In Vivo Imaging of Liver Spheroids Engrafted in the Anterior Chamber of the Mouse Eye
* These authors contributed equally
In This Article
Summary
Here, we describe a platform that allows noninvasive in vivo imaging of liver spheroids engrafted in the anterior chamber of the mouse eye. The workflow spans from generating spheroids from primary liver cells to transplantation into the mouse eye and in vivo imaging at cellular resolution by confocal microscopy.
Abstract
Biomedical studies of the liver in mammals are hindered by the lack of methods for in vivo noninvasive longitudinal imaging at cellular resolution. Until now, optical imaging of the liver in situ is possible by intravital imaging, which offers high-resolution imaging at the cellular level but cannot be performed multiple times and, therefore, longitudinally in the same animal. Noninvasive imaging methods, such as bioluminescence, allow repeated imaging sessions on the same animal but do not achieve cell resolution. To address this methodology gap, we have developed a platform for noninvasive in vivo imaging of liver spheroids engrafted in the anterior chamber of the mouse eye. In the workflow described in this study, primary mouse liver spheroids are generated in vitro and transplanted into the anterior chamber of the eye of recipient mice, where they engraft on the iris. The cornea acts as a natural body window through which we can image the engrafted spheroids by conventional confocal microscopy. The spheroids survive for months in the eye, during which the cells can be studied in contexts of health and disease, as well as being monitored in response to different stimuli over repeated imaging sessions using appropriate fluorescent probes. In this protocol, we provide a breakdown of the necessary steps to implement this imaging system and explain how to best harness its potential.
Introduction
The monitoring of liver function in mammals during health and disease is limited by the lack of high-resolution, noninvasive in vivo imaging techniques. The visualization of this organ is hindered by its inaccessible location, and in order to piece together cellular processes, in vivo studies rely on the sacrifice of animals at different time points. To circumvent this imaging limitation, much work relies on in vitro models, in which liver-like microtissues are visualized and studied in a controlled environment.
In recent years, the development of three-dimensional culture systems, such as liver spheroids, has aided and advanced liver research. Liver spheroids are multicellular aggregates that mimic the microenvironment and complex cell-cell interactions of liver tissue to a certain extent1 and offer clear advantages over traditional monolayer cultures2,3. Liver spheroids are also used as models for different liver diseases4,5,6 and have been instrumental to understanding disease mechanisms. Still, key limitations of the current in vitro liver models are the lack of a physiological in vivo milieu and the limited utilization time in culture (around 20 days)3. Liver spheroids have been previously transplanted to different sites in vivo, such as under the kidney capsule7 or intraperitoneally8, which are not accessible for optical imaging. Intravital liver imaging is a state-of-the-art technique offering cell-resolution real-time imaging. Currently, this in situ liver imaging is only possible on the exteriorized organ, which is highly invasive and often terminal9. Although the fitting of an abdominal window would allow repeated liver imaging sessions, it entails complex surgery and after-care.
To perform longitudinal monitoring at cellular resolution, we explored transplantation of liver spheroids into the anterior chamber of the eye (ACE) of mice, where the liver-like tissue is engrafted in a physiological milieu, connected to the body stimuli, and accessible for optical imaging. The cornea is a transparent tissue and acts as a window through which microtissues engrafted on the iris can be imaged non-invasively and longitudinally by confocal microscopy. Here, we present a workflow of this newly developed platform for in vivo imaging of liver spheroids10. This protocol is a step-by-step guide for its implementation, divided into (1) the extraction of primary mouse liver cells and in vitro formation of liver spheroids, (2) the transplantation of liver spheroids into the ACE of recipient mice, and (3) the in vivo imaging of engrafted liver spheroids in anesthetized mice. Furthermore, we will showcase some of the possibilities and applications of this imaging platform.
Protocol
All procedures performed on animals were approved by the Animal Experiment Ethics Committee at Karolinska Institutet.
1. Extraction of primary mouse liver cells and generation of liver spheroids in vitro
- Preparation
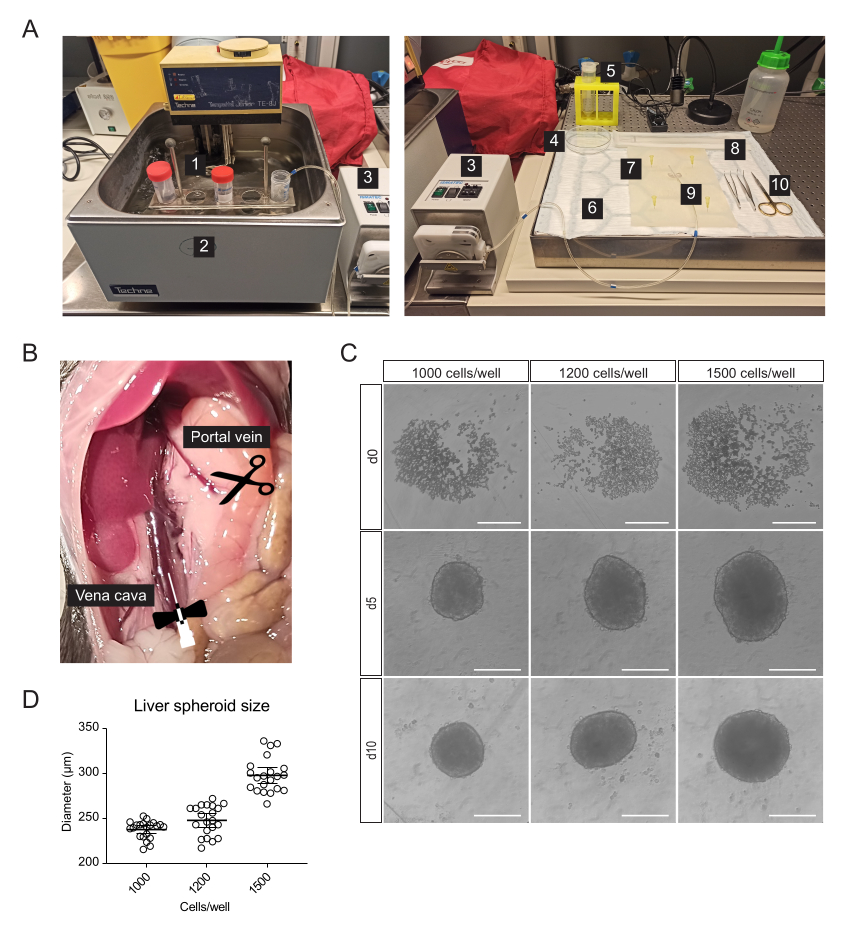
- For cannulation, liver resection, and isolation of primary liver cells, prepare the following sterile or single-use materials in addition to serological pipettes and centrifuge tubes (Figure 1A): peristaltic pump, swing-bucket centrifuge, absorbent pad, dissection mat, two curved-tip forceps, surgical scissors, one 27 G butterfly needle, one 70 µm cell strainer, one cell lifter, one 100 mm Petri dish, cell-counting chamber, and 96-Well, U-shaped-bottom microplates.
- Prepare the solutions used in the liver perfusion, isolation of primary liver cells, and generation of liver spheroids as listed in Table 1.
NOTE: The Digestion buffer and the Gradient solution should be prepared fresh. - Set up the perfusion system consisting of a peristaltic pump, which conducts solutions from the 42 °C water bath to the liver (Figure 1A). The higher temperature of the water bath ensures that the buffers reach the liver at the optimal temperature of 37 °C. Customize this depending on the tube length and the room temperature (RT).
- For the cannulation, fit a 27 G butterfly needle to the end of the tube. Keep the plating media used in the final isolation steps at 4 °C.
- Procedure
- Pre-heat the following solutions in the 42 °C water bath in 50 mL centrifuge tubes: 40 mL of PBS, 20 mL of perfusion buffer, and 12 mL of digestion buffer.
- To clean and warm the pump tubes, circulate approximately 20 mL of the pre-heated PBS.
- Change the tubing to the perfusion buffer, prime the tube and butterfly needle, and set the flow rate to 4 mL/min. Ensure no bubbles in the tubing during buffer changes throughout the protocol.
- Euthanize the mouse by cervical dislocation and use needles to fix the limbs to the dissecting board.
- Wet the abdomen fur with 70% ethanol and dissect it to access the digestive organs.
- Move the intestines to the right to expose the portal vein and inferior vena cava (Figure 1B).
- Cannulate the vena cava at approximately halfway of its length with the needle in a horizontal position, ensure it is stable, and start the pump.
- When the liver starts to inflate, or white dots appear in the closer lobes, cut the portal vein to allow the blood and perfusion buffer to drain out.
- The liver should start blanching immediately. Encourage clearing using curved forceps to clamp the portal vein at 5 s intervals.
- Repeat step 1.2.9 until the liver is yellow and cleared of blood (approx. 15-20 mL of perfusion buffer).
- Stop the peristaltic pump to change the tubing to the digestion buffer and restart the pump. When the digestion buffer reaches the liver, reduce the flow rate to 2.5 mL/min.
NOTE: The phenol red in the digestion buffer allows to discriminate its arrival to the liver and enables the adjustment of pump parameters during the treatment. - To encourage the buffer to reach all hepatic lobes and ensure proper digestion, repeat step 1.2.9 several times.
- Stop the flow of the peristaltic pump when the digestion buffer is depleted or the liver looks sufficiently digested.
NOTE: The degree of digestion can be visually monitored by gently pinching the liver lobes with forceps and checking if small marks appear on the tissue. The liver will also become flimsy. - To extract the liver, cut the hepatic ligaments and connections within the abdominal cavity, aiming to remove it entirely, and place it in the Petri dish containing 10 mL of cold plating medium (Table 1).
- After removing the gallbladder, make small pinches on the lobes using forceps, lightly tearing the hepatic capsule. By shaking the liver in the dish, observe cells pouring out into the medium.
- Holding the liver steady with forceps, gently drag the cell lifter along the lobes to release the cells.
NOTE: Correct interlobular digestion will lead to cell suspension in the media, not tissue fragments. - Using a serological pipette, collect the cell suspension from the Petri dish and filter through the 70 µm cell strainer placed on a 50 mL centrifuge tube. Use fresh plating media to wash the dish of digested liver cells and transfer them to the filter.
- Centrifuge at 50 x g for 5 min at 4 °C to pellet the cells.
- Remove the supernatant, leaving approximately 1 mL to cover the cell pellet, swirl the tube to resuspend the cells, and then gradually add 10 mL of cold plating medium.
- Add the 10 mL of gradient solution to the cell suspension and gently invert the tube 10 times.
- Centrifuge at 200 x g for 10 min at 4 °C.
- The pellet contains viable liver cells enriched for hepatocytes, while the supernatant contains dead cells and debris. Discard the supernatant using a serological pipette, leaving approximately 1 mL, and resuspend the pellet by gentle swirling.
- Add 20 mL of cold plating medium to the cell suspension and centrifuge at 50 x g for 5 min at 4 °C to wash off the gradient solution.
- Remove the supernatant, leaving approximately 1 mL above the pellet, and resuspend the cells in 20 mL of cold plating medium.
NOTE: Here, the cell pellet can be compacted, so if needed, use a 10 mL serological pipette to gently dissociate the cells. - Manually determine the cell number and viability using a cell counting chamber and Trypan Blue.
NOTE: The hepatocyte cells will quickly precipitate in the tube; to resuspend them gently invert the tube a few times. - Seed the liver cells in 200 µL/well of plating medium at 1200 cells/well into 96-well ultra-low adherence plates.
NOTE: The optimal volume of media per well is 200 µL; however, it is possible to seed cells in 100 µL/well. - Spin the plates at 200 x g for 3 min to gather the cells in the center of the wells.
- Incubate (37 °C, 5% CO2) the cells and leave them to form spheroids for 5 days (Figure 1C) naturally.
- On day 5, carefully remove half of the media in the well and replace it with serum-free maintenance media (Table 1). Repeat this step every 48 h until day 10, when the liver spheroids are ready to be transplanted.
NOTE: The formation of a capsule-like structure in cultured liver spheroids shows good aggregation and viability.
2. Transplantation of liver spheroids into the anterior chamber of the eye (ACE)
- Preparation
- For transplantation of liver spheroids into the ACE, ensure to arrange the following resources (Figure 2A): stereomicroscope, isoflurane anesthesia unit, induction chamber, isoflurane, heating pad, custom-made metal base plate, mouse head-holder and gas mask, forceps attached to the solid universal joint, Hamilton 500 µL threaded plunger syringe, silicone, polyethylene and pump tubing, custom-made blunt glass cannula or 24 G catheter, ethanol 70%, sterile saline, sterile 23 G needles, eye ointment (liquid paraffin and Vaseline at 1:1 ratio), disposable 1 mL syringe, and cell suspension dish 35 mm.
- Clean the Hamilton syringe, tubing, and cannula by passing 70% ethanol and saline.
- Fill the Hamilton syringe, tubing, and glass cannula with saline and fix the Hamilton syringe to the bench in a horizontal position with tape (Figure 2A).
- Use a custom-made blunt glass cannula.
- Elongate a borosilicate glass capillary using a dual-stage micropipette puller to an inner diameter of >300 µm to allow the aspiration of the spheroids.
- Bevel and blunt the tip using a microelectrode beveler and expose the cannula tip to a flame for a few seconds to soften the edges.
NOTE: The microelectrode beveler consists of a rotating sanding stone that is operated manually; therefore, specific settings are non-applicable.
- Alternatively, construct a cannula using the plastic portion of a 24 G catheter (Figure 2B).
- Prepare the anesthesia isoflurane unit and warm the heating pad to 37 °C.
- Cover the tips of the forceps attached to the solid universal joint with a piece of polyethylene tubing to form a loop, which helps to stabilize the eye.
- Transfer the liver spheroids from the 96-well plate into a 35 mm cell suspension dish with maintenance media using a pipette and 200 µL tip.
- Procedure
- Anesthetize the mouse in the induction chamber using a dose of 2.5% isoflurane and 280 mL/min air.
- When the mouse is unconscious, lower the anesthesia to 1.8% isoflurane and 280 mL/min air, connect the anesthesia tube to the head-holder, and quickly transfer the animal onto the heating pad, positioning the nose inside the head-holder.
- Immobilize the head with the screws, gently pop the eye out of the socket and secure it with the forceps and place a drop of saline on both eyes to prevent drying.
- Under the stereoscope, use the Hamilton syringe to aspirate and collect the liver spheroids into the tip of the cannula and leave them to rest horizontally on a clean surface.
NOTE: Aspirating media along with the liver spheroids helps to prevent them from sticking to the cannula walls. - Carefully puncture the cornea using a 23 G needle and dry the seeping aqueous humor with a tissue. If needed, to make the incision wider, carefully slide the needle sideways to slice the cornea.
NOTE: A single-use sterile needle is used to perform the corneal puncture, so the cornea is not disinfected prior to incision. - Add saline drops to the eye to avoid drying.
- Take the cannula containing the liver spheroids and hold it vertically to allow the spheroids to gravitate toward the tip of the cannula.
- Gently insert the cannula into the hole, and with the bevel directed towards the pupil, use the Hamilton syringe to slowly expulse the liver spheroids into the ACE (Figure 2C).
NOTE: Before removing the cannula, waiting a few seconds for the liquid pressures inside and outside the eye to readjust and avoid the spheroids escaping back out of the eye is recommended. - From outside the cornea, position the liver spheroids around the pupil and away from the incision by gently prodding the cornea with the tip of the cannula (Figure 2C).
- Wait ~5-10 min for the liver spheroids to settle onto the iris before releasing the eye from the forceps.
- Apply vaseline eye ointment to the operated eye, which helps lubricate and heal the cornea.
- If desired, proceed to operate on the second eye following the same method.
- Before awakening the mouse, administer an analgesic to avoid post-operative discomfort, for example, 0.1 mg/kg buprenorphine in sterile saline, administered subcutaneously.
NOTE: Only one dose of analgesics was administered to the mice as they recovered quickly from this small procedure and did not show any signs of pain or altered behavior. Since this procedure is very quick (taking under 10 min) and causes only minor discomfort, the mice require no post-operative care, other than the post-operative analgesia administered before awakening the animal.
3. In vivo imaging of engrafted liver spheroids in the ACE
- Preparation
- Prepare the following materials and instruments for noninvasive in vivo imaging of liver spheroids engrafted in ACE (Figure 3A): upright confocal microscope, long working-distance water-dipping objective, isoflurane anesthesia unit, induction chamber, isoflurane, heating pad, custom-made metal base plate, mouse head-holder and gas mask, forceps attached to the solid universal joint, artificial-tear gel, eye ointment (liquid paraffin and Vaseline at 1:1 ratio).
- Optional materials include injectable fluorescent probes, disposable syringes, and 27 G needles for tail intravenous injection.
- Procedure
- Anesthetize the mouse in the induction chamber using a dose of 2.5% isoflurane and 280 mL/min air.
- When the mouse is unconscious, lower the anesthesia to 1.8% isoflurane and 280 mL/min air, connect the anesthesia tube to the head-holder, and quickly transfer the animal onto the heating pad, positioning the nose inside the head-holder.
- Immobilize the head in the head-holder by using the screws.
- Place a drop of artificial tear gel on both eyes to prevent drying.
- At this point, intravenously inject fluorescent probes through the tail vein and image immediately after.
- Tilt the head, gently pop the eye out of the socket, and secure it with forceps and into position under the objective.
- Apply a generous amount of artificial tear gel to fill the space between the cornea and the objective and focus on the liver spheroids through the eyepiece.
NOTE: If possible, remove the eyepiece of one of the oculars to gain non-magnified vision and more easily locate the spheroids on the iris. - In order to obtain high-resolution imaging despite the breathing movements of the animal, use 25x objectives and the following imaging settings: format 512 x 512 pixels, scan speed of 600 Hz, and a Z-stack thickness of 3 µm. See detailed imaging settings in Table 2.
NOTE: Throughout the imaging, the anesthesia concentration is adjusted from 1.6-2.2 mL/h isoflurane to achieve a shallow, controlled breathing rhythm and thereby minimize the movement of the animal. - At the end of the imaging session, treat the imaged eyes with vaseline eye ointment before removing isoflurane and awakening the animal.
Representative Results
Primary liver cells, enriched for hepatocytes, were isolated from the mouse liver by two-step collagenase perfusion, using a peristaltic pump to circulate warm buffers through the liver taking advantage of the organ's vasculature to deliver dissociation enzymes to all cells (Figure 1A). For this, the inferior vena cava was cannulated, and the portal vein was snipped to allow the flow-through of buffers (Figure 1B). First, an HBSS-based buffer was flushed through the liver to clear the blood. If the cannulation is successful and there are no blood clots, the liver blanches and becomes yellow within a few seconds. Secondly, a Digestion buffer containing the Liberase enzyme blend was circulated through the liver to dissociate the tissue into a single-cell suspension. The cells were manually counted and seeded into 96-well ultra-low-adherence (ULA) plates, which enable the self-assembly into spheroids within a few days. On day 5, the spheroids are formed, and the thin capsule bordering the spheroids indicates successful aggregation (Figure 1C). We wait until day 10 to transplant, at which point the spheroids are compact and have developed strong cell-cell connections. The number of seeded cells per well determined the size of the liver spheroid, with 1000, 1200, and 1500 cells/well yielding spheroids of 238 µm ± 10 µm, 248 µm ± 17 µm, and 298 µm ± 19 µm (mean ± SD) diameters, respectively (Figure 1C,D). For transplantation, we select spheroids of approximately 250 µm diameter for the following reasons: (1) the spheroids size should not be too large to avoid hypoxia and necrotic core, but should contain enough cells to support cell-cell communications and to allow graft remodeling in the eye, (2) the weight of spheroids of this size allows them to gravitate towards the iris and improve their engraftment, (3) this size is appropriate in relation to transplanting 5-10 spheroids per mouse eye.
The transplantation surgery requires a manual-threaded syringe connected to a glass cannula (Figure 2A). The glass cannula consists of a borosilicate glass capillary modified in-house to have a fine blunt tip using a micropipette puller and beveler. A simpler alternative cannula can be created using a commercially available plastic catheter connected to the syringe tubing and stabilized in a pipette tip (Figure 2B). The surgery consists of the inoculation of liver spheroids into the ACE through an incision in the cornea (Figure 2C). The spheroids were positioned on the borders of the pupil to make them better accessible for imaging and avoid them moving into the ocular angle. Albino mice were used for transplantation, as their non-pigmented iris allow the in vivo imaging of the engrafted liver spheroids. Recipient mice were transplanted into both eyes with 7-10 spheroids/eye, and stereoscopic images were taken at 3 days post-transplantation (post-Tx) as well as at 1 week and 1 month post-Tx to document the cornea healing and spheroid engraftment success (Figure 2D). Of note, the change in appearance of the liver spheroids in the ACE between when freshly transplanted and fully engrafted is due to the settlement of the graft onto the iris, as well as to the growth of a monolayer of iris cells over the spheroid. The engraftment success rate of liver spheroids in the ACE is 70% (n = 9 eyes in both male and female mice) (Figure 2E). The first days post-Tx are the most critical for survival and engraftment, likely due to the recipient animal rubbing its eyes and dislodging the spheroids before the cornea has healed. The size of the liver spheroids does not differ significantly post-Tx and changes to shape are attributed to graft remodeling and engraftment (Figure 2F). At 1 month post-Tx, all engrafted spheroids present on the iris were vascularized and innervated, as shown by immunofluorescence staining (Figure 2G).
Noninvasive in vivo imaging is performed on anesthetized recipient mice using an upright confocal microscope and long-distance dipping objective (Figure 3A, Table 2). The fluorescence imaging in the ACE can be achieved through different approaches, as depicted in Figure 3B. The injection of fluorescent probes into the circulation of the recipient mouse allows the visualization of different cell types and structures within the spheroids. We used lectin to mark blood vessels (Figure 3C), CMFDA to observe the bile canaliculi network (Figure 3D) and pHrodo-LDL, which confirmed active LDL-uptake into spheroid cells (Figure 3E). Liver spheroids generated from reporter mouse models can also be used. Albumin-Cre:tdTomato spheroids allowed labeling and tracking of hepatocytes (Figure 3F), and spheroids expressing the Fluorescent Ubiquitin Cell Cycle Indicator (FUCCI) biosensor were used to visualize cell cycle dynamics at single-cell resolution (Figure 3G). Finally, liver spheroids can be genetically modified in vitro prior to transplantation, and, in the case of adeno-associated virus (AAV)-GFP transduction, the expression was observed in vivo for over 6 months (Figure 3H).

Figure 1: Isolation of primary mouse hepatocytes and generation of liver spheroids. (A) Material and equipment used for isolation of primary mouse hepatocytes: 1. Isolation buffers; 2. Water bath; 3. Peristaltic pump; 4. Petri dish; 5. Cell strainer; 6. Absorbent pad; 7. Dissection mat; 8. Cell Lifter; 9. Butterfly needle 27 G; 10. Dissection tools. (B) Abdominal cavity during surgery: the vena cava is cannulated and perfused, and the portal vein is snipped to allow flow-through of the buffers. (C) Brightfield images of the formation of hepatic spheroids in vitro at 0 (d0), 5 (d5), and 10 (d10) days post-seeding, scale bars = 200 µm. (D) Liver spheroid size at different cell-seeding concentrations, n = 21 spheroids. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Transplantation and engraftment of liver spheroids into the ACE of mice. (A) Materials and equipment used for transplantation (Tx) of liver spheroids into the ACE: 1. Liver spheroids in culture dish; 2. Sterile saline; 3. Eye ointment; 4. Needles 23 G; 5. Cannula; 6. Hamilton syringe; 7. Anesthesia gas tube; 8. Head-holder and gas mask; 9. Heating pad; 10. Custom-made metal base plate; 11. Forceps and solid universal joint. (B) Cannula and Hamilton syringe setup: 1. Glass cannula connected to the Hamilton syringe via Portex tubing and a 27G needle; 2. Glass cannula is connected to the Portex tubing via additional segments of Silicone tubing and PharMed tubing; 3. Alternative assembled plastic cannula; 4. Parts forming the plastic cannula: 24G BD Insyte plastic catheter connected via PharMed tubing and sheathed in a cut-off 10 µl pipette tip for stability and grip. (C) Illustration of Tx surgery steps: 1. The spheroids are collected into the cannula; 2. The cornea is punctured with a needle; 3. The cannula is inserted into the incision, and the spheroids are released into the ACE; 4. From the outside of the eye, the spheroids are positioned close to the pupil and away from the incision. (D) Stereoscopic images of liver spheroids (sph) in the mouse eye on the day of surgery and at 3-, 7-, and 30-days post-Tx. Arrows indicate viable spheroids. (E) Liver spheroid (size of 1200 cells/well) engraftment rate post-Tx, n= 9 eyes in 6 recipient mice. (F) Size of liver spheroids in culture, prior to transplantation (in vitro, n= 20 spheroids from single preparation) and at 1-month post-Tx in the ACE (in vivo, n= 16 spheroids in 3 recipient mice), calculated by averaging vertical and horizontal diameters. (G) Immunofluorescence staining of engrafted liver spheroids at 2 months post-Tx, showing vascularization (CD31, pink, dashed line delineates the spheroid mass) and sympathetic innervation (tyrosine hydroxylase (TH), orange), scale bar = 100 µm. Data for panel F has been adapted with permission from Lazzeri-Barcelo et al.10. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Noninvasive intraocular in vivo imaging of engrafted liver spheroids. (A) Material and equipment used for in vivo ACE imaging: 1. Upright laser scanning confocal microscope; 2. Dark box; 3. Motorized XYZ stage; 4. Dipping-objective; 5. Head-holder and gas mask; 6. Forceps and solid universal joint; 7. Heating pad; 8. Custom-made metal base plate. (B) Diagram depicting different approaches used for in vivo imaging of fluorescent readouts in liver spheroids engrafted in the eye. (C-H) Representative images of ACE-liver spheroids during in vivo imaging by confocal microscopy. The backscatter signal is used to observe the spheroid volume and structure; (C) Blood vessels labeled by i.v. injection of fluorescent lectin, scale bar = 100 µm; (D) Bile canaliculi network labeled by injection of fluorescent CMFDA, scale bar = 50 µm; (E) LDL uptake by injection of fluorescent pHrodo-LDL probe, scale bar= 100 µm; (F) Td-Tomato-expressing hepatocytes, arrowheads indicate nuclei and asterisks indicate intra-spheroid vasculature, scale bar = 50 µm; (G) Monitoring of cell cycle dynamics in FUCCI-expressing liver spheroids, scale bars = 50 µm (main image) and 20 µm (blow-up). (H) liver spheroids transduced in vitro with AAV8-GFP prior to Tx and imaged in the eye at 6 months post-Tx, scale bar = 50 µm. The image in panel G has been adapted with permission from Lazzeri-Barcelo et al.10. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Solutions used for the isolation of primary mouse hepatocytes. Composition of solutions and buffers needed for mouse hepatocyte isolation. The Digestion buffer and Gradient solution components should be mixed fresh on the day of isolation. Please click here to download this Table.
Table 2. Confocal Leica SP5 microscope settings used for intraocular in vivo imaging of liver spheroids. The table has been adapted with permission from Lazzeri et al.10. Please click here to download this Table.
Discussion
This protocol describes a novel platform for intraocular in vivo imaging of liver spheroids engrafted in the ACE. The ACE has previously been used as a transplantation site of other organ-derived microtissues, such as pancreatic islets11,12, due to its unique engraftment microenvironment, being rich in vessels, nerves, and oxygen, and the access to imaging through the cornea. While intravital liver imaging enables the visualization of cells and processes in situ, longitudinal monitoring is not possible. Liver imaging through an abdominal window entails complex surgery, and the movement of the organ within the body makes single-cell tracking over time difficult. Hence, this novel imaging method enables noninvasive, longitudinal monitoring of liver cells at single-cell resolution.
This protocol is divided into three parts. First is the isolation of primary hepatocytes via two-step collagenase perfusion, adapted from Charni-Natan et al.13, with the difference that we perform the liver perfusion on the dead mouse instead of the anesthetized live animals. This variation brings certain advantages, such as fewer ethical considerations and the avoidance of anesthesia residue in the organism. In this work, we generate liver spheroids from the hepatocyte-enriched fraction of the isolation, but this does not exclude the potential to isolate other nonparenchymal cell populations using other specialized protocols to make coculture spheroids of diverse composition14,15.
The second part of this protocol involves the transplantation of the liver spheroids into the ACE of recipient mice. This is a quick (under 10 min) and simple surgery performed in anesthetized mice and does not require any post-operative treatment. The cornea puncture self-seals and heals over 3-5 days. Occasionally, during the healing process, some hazing is observed around the incision, but this clears within a few days. We have not experienced cases of anterior synechia in the eyes of operated animals. We perform the transplantation procedures in a clean but open-air laboratory and without issues with infections in the operated eyes. The inoculation and engraftment of spheroids in the eye do not compromise the vision or alter the behavior of the recipient animal. In this protocol, we use isoflurane anesthesia for both transplantation surgery and in vivo imaging, which is well tolerated in mice. Due to its dose-dependent effect, it can be easily adjusted throughout the procedures and brings the advantage of reducing sleeping and awakening times. However, alternative injectable anesthetics can be used. After transplantation, we generally allow 1 month for the spheroids to fully engraft, become vascularized, and innervated, before performing treatment interventions and in vivo imaging. We have also shown that transplantation and engraftment is possible using human liver spheroids and immunocompromised recipient mice10.
The third part of this method is the in vivo imaging of the engrafted liver spheroids in the ACE. This protocol describes the in vivo imaging setup, which uses microscopy equipment commonly found in research imaging facilities. Moreover, the specialized materials, such as the mouse head-holder and the plastic cannula, are now commercially available. With this imaging setup, we are able to capture z-sections and obtain a three-dimensional reconstruction of the spheroid architecture, depending on the depth of laser penetration and fluorescence detection. Monitoring cellular function in the engrafted liver spheroids relies on the visualization of fluorescent proteins that report on cell types, cellular functions, and dynamics. Thus, this imaging platform can be exploited using different modalities, alone or in combination: (1) Fluorescent probes can be administered intravenously, e.g., antibodies to label and track cells as well as functional dyes; (2) Liver spheroids can be generated from cells isolated from reporter mouse models that express liver-specific fluorescent proteins, e.g., FUCCI liver spheroids that report on cell cycle dynamics; (3) The formation of liver spheroids in vitro can be combined with transfection or transduction, to equip the spheroids with fluorescent proteins and biosensors. e.g., adeno-associated viruses. In our experimental settings and by using a single photon for excitation, the imaging depth that is possible to achieve is roughly 60-100 µm. However, this is dependent on the laser power and multiphoton imaging availability, fluorescence probe emission characteristics and sensitivity of the detectors, as well as the angle of the eye in which the spheroid is engrafted. After imaging acquisition, the downstream image analysis can be performed using popular programs such as Image J and Imaris. For example, in the case of the FUCCI reporter, cell-cycle active cells in green can be counted and contrasted to the number of total red cells to assess cell-cycle activity within the engrafted spheroid. Additionally, the ACE-imaging platform allows substances to be applied to the eye (in the form of eye drops) or injected directly into the ACE to treat the graft and monitor its reaction. Post-mortem, the transplanted spheroids can easily be retrieved by manual microdissection and can provide valuable information by ex vivo techniques, such as immunofluorescence staining, transcriptomic analysis, etc.10.
This technique has certain limitations. The first is that from our experience, the recipient mice must be albino, i.e., have non-pigmented iris. Upon engraftment, the liver spheroids become covered by a monolayer of iris cells, which does not affect the viability or function of the spheroids, but the pigment in the iris cells prevents imaging. A second consideration is the stability during intraocular imaging in anesthetized mice. During the in vivo imaging sessions, the anesthesia concentration and breathing of the animal must be closely monitored to minimize movement. Nevertheless, using the imaging settings indicated here, we are able to achieve high-resolution imaging at the single-cell level.
To summarize, this protocol describes the implementation of a noninvasive in vivo imaging platform of liver-like tissue engrafted in the eyes of mice. We use easy procedures, common equipment, and affordable materials, making it an attainable approach for many investigators. This model combines the advantages of in vitro 3D-liver spheroids with the in vivo milieu and optical accessibility provided by the ACE to create a valuable platform for studying liver physiology and pathology in basic research and pre-clinical settings.
Acknowledgements
This work was supported by the Swedish Diabetes Association, Funds of Karolinska Institutet, The Swedish Research Council, Novo Nordisk Foundation, The Family Erling-Persson Foundation, Strategic Research Program in Diabetes at Karolinska Institutet, The Family Knut and Alice Wallenberg Foundation, The Jonas & Christina af Jochnick Foundation, Swedish Association for Diabetology and ERC-2018-AdG 834860-EYELETS. The figure drawings were created by FL-B using BioRender.com.
Materials
| Name | Company | Catalog Number | Comments |
| 27 G butterfly needle | Venofix | 4056388 | |
| AAV8-CAG-GFP | Charles River | CV17169-AV9 | Incubated with isolated hepatocytes at 1 µL/mL during liver spheroid formation |
| Absolute and 70% ethanol | N/A | N/A | |
| Absorbent pad | Attends | 203903 | |
| Albumin-Cre;RCL-tdTomato (B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J ; B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J) | Jackson | #003574 and #007914 | Mice obtained from in-house breeding |
| B6 albino mice (B6(Cg)-Tyrc-2J/J) | Jackson | #000058 | Mice obtained from in-house breeding |
| B6;129P2-Gt(ROSA)26Sor[tm1(CAG-Venus/GMNN,-Cherry/CDT1)Jkn]/JknH | INFRAFRONTIER/EMMA | EM:08395 | Mice obtained from in-house breeding |
| BD Insyte IV Catheter 24 G x 0.75 in | BD Medical | 381212 | |
| Borosillicate standard glass cappilaries | World Precision Instruments | 1B150-4 | |
| Cell lifter | Corning | 3008 | |
| Cell strainer, 70 µm | Falcon | 352350 | |
| Custom-made metal plate | Hardware store | N/A | |
| Dexamethasone | Sigma-Aldrich | D4902 | |
| Dual-Stage Glass Micropipette Puller | Narshige | Model PC-100 | |
| EDTA | Sigma-Aldrich | E9884 | |
| Electric heating pad | Hardware store | N/A | |
| FBS | Gibco | N/A | |
| GlutaMAX | Gibco | 35050061 | |
| Green CMFDA | Abcam | ab145459 | Reconstituted in DMSO, administered at 100 µg/mouse in PBS 10% FBS |
| Hamilton syringe | Hamilton | 81242 | Model 1750 Luer Tip Threaded Plunger Syringe, 500 µL |
| HBSS; no calcium, no magnesium and no phenol red | Gibco | 14175095 | |
| HCX IRAPO L 25x/0.95 W objective | Leica | N/A | |
| HEPES | Gibco | 15630080 | |
| Induction chamber 0.8 L | Univentor | 8329001 | |
| Insulin-Transferrin-Selenium (ITS-G) | Gibco | 41400045 | |
| Isoflurane | Baxter | N/A | |
| Lectin DyLight-649 | Invitrogen | L32472 | Administered at 1 mg/mL and 100 µL/mouse |
| Liberase TM Research Grade | Sigma-Aldrich | 5401127001 | |
| Microelectrode beveler | World Precision Instruments | Model BV-10 | |
| Mouse head-holder and gas mask | Narshige | Model SGM-4 | |
| Nunclon Sphera 96-Well, U-Shaped-Bottom Microplate | Thermo Fisher | 174929 | |
| Oculentum simplex | APL | N/A | |
| PBS 10x | Gibco | 14080055 | |
| PBS 1x; no calcium, no magnesium | Gibco | 14190144 | |
| Penicillin-Streptomycin | Gibco | 15140122 | |
| Percoll | Sigma-Aldrich | P1644 | |
| Peristaltic pump | Ismatec | Model ISM795 | |
| PharMed BPT Pump Tubing | VWR | VERN070540-07 | Inner diameter 0.76 mm, outer diameter 2.46 mm |
| pHrodo Red-LDL | Invitrogen | L34356 | Administered at 1 mg/mL and 100 µL/mouse |
| Portex Fine Bore Polyethylene Tubing | Smiths Medical | 800/100/140 | Inner diameter 0.4 mm, outer diameter 0.8 mm |
| Silicone dissection mat | Hardware store | N/A | |
| Sodium chloride 0.9% | Braun | N/A | |
| Solid Universal Joint | Narshige | Model UST-2 | |
| Stereomicroscope | Leica | Model M80 | |
| Suspension culture dish 35 mm | Sarstedt | 833900500 | |
| Temgesic | Indivor | N/A | Administered s.c. at 0.05 mg/mL and 2 µL/g mouse |
| Translucent Silicone Tubing | VWR | 228-1450 | Inner diameter 1.5 mm, outer diameter 3 mm |
| Trypan Blue | Sigma-Aldrich | T8154 | |
| Univentor 400 Anesthesia unit | Univentor | 8323001 | |
| Upright laser scanning confocal microscope | Leica | Model TCS SP5 II | |
| Viscotears | Novartis | N/A | |
| William's E Medium; no glutamine, phenol red | Gibco | 22551089 |
References
- Bell, C. C., et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci Rep. 6, 25187 (2016).
- Lauschke, V. M., et al. Massive rearrangements of cellular MicroRNA signatures are key drivers of hepatocyte dedifferentiation. Hepatology. 64 (5), 1743-1756 (2016).
- Oliva-Vilarnau, N., Vorrink, S. U., Ingelman-Sundberg, M., Lauschke, V. M. A 3D cell culture model identifies Wnt/beta-catenin mediated inhibition of p53 as a critical step during human hepatocyte regeneration. Adv Sci (Weinh). 7 (15), 2000248 (2020).
- Hurrell, T., et al. Human liver spheroids as a model to study aetiology and treatment of hepatic fibrosis. Cells. 9 (4), 964 (2020).
- Kozyra, M., et al. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci Rep. 8 (1), 14297 (2018).
- Lauschke, V. M., Shafagh, R. Z., Hendriks, D. F. G., Ingelman-Sundberg, M. 3D primary hepatocyte culture systems for analyses of liver diseases, drug metabolism, and toxicity: Emerging culture paradigms and applications. Biotechnol J. 14 (7), e1800347 (2019).
- Shibuya, K., et al. The efficacy of the hepatocyte spheroids for hepatocyte transplantation. Cell Transplant. 30, 9636897211000014 (2021).
- Hamazaki, K., Doi, Y., Koide, N. Microencapsulated multicellular spheroid of rat hepatocytes transplanted intraperitoneally after 90% hepatectomy. Hepatogastroenterology. 49 (48), 1514-1516 (2002).
- Marques, P. E., et al. Imaging liver biology in vivo using conventional confocal microscopy. Nat Protoc. 10 (2), 258-268 (2015).
- Lazzeri-Barcelo, F., et al. Intraocular liver spheroids for noninvasive high-resolution in vivo monitoring of liver cell function. Nat Commun. 15 (1), 767 (2024).
- Speier, S., et al. Noninvasive in vivo imaging of pancreatic islet cell biology. Nat Med. 14 (5), 574-578 (2008).
- Leibiger, I. B., Berggren, P. O. Intraocular in vivo imaging of pancreatic islet cell physiology/pathology. Mol Metab. 6 (9), 1002-1009 (2017).
- Charni-Natan, M., Goldstein, I. Protocol for Primary Mouse Hepatocyte Isolation. STAR protocols. 1 (2), 100086 (2020).
- Baze, A., et al. Three-dimensional spheroid primary human hepatocytes in monoculture and coculture with nonparenchymal cells. Tissue Eng Part C Methods. 24 (9), 534-545 (2018).
- Mohar, I., Brempelis, K. J., Murray, S. A., Ebrahimkhani, M. R., Crispe, I. N. Isolation of nonparenchymal cells from the mouse liver. Methods Mol Biol. 1325, 3-17 (2015).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved