythrocytes and collagen.
- In the Edit Markers and Colors… section, assign marker names that correspond to the Opal fluorophore and adjust the color to the preferred one.
- To unmix the fluorophores, select Prepare All in the lower left corner.
- Go through the images and check if all signals are visible in the images and if the unmixing went well. Select the eyeball icon to turn off and on all the markers one by one to check the quality.
- Optionally, train the algorithms for tissue segmentation, cell segmentation, and phenotyping.
- Go to the Export tab and make a new empty export directory by clicking the Browse… button under the Export Directory.
- Under the Images to export:, select Composite Image and Component Images (multi-image TIFF).
- Select File | Save | Project to save the algorithm at a certain location.
- Go to Batch Analysis tab vertically on the left for the batch processing of slides.
- Select Create separate directories for each item under the Export Options.
- To add slides for analysis, select .qptiff files under the Add Slides… button and load them into the batch analysis.
- Select Run to start the batch processing of slides.
7. ROI drawing
- Create a folder with only the component files from section 6, but keep the hierarchical folder structure intact (component files being in folders named by sample/slide).
- Open the QuPath whole slide viewer software.
- Click Create project on the left and select/make a new empty folder with a suitable name.
- Click Automate and select Show script editor.
- Copy-paste the script that is available in Supplemental File 7. At line 34, change the location to where the slide folders containing all the component files are (the folder created in step 7.1.
- Select Run and return when the batch stitching of slides is finished (the next day or later) to continue.
- Drag the generated .ome.tif files into the QuPath project and save them as a project.
- When a new window automatically pops up, select Set image type | Fluorescence and click Import.
- In the menu to the left, observe the list of samples; double-click on one to open the sample (Figure 5A).
- To adjust the intensity of the channels to make them better visible, click the contrast icon.
- Select all channels and click Reset.
- Toggle off autofluorescence.
- To start drawing an ROI for the tumor, click the contrast icon and select Show grayscale. Select the tumor marker channel and adjust the intensity to make it optimally visible (Figure 5B).
- Click the brush tool to draw a tumor ROI roughly.
- While selecting the wand tool, click outside the ROI while pressing the alt key to smooth the ROI from the outside (Figure 5C).
- Merge separated tumor pieces with the same ROI.
- Give the ROI a suitable name such as tumor by right-clicking the annotation in the list on the left; select Set properties and enter the name.
- To make an ROI for the IM, expand the existing ROI from the tumor region by selecting: Objects | Annotations… | Expand annotations.
- Select what size the expansion radius should be and select Remove interior and Constrain to parent (Figure 5D).
- Click the contrast icon, select the autofluorescence channel, and adjust the intensity to make it optimally visible.
- Click the wand and adjust the ROI while pressing the alt key to smooth the ROI from the outside and remove any background that should not be part of this ROI.
- Give the ROI a suitable name such as invasive margin or IM by right-clicking the annotation in the list on the left, select Set properties, enter the name, and optionally change its color to green.
- Save the annotations: File | Export objects | Export All objects and click OK with the default selection on Export as FeatureCollection and save it at a preferred location.
8. Immune cell detection
- As ImmuNet uses component data (multi-channel TIFF files) for both training and inference, split annotations into training and validation sets. To train the model, follow the steps described in the Readme file of the repository, substituting the example dataset and annotations with the desired data. Apart from different immune cells, provide the model with negative examples by making background annotations at sites that should not be recognized as a cell of interest: tumor cells, other cells, or "no cells" (structures that could be confused with cells of interest); see the ImmuNet publication for details32.
- Using the validation annotations, ensure that the performance is satisfactory. Look at the error rate per annotation type - a share of validation annotations that the model has not detected - the most straightforward evaluation metric. Evaluate the performance with respect to false positives by making a few fully annotated ROIs and calculating the F-scores.
- In addition to quantitative evaluation, visually inspect the prediction to get a qualitative sense of the errors the model tends to make (Figure 6, Supplemental File 6: Supplemental Figure S4, and Supplemental Figure S5). If the model performance is judged to be insufficient, visualize the prediction for some tiles as described in the repository and check which sites are the most error-prone. Make more annotations at such sites and re-run model training and evaluation.
- When the target performance is achieved, run the inference for the whole dataset as described in the Inference for the whole dataset section of the repository Readme. Use the obtained .csv files with the model prediction as input for data analysis (write a Python or R script for that).
9. Prediction phenotyping and data analysis
NOTE: In this section, we give an example of simple data analysis for a single melanoma sample stained with the lymphocyte panel, which combines the locations of immune cells identified by ImmuNet (section 8) and ROIs delineated with QuPath (section 7). The analysis has been performed in R 4.1.1 (a script is provided as Supplemental File 8). The script requires the packages: plyr 1.8.8, dplyr 1.0.8, tidyr 1.2.0, sf 1.0-7, ggplot2 3.4.0, RANN 2.6.1, and RColorBrewer 1.1-2, which can be installed with the install.packages() command. As an input, it takes a .csv file with ImmuNet's prediction of a sample and a file with ROIs exported from QuPath. Steps 9.1-9.6 describe the analysis of a single sample performed in the provided script, and sections 9.7-9.9 describe options for the analysis of multiple samples.
- After loading ImmuNet's prediction into R, determine the thresholds for predicted marker expression by plotting the markers that define phenotypes against each other and selecting the thresholds that separate the populations best.
NOTE: The gating strategy used for the given sample is shown in Figure 7B. Gating strategies for the myeloid and dendritic cell panels are shown in Supplemental File 6: Supplemental Figure S6 and Supplemental Figure S7. - After determining the thresholds, use them to assign each ImmuNet prediction a phenotype defined in a panel. In some predictions, observe that neither of the predicted markers is above the threshold or the combination of markers considered expressed after thresholding may be inconsistent (e.g., CD3+ CD20+ predictions in the lymphocyte panels). If good model performance is achieved at step 8.3, the fraction of such predictions will be small; filter them out before the analysis.
- To separately analyze ROIs for the tumor and its invasive margin up to 100 µm drawn in QuPath, load the corresponding GeoJSON files in R, and for each prediction, determine the ROI into which the prediction falls.
- For a sanity check and as a part of the exploratory data analysis, visualize the immune cells found in a sample separately in the corresponding ROIs together with ROIs' boundaries (Figure 7A).
- Now, calculate the densities of different immune cells separately for each ROI. The densities found in the given sample are shown in Table 1.
- If multiple samples are available, visualize the distribution of cell densities. Log-transform the density values to achieve normally distributed values.
NOTE: When counts of certain phenotypes are 0, these cannot be Log-transformed, leading to missing values. To overcome this problem, LaPlacian smoothing can be applied by adding 0.5 to all cell counts first before dividing by the surface area. - Analyze the density values and plot them using the software of choice (Figure 8).
- Preserved locations of cells enable spatial analysis. For instance, for each detected immune cell, find a nearest neighbor, and then for each phenotype, calculate the percentage of cases when the different phenotypes occur as the nearest neighbor.
NOTE: Since the number of natural killer (NK) cells found in this sample was very small, we excluded them from this analysis. The obtained results for Tumor and IM ROIs are given in Figure 9.
Representative Results
FFPE blocks containing tumor tissue were selected on the basis of pathology reports and HE-stained slides. When multiple tumor lesions are resected from the patient and/or tumor samples are large, these are divided over multiple FFPE blocks. We prefer analyzing immune cells in both the tumor compartment and what is known as the invasive margin (IM) of the tumor. The IM is non-cancerous stromal tissue that is adjacent to the tumor. Therefore, when there are multiple FFPE blocks available for one tumor sample, the FFPE blocks that contain both tissue types are selected. As seen on the HE-stained slides, one FFPE block contained tumor tissue and stromal tissue adjacent to the tumor (Figure 1A). Another FFPE block from the same tumor contained much less surrounding stromal tissue (Figure 1B). However, for some tissue samples there is no choice in FFPE blocks or the IM is not present in any of the FFPE blocks. This is often the case for (needle) biopsies, which must be kept in mind during data interpretation.

Figure 1: HE-stained slides of a melanoma tumor specimen. (A) An example of a tumor sample with stromal tissue adjacent to the tumor (IM) in the upper right corner of the sample (indicated with black arrowheads). (B) Another sample from the same tumor lesion with little to no stromal tissue present in the sample. Scale bars = 5 mm. Abbreviations: HE = hematoxylin and eosin; IM = invasive margin. Please click here to view a larger version of this figure.
{kind=link}
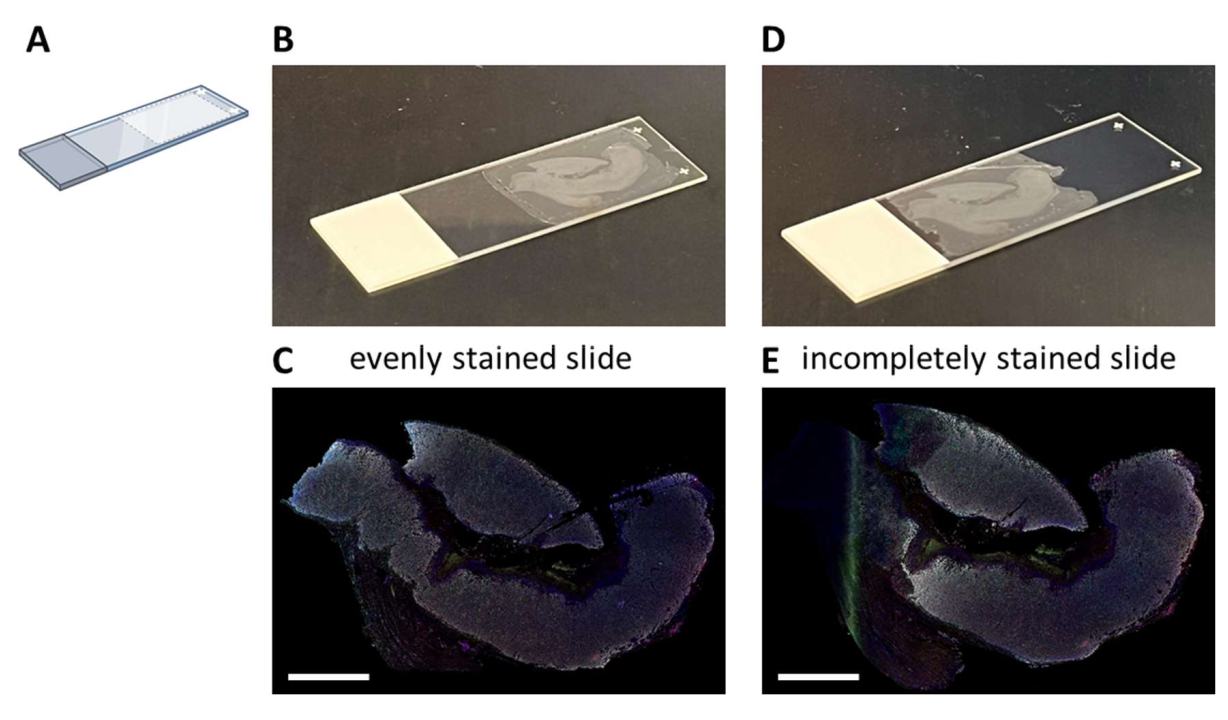
Multiplex IHC staining with a proposed seven-color panel (Supplemental File 4) can be performed either manually in a 3-day staining process (considering normal working hours) or overnight in an autostainer. When making use of the autostainer, sections have to be mounted on a particular location on the glass slide that enables optimal fluidics of the system (Figure 2A). When sections are correctly mounted on slides (Figure 2B), they will be evenly stained (Figure 2C). If sections are not optimally mounted on the glass slide (Figure 2D), it often results in a suboptimal staining pattern (Figure 2E) because the fluidics of the autostainer do not reach the (complete) tissue. This can happen when samples are very large, or when mounted slides are provided by someone who is not aware of this issue. In these cases, only the well-stained part of the slide should be selected for analysis. Another choice for these types of samples could be to stain them manually to spread the liquids optimally.

Figure 2: Mounting of the FFPE section to the glass slide and impact. (A) Schematic of where to mount on the glass slide for optimal staining on the autostainer. (B) Example of a correctly mounted slide. (C) Correctly mounted slides result in an evenly stained tissue section. (D) Example of a suboptimal mounted slide. (E) Suboptimal mounted slides can result in an incomplete stained tissue section as seen on the left side of this picture. Scale bars = 5 mm. Abbreviation: FFPE = formalin-fixed and paraffin-embedded. Please click here to view a larger version of this figure.
{kind=link}
When large multiplex IHC experiments are performed in multiple staining rounds and large quantities of solutions need to be prepared, it is best to first test these reagents in a monoplex IHC run before proceeding to the multiplex IHC. Monoplex IHC is checked with the digital pathology imager for expected staining patterns and exposure times are set with the corresponding filters on control slides (Figure 3A-H). Tonsil tissue is used as a positive control for most immune cell markers. As DAPI exposure time in tonsil control tissue is always higher than in other tissues (Figure 3G), DAPI exposure time has to be set on the tissue type to be studied. Regular exposure times with this type of scanning are between 1 ms and 30 ms, depending on the fluorophore and filter (Figure 3I). When a monoplex IHC exceeds these numbers or the staining pattern is not as clear as expected, the antibody solution should be adjusted or replaced. In the example shown here, we decided to increase the concentration of FOXP3 (Figure 3C and Figure 3I) to have the intensity more in range with the other markers. Autofluorescence may also be stronger in other tissues than in tonsil control tissue. In our setting, the exposure time for the Sample AF filter is between 25 ms and 50 ms (Figure 3H,I).

Figure 3: Setting of exposure times on monoplex IHC and unstained control samples. (A) CD20 - Opal 480 signal in tonsil control tissue. (B) CD3 - Opal 520 signal in tonsil control tissue. (C) FOXP3 - Opal 570 signal in tonsil control tissue. (D) CD56 - Opal 620 signal in tonsil control tissue (E) CD8 - Opal 690 signal in tonsil control tissue. (F) Tumor marker - Opal 780 signal in tonsil control tissue. (G) DAPI signal in tonsil control tissue is often weaker than the tissue type of interest. (H) Autofluorescence - sample AF signal in tumor control tissue. (I) Screenshot of exposure times before adjusting it with 10% and checking on multiplex IHC stained slides. Scale bars = 100 µm. Abbreviations: AF = autofluorescence; IHC = immunohistochemistry; DAPI = 4'6-diamidino-2-phenylindol. Please click here to view a larger version of this figure.
{kind=link}
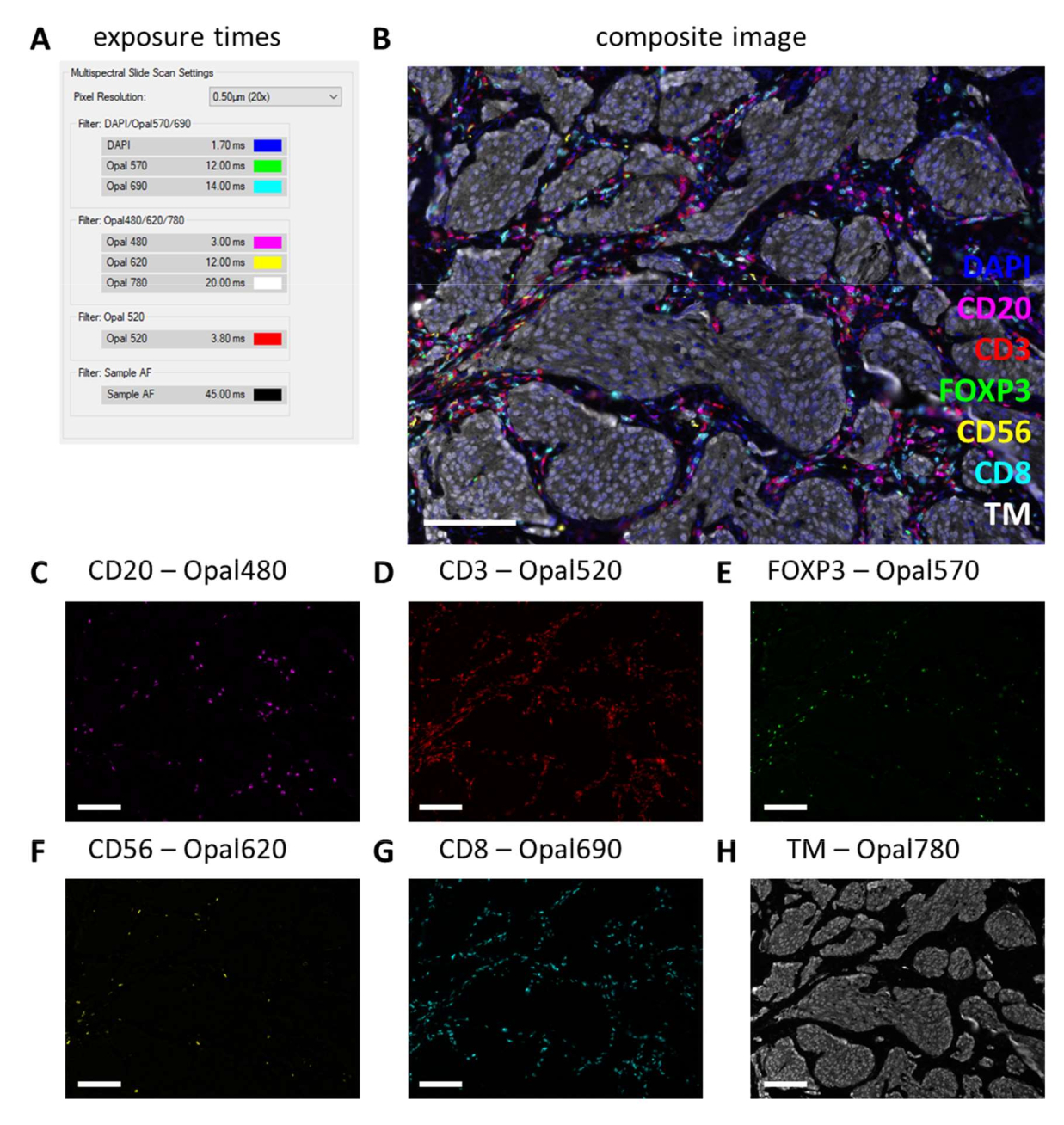
After multiplex IHC is performed, exposure times are adjusted from the monoplex IHC settings by checking a few multiplex IHC slides and selecting auto-expose. With this type of scanning, there is no saturation protection option and therefore, it is extremely important to avoid setting the exposure too high, thereby avoiding overexposure. Overexposure hampers the spectral unmixing of the fluorophores. We often do not set exposure times exceeding the exposure times that were based on the monoplex IHC and we only decrease exposure times for markers that are stronger in the multiplex IHC (Figure 3G and Figure 4A). By auto-exposing on different locations on a few slides, it can be observed that the exposure times of a few filters are still too high. These must be adjusted to the lowest number that is observed when using the auto-exposure setting and subtract another 10% of the value to prevent overexposure in other unseen locations (Figure 4A). With this method, the exposure times can be lower for certain filters than the ones that were set on monoplex IHC. However, with a successful multiplex IHC experiment, all the markers should be observable, at least on the control slide (Figure 4B-H, Supplemental File 6: Supplemental Figure S1, and Supplemental Figure S2). Consider that certain markers may not be present in every sample. By including a control slide containing at least a tonsil section, successful staining of all markers of the standard panels and signal strength can be verified.

Figure 4: Example of a successfully stained section with the lymphocyte panel in a melanoma tumor specimen. (A) Exposure times used to record this multiplex IHC sample. (B) Composite image of multiplex IHC lymphocyte panel within tumor tissue. (C) CD20 - Opal 480 signal in magenta. (D) CD3 - Opal 520 signal in red. (E) FOXP3 - Opal 570 signal in green. (F) CD56 - Opal 620 signal in yellow. (G) CD8 - Opal 690 signal in cyan. (H) TM - Opal 780 in white. Scale bars = 100 µm. Abbreviation: TM = tumor marker; IHC = immunohistochemistry. Please click here to view a larger version of this figure.
{kind=link}
Multiplex IHC slides are fully scanned by the digital imager. Tiles for subsequent analysis are selected in the slide viewer. However, when more specific regions need to be analyzed such as tumor versus IM, these regions of interest (ROIs) can be drawn using QuPath. After batch processing of the tiles that are selected in the slide viewer is completed, component files are merged back together (Figure 5A and Supplemental File 7). Using the tumor marker channel (Figure 5B) and the magic wand tool in QuPath, the tumor outline can be traced to form the "Tumor ROI" (Figure 5C). Next, the Tumor ROI can be expanded with a certain distance, in this case, 500 µm, to create an "invasive margin ROI" (Figure 5D). Any unwanted background (non-tissue) is removed from this ROI with the magic wand tool by looking at the autofluorescence signal (Figure 5E). Both Tumor ROI and IM ROI are saved as a GeoJSON file for further processing (Figure 5F).

Figure 5: Tumor ROI and invasive margin ROI drawing process in QuPath. (A) Merged component files. (B) Grayscale image showing only the tumor marker channel. (C) Tumor ROI is drawn around the tumor marker signal. (D) A new ROI is made by expanding the Tumor ROI by 100-500 µm to form the IM ROI. (E) The IM ROI is adjusted to only include stromal tissue by excluding background (negative signal) and other large tissue structures such as fat, blood vessels, and hair follicles. (F) The resulting tumor ROI and IM ROI are saved and exported into GeoJSON files for further processing of the regions. Tumor ROI is displayed with a red outline and the IM ROI with a green outline. Scale bars = 2 mm. Abbreviations: ROI = region of interest; IM = invasive margin; GeoJSON = Geographic JavaScript Object Notation. Please click here to view a larger version of this figure.
{kind=link}
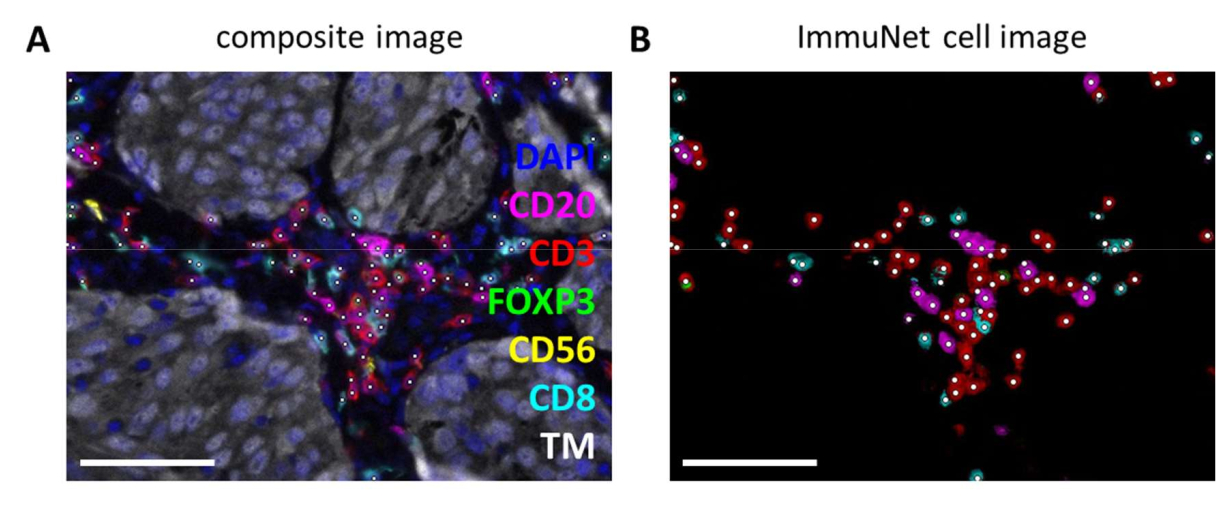
ImmuNet networks can be used to detect immune cells. For the lymphocyte panel, the experimental composite image (Figure 6A) can be visually compared with the immune cells detected by the software (Figure 6B). Similar visual comparisons can be made for the myeloid panel (Supplemental File 6: Supplemental Figure S4) and the dendritic cell panel (Supplemental File 6: Supplemental Figure S5).

Figure 6: Lymphocytes recognized by ImmuNet. (A) Composite image of Figure 4B showing cells recognized by ImmuNet with white dots. (B) Cells recognized by ImmuNet and subsequent detected marker expression. Scale bars = 50 µm. Abbreviation: TM = tumor marker. Please click here to view a larger version of this figure.
{kind=link}
Immune cells detected by ImmuNet and saved in .csv format can be imported into any programming language for further analysis. We performed spatial visualization and gating in R (Supplemental File 8). The detected cells can then be spatially visualized (Figure 7A, Supplemental File 6: Supplemental Figure S6 and Supplemental Figure S7). Gating on pseudomarker expression can be performed to phenotype the individual immune cells (Figure 7B).

Figure 7: Gating strategy of lymphocyte panel. (A) Immune cells detected in Tumor and Invasive Margin regions of interest delineated with QuPath. (B) Gating of all cells detected by ImmuNet from part A. Lymphocytes are first gated on CD20+ B cells and CD3+ T cells. CD3+ T cells are further gated for CD8 and FOXP3 expression. The CD20-CD3- population is gated for CD56+ natural killer cells. Please click here to view a larger version of this figure.
{kind=link}
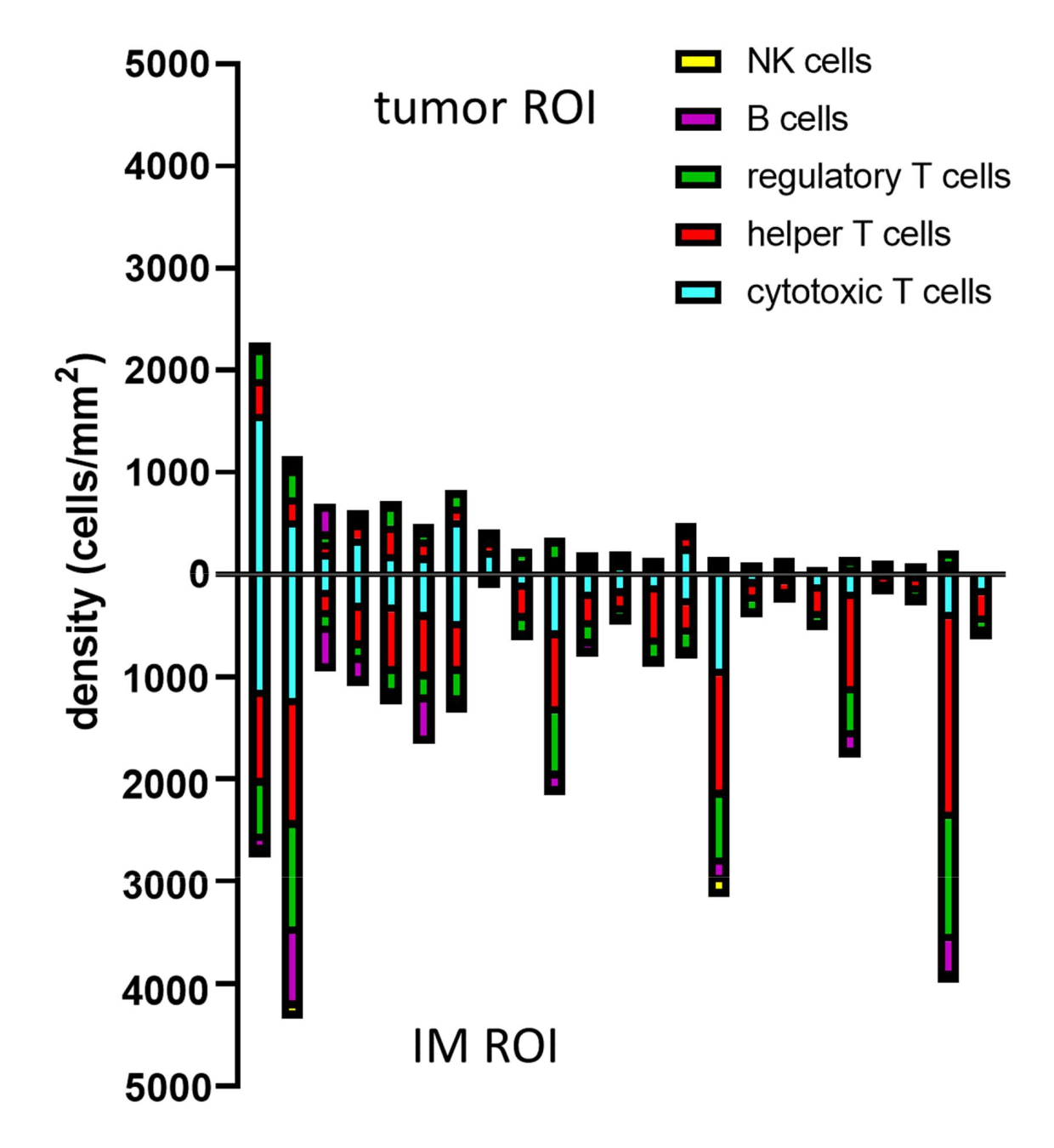
When phenotypes of the predicted cells are determined with gating, cell densities of different phenotypes can be calculated within different ROIs. This is calculated by dividing the total number of cells per phenotype by the surface area of the ROI (Table 1, Figure 8, and Supplemental File 8). Here, B cells are defined as CD3-CD20+, helper T cells as CD3+CD20-CD8-FoxP3-, regulatory T cells as CD3+CD20-CD8-FoxP3+, cytotoxic T cells as CD3+CD20-CD8+FoxP3-, and NK cells as CD3-CD20-CD56+.
| Phenotype | Density in Tumor (cells/mm2) | Density in IM (cells/mm2) |
| B cell | 185.74 | 145.62 |
| Helper T cell | 301.46 | 157.51 |
| Regulatory T cell | 38.53 | 19.53 |
| Cytotoxic T cell | 185.35 | 83.21 |
| NK cell | 0.18 | 0 |
Table 1: Densities of phenotypes in ROIs. Densities of cells of different phenotypes found in a single melanoma sample stained with the lymphocyte panel. Densities are calculated separately in Tumor and IM ROIs. Abbreviations: IM = invasive margin; ROI = region of interest.

Figure 8: Example of data analysis for multiple samples. Density analysis of different lymphocyte phenotypes in tumor and IM of 23 primary melanoma tumors. Abbreviations: IM = invasive margin; ROI = region of interest. Please click here to view a larger version of this figure.
{kind=link}
To dive more into the spatial information of these immune cells, it is also possible to determine distances between identified phenotypes or percentages of phenotypes of nearest neighbors in a sample (Figure 9).

Figure 9: Example of nearest-neighbor analysis for a single sample. Percentage of nearest-neighbor phenotypes for different cell types in (A) Tumor and (B) IM ROIs found in a single melanoma sample stained with the lymphocyte panel. Abbreviations: IM = invasive margin; ROI = region of interest. Please click here to view a larger version of this figure.
{kind=link}
Supplemental File 1: Multiplex IHC summarizing protocol specifications. Please click here to download this File.
Supplemental File 2: Autostainer protocol for monoplex. Please click here to download this File.
Supplemental File 3: Autostainer protocol for autofluorescence compensation. Please click here to download this File.
Supplemental File 4: Autostainer protocol for multiplex immunohistochemistry. Please click here to download this File.
Supplemental File 5: Template .csv file. Please click here to download this File.
Supplemental File 6: Myeloid and dendritic cell panels in a melanoma tissue sample; marking slides in case of scanning failure; myeloid and dendritic cells recognized by ImmuNet; gating strategies of myeloid and dendritic cell panels. Please click here to download this File.
Supplemental File 7: QuPath stitch script. Please click here to download this File.
Supplemental File 8: Data analysis script. Please click here to download this File.
Discussion
Spatial analysis of the TME is a sought-after technique to learn more about the immune cell compartment and discover new prognostic and predictive biomarkers, particularly in the field of immuno-oncology16. Many different techniques are being developed for this purpose, involving the detection of proteins, mRNA transcripts, or a combination of the two, with estimations up to 100-1,000 targets. However, higher multiplexing comes at the cost of less high-throughput experiments, higher experimental costs, and technical challenges, and often, only a small part of the TME can be analyzed. Multiplex IHC using the TSA-based method that we describe here, detects six different markers + DAPI simultaneously, is relatively less expensive to perform, and whole tissue sections are imaged in under 20 min, ready to be analyzed fully. This technique has become less complex with the automation of the staining procedure. Improvements in the multispectral microscope, which include the addition of two extra filters, have improved spectral unmixing and scanning times tremendously. It is possible to detect up to eight different markers + DAPI simultaneously. However, by expanding the multiplexing with more markers, the aforementioned benefits disappear as spectral unmixing becomes more challenging and scanning times for whole slides increase substantially. Efforts are being undertaken to standardize multiplex IHC between different institutions to facilitate implementation in the diagnostic setting more easily. For this standardization of multiplex IHC, we advise users to adhere to the more accessible protocol with six different markers + DAPI. Nevertheless, still quite some technical know-how is necessary and downstream analysis can be challenging, for which we have developed methodologies that are described in this protocol.
Standardization begins with multiplex IHC panel development. The importance of the choice of primary antibodies detecting particular protein targets has been emphasized before17. Our multiplex IHC panels are mostly developed with primary antibody clones that are also used and validated for IHC at our diagnostics department. However, in the case of the dendritic cell multiplex IHC panel, most antibodies were not used in the diagnostic setting (van der Hoorn et al., manuscript in submission). To ensure specificity and minimize batch differences, we chose to use monoclonal antibodies over polyclonal antibodies and have also validated most antibodies using transfected cell lines and primary cells. Over the years, different versions of multiplex IHC panels have been used in numerous studies using the Vectra 3 system18,21,23,24,25,26,27,28,29,30,31,32. To implement these multiplex IHC panels optimally on the PhenoImager HT system, some adjustments had to be made in primary antibody and fluorophore combinations. To benefit from better spectral unmixing and faster scanning times of whole tissue sections, implementation of the latest Opal480 and Opal780 fluorophores and avoiding the use of Opal540 and Opal650 fluorophores in seven-color multiplex IHC panels is necessary. Scanning times are ~3-10 times faster depending on the size of the tissue section. Multiplex IHC panel adjustments were quite easy to achieve, but some considerations need to be kept in mind. The fluorescent spectrum of Opal480 overlaps a lot with the autofluorescence spectrum and therefore, interferes with the spectral unmixing of erythrocytes and other autofluorescent structures. Using an increased concentration of the primary antibody paired with Opal480 solved this issue in most cases. The implementation of the proprietary Sample AF filter on the PhenoImager HT facilitates the unmixing of Opal480 and autofluorescence. However, it is best to use a primary antibody that yields a clear signal when used with Opal480 so that its signal is higher than the autofluorescence.
Even though these multiplex IHC panels are established, batch-to-batch variation is something that needs to be considered. By performing monoplex IHC controls before starting the full multiplex IHC experiment, we sometimes observed that primary antibodies perform either stronger or weaker from experiment to experiment. The reasons for this could be pipetting errors, suboptimal reagent storage conditions, and shelf life. We solved this by adjusting the primary antibody solution based on our experience. Even when none of the aforementioned adjustments had to be made, with every multiplex IHC batch experiment, it is important to set exposure times based on monoplex IHC-stained control slides.
Because our research was initially focused on different types of carcinomas and melanoma, multiplex IHC panels were required to be interchangeable between tumor types with minimal adjustments. Therefore, we always included multiple (tumor) tissue types in the optimization process and observed that dilutions for primary antibodies for immune cell markers can be kept similar between different tumor types. However, tumor tissue detection between carcinomas and melanoma needs different tumor markers. Accordingly, the tumor marker was always optimized to work at the end of each multiplex IHC panel and is currently always used in conjunction with Opal780, which coincidentally also has to be at the last fluorophore in a multiplex IHC staining procedure. By using the tumor marker consequently at the end of the multiplex IHC, these multiplex IHC panels can be easily exchanged for other tumor types, such as glioblastoma (i.e., GFAP) and Hodgkin lymphoma (i.e., CD30). For angiosarcoma, we used this lymphocyte multiplex IHC panel with erythroblast transformation-specific-related gene (ERG) as the tumor marker with only two optimization experiments25. The optimization included titration of the ERG primary antibody and testing the multiplex IHC panel with ERG at the end.
Other adjustments to these multiplex IHC panels can also be made by exchanging a certain immune cell marker for another immune or functional marker. Every change requires optimization. The protocol for optimization could be followed as described previously17. Certain changes to the proposed multiplex IHC panels will interfere with the ImmuNet algorithms that we have created. Sufficient data must be generated and time has to be spent to implement these changes into the algorithm (at least 750 annotations for every new marker and/or cell phenotypes, and 150 annotations for validation of previously trained markers). The panels presented here do not contain functional markers, although the implementation of immune checkpoint markers such as PD-1 and PD-L1 into multiplex IHC panels is performed in our laboratory. However, the analysis of markers that are less binary in negative and positive signals has proven to be more difficult and is an area of active research in our group.
The number of markers that can be simultaneously assessed with multiplex IHC is limited compared to other novel techniques. While this can be circumvented by analyzing different panels on consecutive slices of an FFPE block, it will be hard to compare these slices spatially. Orientation and folded artifacts are likely not the same after slide preparation. Nevertheless, multiplex IHC is quite accessible, which makes it an attractive tool for more institutions and researchers and therefore, more suitable for future implementation in a diagnostic setting. With the standardization of multiplex IHC immune cell panels for multiple tumor types and downstream analysis pipelines, more knowledge could be gained about differences in TME between patients and tumor types. This can, for instance, lead to more insights into the role of the TME in antitumor response to specific treatments. This may even give rise to new biomarkers to predict factors such as response to treatment and expected survival. Overall, this can enable multiplex IHC to become a clinical tool to aid with clinical decision-making, in a personalized medicine approach. Admittedly, more steps of the analysis procedure should probably be automated and standardized for it to be feasible for use in a daily diagnostic setting, so as of yet, it is mostly a futuristic perspective.
Analysis of multiple markers on a single sample slide can be a very powerful tool in spite of its technical challenges. With standardized experimental protocols and a robust analysis method, as we described here using ImmuNet, the quantification of multiple markers makes it more informative than classical IHC, while multiplex IHC remains relatively high-throughput compared to novel higher plex experimental methods.
Acknowledgements
The PhenoImager HT was purchased through funding provided by the Radboud University Medical Center and Radboud Technology Center for Microscopy. CF is financially supported by a Dutch Cancer Society grant (10673) and ERC Adv grant ARTimmune (834618). JT is financially supported by an NWO Vidi grant (VI.Vidi.192.084). The authors would like to thank Eric van Dinther and Ankur Ankan for their assistance in creating workflows to store multiplex IHC data and Bengt Phung is thanked for instructions on how to implement multiplex IHC data in QuPath for ROI drawing.
Materials
| Name | Company | Catalog Number | Comments |
| anti-CD14 | Cell Marque | 114R-16 | section 3, clone EPR3653 |
| anti-CD163 | Cell Marque | 163M-15 | section 3, clone MRQ-26 |
| anti-CD19 | Abcam | ab134114 | section 3, clone EPR5906 |
| anti-CD1c (BDCA1) | Thermo Scientific | TA505411 | section 3, clone OTI2F4 |
| anti-CD20 | Thermo Scientific | MS-340-S | section 3, clone L26 |
| anti-CD3 | Thermo Scientific | RM-9107 | section 3, clone sp7 |
| anti-CD303/BDCA2 | Dendritics via Enzo Lifesciences/Axxora | DDX0043 | section 3, clone 124B3.13 |
| anti-CD56 | Cell Marque | 156R-94 | section 3, clone MRQ-42 |
| anti-CD66b | BD Biosciences | 555723 | section 3, clone G10F5 |
| anti-CD68 | Dako Agilent | M087601 | section 3, clone PG-M1 |
| anti-CD8 | Dako Agilent | M7103 | section 3, clone C8/144B |
| anti-Foxp3 | Thermo Scientific | 14-4777 | section 3, clone 236A/E7 |
| anti-Gp100 | Dako Agilent | M063401 | section 3, clone HMB45 |
| anti-HLA-DR, DP, DQ | Santa Cruz | sc-53302 | section 3, clone CR3/43 |
| anti-MART-1 | Thermo Scientific | MS-799 | section 3, clone A103 |
| anti-pan cytokeratin | Abcam | ab86734 | section 3, clone AE1/AE3 + 5D3 |
| anti-SOX10 | Sigma Aldrich | 383R | section 3, clone EP268 |
| anti-Tyrosinase | Sanbio | MONX10591 | section 3, clone T311 |
| anti-XCR1 | Cell Signaling Technologies via Bioké | 44665S | section 3, clone D2F8T |
| antibody diluent | Akoya BioSciences | SKU ARD1001EA | section 3, from Opal 7-Color Automation IHC Kit 50 slide (can optionally also be replaced by TBST with 10% BSA) |
| Bond Aspirating Probe | Leica Biosciences | S21.0605 | section 3 |
| Bond Aspirating Probe Cleaning | Leica Biosciences | CS9100 | section 3 |
| Bond Dewax Solution | Leica Biosciences | AR9222 | section 3 |
| Bond Objectglas label + print lint | Leica Biosciences | S21.4564.A | section 3 |
| Bond Research Detection System 2 | Leica Biosciences | DS9777 | section 3 |
| Bond RX autostainer | Leica Biosciences | - | section 3, automated platform |
| Bond TM Epitope Retrieval 1 - 1 L | Leica Biosciences | AR9961 | section 3 |
| Bond TM Epitope Retrieval 2 - 1 L | Leica Biosciences | AR9640 | section 3 |
| Bond TM Wash Solution 10x - 1 L | Leica Biosciences | AR9590 | section 3 |
| BOND Universal Covertile | Leica Biosciences | S21.4611 | section 3 |
| Bond(TM) Titration Kit | Leica Biosciences | OPT9049 | section 3 |
| Coverslip 24 x 32 mm #1 (0.13-0.16 mm) | Fisher Scientific | 15717592 | section 2 |
| coverslip 24 x 50 mm | VWR | 631-0146 | section 2 |
| DAPI Fluoromount-G | VWR | 0100-20 | section 3, whenever monoplex slides need to be quickly checked, not for official analysis, then DAPI is stained seperately for better results |
| Eosine | section 2, home made | ||
| Ethanol 99.5% | VWR | 4099.9005 | section 2 |
| Fluoromount-G | VWR | 0100-01 | section 3 |
| haematoxyline | - | section 2, home made | |
| ImmuNet | - | immune cell detection and phenotyping pipeline | |
| inForm software 2.4.10 | Akoya BioSciences | - | section 4 & 6 |
| OPAL 480 reagent pack | Akoya BioSciences | FP1500001KT | section 3 |
| OPAL 520 reagent pack | Akoya BioSciences | FP1487001KT | section 3 |
| OPAL 570 reagent pack | Akoya BioSciences | FP1488001KT | section 3 |
| OPAL 620 reagent pack | Akoya BioSciences | FP1495001KT | section 3 |
| OPAL 690 reagent pack | Akoya BioSciences | FP1497001KT | section 3 |
| OPAL 780 reagent pack | Akoya BioSciences | FP1501001KT | section 3 |
| Opal 7-Color Automation IHC Kit 50 slide | Akoya BioSciences | NEL821001KT | section 3 |
| PhenoChart 1.1.0 | Akoya BioSciences | - | section 5 |
| PhenoImagerHT | Akoya BioSciences | CLS143455 | section 4, digital pathology imager with slide viewer and imaging software (formerly known as Vectra Polaris) |
| Quick-D mounting medium | Klinipath | 7280 | section 2 |
| QuPath 0.3.2 | whole slide image analysis software platform | ||
| R 4.1.1 | |||
| Slide boxes | VWR | 631-0737 | section 1 |
| SuperFrost Plus | Thermo Scientific through VWR | 631-9483 | section 1 |
| Vectra Polaris software 1.0.13 | Akoya BioSciences | - | section 4 |
| Xylene | VWR | 4055-9005 | section 2 |
References
- Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunology. 3 (11), 991-998 (2002).
- van der Woude, L. L., Gorris, M. A. J., Halilovic, A., Figdor, C. G., de Vries, I. J. M. Migrating into the tumor: a roadmap for T cells. Trends in Cancer. 3 (11), 797-808 (2017).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Fridman, W. H., et al. The immune microenvironment of human tumors: general significance and clinical impact. Cancer Microenvironment. 6 (2), 117-122 (2013).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Angell, H. K., Bruni, D., Barrett, J. C., Herbst, R., Galon, J. The Immunoscore: colon cancer and beyond. Clinical Cancer Research. 26 (2), 332-339 (2020).
- Angell, H., Galon, J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Current Opinion in Immunology. 25 (2), 261-267 (2013).
- Galon, J., et al. Cancer classification using the Immunoscore: a worldwide task force. Journal of Translational Medicine. 10, 205 (2012).
- Galon, J., et al. World-wide Immunoscore Task Force: meeting report from the 34;Melanoma Bridge", Napoli, November 30th-December 3rd, 2016. Journal of Translational Medicine. 15 (1), 212 (2017).
- Tumeh, P. C., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 515 (7528), 568-571 (2014).
- Creemers, J. H. A., et al. A tipping point in cancer-immune dynamics leads to divergent immunotherapy responses and hampers biomarker discovery. Journal for Immunotherapy of Cancer. 9 (5), e002032 (2021).
- Blank, C. U., Haanen, J. B., Ribas, A., Schumacher, T. N. CANCER IMMUNOLOGY. The "cancer immunogram". Science. 352 (6286), 658-660 (2016).
- Teruya-Feldstein, J. The immunohistochemistry laboratory looking at molecules and preparing for tomorrow. Archives of Pathology & Laboratory Medicine. 134 (11), 1659-1665 (2010).
- Ramos-Vara, J. A., Miller, M. A., et al. When tissue antigens and antibodies get along: revisiting the technical aspects of immunohistochemistry--the red, brown, and blue technique. Veterinary Pathology. 51 (1), 42-87 (2014).
- Hegde, P. S., Karanikas, V., Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clinical Cancer Research. 22 (8), 1865-1874 (2016).
- Parra, E. Novel platforms of multiplexed immunofluorescence for study of paraffin tumor tissues. Journal of Cancer Treatment and Diagnosis. 2 (1), 43-53 (2018).
- Gorris, M. A. J., et al. Eight-color multiplex immunohistochemistry for simultaneous detection of multiple immune checkpoint molecules within the tumor microenvironment. Journal of Immunology. 200 (1), 347-354 (2018).
- Roelofsen, T., et al. Spontaneous regression of ovarian carcinoma after septic peritonitis; a unique case report. Frontiers in Oncology. 8, 562 (2018).
- van den Brand, D., et al. Peptide-mediated delivery of therapeutic mRNA in ovarian cancer. European Journal of Pharmaceutics and Biopharmaceutics. 141, 180-190 (2019).
- van den Brand, D., et al. EpCAM-binding DARPins for targeted photodynamic therapy of ovarian cancer. Cancers. 12 (7), 1762 (2020).
- Di Blasio, S., et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nature Communications. 11 (1), 2749 (2020).
- van Beek, J. J. P., et al. Human pDCs are superior to cDC2s in attracting cytolytic lymphocytes in melanoma patients receiving DC vaccination. Cell Reports. 30 (4), 1027-1038 (2020).
- Rodriguez-Rosales, Y. A., et al. Immunomodulatory aged neutrophils are augmented in blood and skin of psoriasis patients. Journal of Allergy and Clinical Immunology. 148 (4), 1030-1040 (2021).
- Hoeijmakers, Y. M., et al. Immune cell composition in the endometrium of patients with a complete molar pregnancy: Effects on outcome. Gynecologic Oncology. 160 (2), 450-456 (2021).
- van Ravensteijn, S. G., et al. Immunological and genomic analysis reveals clinically relevant distinctions between angiosarcoma subgroups. Cancers. 14 (23), 5938 (2022).
- van der Woude, L. L., et al. Tumor microenvironment shows an immunological abscopal effect in patients with NSCLC treated with pembrolizumab-radiotherapy combination. Journal for Immunotherapy of Cancer. 10 (10), e005248 (2022).
- Graham Martinez, C., et al. The immune microenvironment landscape shows treatment-specific differences in rectal cancer patients. Frontiers in Immunology. 13, 1011498 (2022).
- Cortenbach, K. R. G., et al. Topography of immune cell infiltration in different stages of coronary atherosclerosis revealed by multiplex immunohistochemistry. International Journal of Cardiology. Heart & Vasculature. 44, 101111 (2023).
- van Wilpe, S., et al. Homologous recombination repair deficient prostate cancer represents an immunologically distinct subtype. Oncoimmunology. 11 (1), 2094133 (2022).
- Gorris, M. A. J., et al. Paired primary and metastatic lesions of patients with ipilimumab-treated melanoma: high variation in lymphocyte infiltration and HLA-ABC expression whereas tumor mutational load is similar and correlates with clinical outcome. Journal for Immunotherapy of Cancer. 10 (5), e004329 (2022).
- van Wilpe, S., et al. Intratumoral T cell depletion following neoadjuvant chemotherapy in patients with muscle-invasive bladder cancer is associated with poor clinical outcome. Cancer Immunology, Immunotherapy. 72 (1), 137-149 (2023).
- Sultan, S., Gorris, M. A. J., Buytenhuijs, F., Lvan de Woude, L. A Segmentation-free machine learning architecture for immune land-scape phenotyping in solid tumors by multichannel imaging. bioRxiv. , (2021).
- Parra, E. R., et al. Immuno-profiling and cellular spatial analysis using five immune oncology multiplex immunofluorescence panels for paraffin tumor tissue. Scientific Reports. 11 (1), 8511 (2021).
- Parra, E. R., et al. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Scientific Reports. 7 (1), 13380 (2017).
- Sun, Z., Nyberg, R., Wu, Y., Bernard, B., Redmond, W. L. Developing an enhanced 7-color multiplex IHC protocol to dissect immune infiltration in human cancers. PLoS One. 16 (2), e0247238 (2021).
- LeicaBiosystems. BOND RX Fully Automated Research Stainer Protocols. , (2022).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved