
0:01
Concepts
3:52
Preparing Cells for the Antibody Staining
5:35
Antibody Staining
6:52
Mounting Coverslips
7:47
Confocal Imaging
11:07
Results
Microscopie confocale à fluorescence : une technique pour localiser les protéines dans les fibroblastes de souris
Source: Dominique R. Bollino1, Eric A. Legenzov2, Tonya J. Webb1
1 Département de microbiologie et d'immunologie, University of Maryland School of Medicine et Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
2 Center for Biomedical Engineering and Technology, University of Maryland School of Medicine, Baltimore, Maryland 21201
La microscopie par fluorescence confocale est une technique d'imagerie qui permet une résolution optique accrue par rapport à la microscopie d'épifluorescence « à champ large » classique. Les microscopes confocals sont capables d'obtenir une meilleure résolution optique x-y grâce à la « numérisation laser » - généralement un ensemble de miroirs à tension contrôlée (miroirs de galvanomètre ou de « galvo ») qui dirigent l'éclairage laser à chaque pixel du spécimen à la fois. Plus important encore, les microscopes confocals atteignent une résolution z-axiale supérieure en utilisant un sténopé pour enlever la lumière de mise au point provenant d'endroits qui ne sont pas dans le z-plan étant numérisé, permettant ainsi au détecteur de recueillir des données à partir d'un z-plan spécifié. En raison de la haute résolution Z réalisable dans la microscopie confocale, il est possible de recueillir des images à partir d'une série de z-planes (également appelé z-stack) et de construire une image 3D à travers un logiciel.
Avant de discuter du mécanisme d'un microscope confocal, il est important de considérer comment un échantillon interagit avec la lumière. La lumière est composée de photons, des paquets d'énergie électromagnétique. Un photon empiéchant sur un échantillon biologique peut interagir avec les molécules qui composent l'échantillon de l'une des quatre façons : 1) le photon n'interagit pas et passe à travers l'échantillon; 2) le photon est réfléchi/dispersé; 3) le photon est absorbé par une molécule et l'énergie absorbée est libérée sous forme de chaleur par des processus collectivement connus sous le nom de carie non radiative; et 4) le photon est absorbé et l'énergie est alors rapidement réémise en tant que photon secondaire par le processus connu sous le nom de fluorescence. Une molécule dont la structure permet l'émission de fluorescence est appelée fluorophore. La plupart des échantillons biologiques contiennent des fluorophores endogènes négligeables; par conséquent, les fluorophores exogènes doivent être utilisés pour mettre en évidence les caractéristiques d'intérêt dans l'échantillon. Pendant la microscopie de fluorescence, l'échantillon est éclairé avec la lumière de la longueur d'onde appropriée pour l'absorption par le fluorophore. Lors de l'absorption d'un photon, un fluorophore est dit être «excité» et le processus d'absorption est appelé «excitation». Lorsqu'un fluorophore abandonne l'énergie sous la forme d'un photon, le processus est connu sous le nom d'« émission », et le photon émis est appelé fluorescence.
Le faisceau lumineux utilisé pour exciter un fluorophore est focalisé par la lentille objective d'un microscope et converge à un « point focal » où il est focalement focal. Au-delà du point focal, la lumière diverge à nouveau. Les poutres entrantes et sortantes peuvent être visualisées comme une paire de cônes touchant au point focal (voir la figure 1, panneau gauche). Le phénomène de diffraction impose une limite à la façon dont étroitement un faisceau de lumière peut être concentré - le faisceau se concentre réellement à un endroit de taille finie. Deux facteurs déterminent la taille de la tache focale : 1) la longueur d'onde de la lumière, et 2) la capacité de collecte de lumière de la lentille objective, qui se caractérise par son ouverture numérique (NA). Le «spot» focal s'étend non seulement dans le plan x-y, mais aussi dans la direction z, et est en réalité un volume focal. Les dimensions de ce volume focal définissent la résolution maximale réalisable par l'imagerie optique. Bien que le nombre de photons soit le plus grand dans le volume focal, les chemins de lumière conique au-dessus et au-dessous de la mise au point contiennent également une densité plus faible de photons. N'importe quel fluorophore dans le chemin de lumière peut ainsi être excité. Dans la microscopie conventionnelle (à champ large), les émissions des fluorophores au-dessus et au-dessous du plan focal contribuent à la fluorescence hors foyer (un « fond brumeux »), ce qui réduit la résolution et le contraste de l'image, comme le montre la figure 1, le cube rouge représentant l'émission de fluorophore au-dessus du plan focal (étoile rouge) qui a comme conséquence la fluorescence hors-focus (en haut à droite). Ce problème est amélioré dans la microscopie confocale, en raison de l'utilisation d'un trou d'épingle. (figure 2, en bas à droite). Tel que représenté à la figure 3, le trou d'épingle permet aux émissions provenant du point focal d'atteindre le détecteur (à gauche), tout en empêchant la fluorescence hors foyer (à droite) d'atteindre le détecteur, améliorant ainsi à la fois la résolution et le contraste.
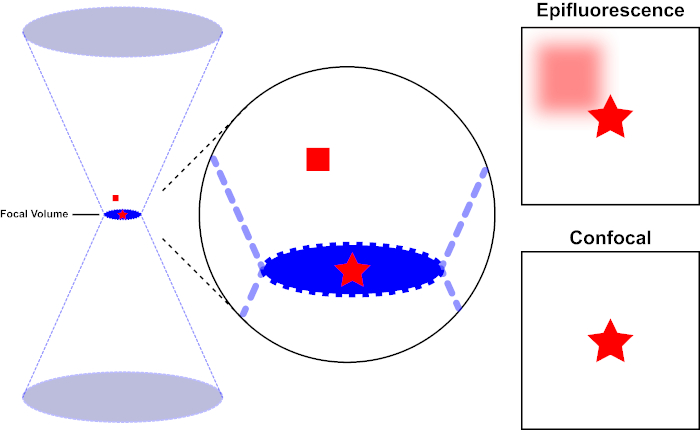
Figure 1. Résolution optique de l'épifluorescence par rapport à la microscopie confocale. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Le faisceau lumineux utilisé pour exciter un fluorophore est focalisé par la lentille objective d'un microscope et converge à un volume focal, puis diverge (à gauche). L'étoile rouge représente le plan focal d'un échantillon qui est représenté tandis que le carré rouge représente l'émission de fluorophore au-dessus du plan focal. Lors de la capture d'une image de cet échantillon à l'aide d'un microscope épifluorescent, l'émission du carré rouge flou sera visible et contribuera à un « fond brumeux » (en haut à droite). Les microscopes confocals ont un trou d'épingle qui empêche la détection de la lumière émise à l'extérieur du plan focal, éliminant le « fond brumeux » (en bas à droite).
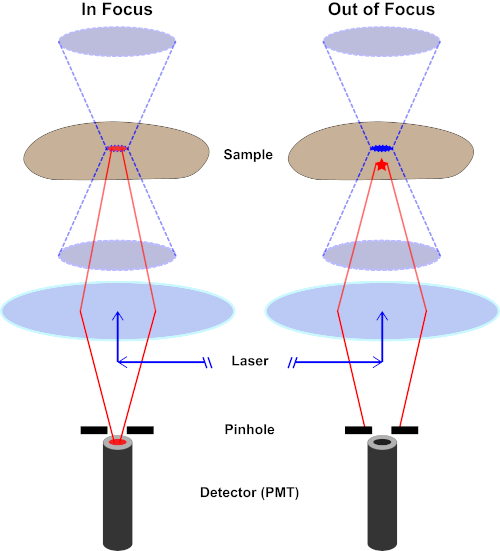
Figure 2. Effet de trou d'épingle dans la microscopie confocale. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Bien que l'intensité la plus élevée de la lumière d'excitation soit au point focal de la lentille (gauche, ovale rouge), d'autres parties de l'échantillon ne se trouve pas dans le point focal (droite, étoile rouge) obtiendront la lumière et la fluoresce. Afin d'éviter que la lumière émise par ces régions floues n'atteigne le détecteur, un écran muni d'un sténopé est présent devant le détecteur. Seule la lumière dans le foyer (à gauche) émise par le plan focal est capable de traverser le sténopé et d'atteindre le détecteur. La lumière hors foyer (à droite) est bloquée avec le sténopé et n'atteint pas le détecteur.
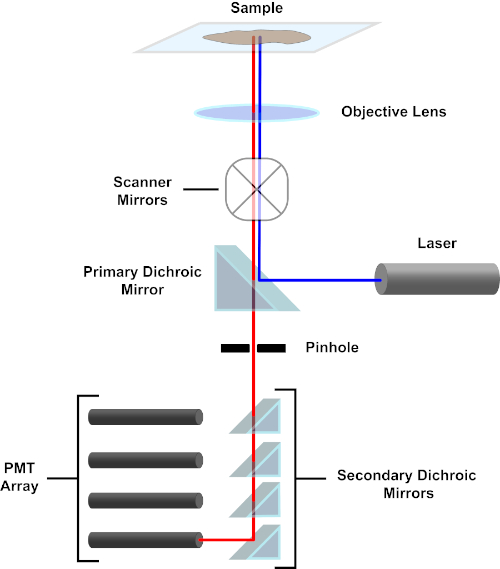
Figure 3. Principaux composants d'un microscope à balayage laser confocal. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Par souci de simplicité, la description mécaniste d'un microscope confocal sera limitée à celle du Nikon Eclipse Ti A1R. Bien qu'il puisse y avoir des différences techniques mineures entre les différents microscopes confocal, l'A1R sert bien comme un bon modèle pour décrire la fonction de microscope confocal. Le faisceau lumineux d'excitation, produit par un tableau de lasers à diodes, est reflété par le miroir dichroïque primaire dans l'objectif, qui concentre la lumière sur le spécimen en cours d'image. Le miroir dichroïque primaire réfléchit sélectivement la lumière d'excitation tout en permettant à la lumière à d'autres longueurs d'onde de passer à travers. La lumière rencontre alors les miroirs de balayage qui balaient le faisceau lumineux à travers le spécimen d'une manière x-y, illuminant un seul (x,y) pixel à la fois. La fluorescence émise par les fluorophores au pixel lumineux est recueillie par l'objectif et passe à travers le miroir dichroïque primaire pour atteindre un tableau de tubes photomultiplicateurs (PMT). Les miroirs dichroiques secondaires dirigent la lumière d'émission vers le PMT approprié. La lumière d'excitation dispersée par l'échantillon dans l'objectif est réfléchie par le miroir dichroïque primaire vers le spécimen, et donc empêchée d'entrer dans la détection voie légère et d'atteindre les PMT (voir la figure 3). Cela permet de quantifier la fluorescence relativement faible sans contamination par la lumière dispersée du faisceau lumineux d'excitation, qui est généralement des ordres de grandeur plus intenses que la fluorescence. Parce que le sténopé bloque la lumière de l'extérieur du volume focal, la lumière arrivant au détecteur provient d'un étroit, sélectionné z-plan. Par conséquent, les images peuvent être recueillies à partir d'une série de z-planesadjacents; cette série d'images est souvent appelée « z-stack ». En utilisant le logiciel approprié, une pile zpeut être traitée pour générer une image 3D du spécimen. Un avantage particulier de la microscopie confocale est la capacité de distinguer la localisation subcellulaire de la coloration. Par exemple, la différenciation entre la coloration de membrane de la coloration intracellulaire, qui est très provocante avec la microscopie conventionnelle d'épifluorescence (1, 2, 3).
La préparation de l'échantillon est une facette importante de l'imagerie confocale. Une force des techniques de microscopie optique est la flexibilité d'imager des cellules vivantes ou fixes. Lorsque vous tentez de produire des images 3D, en raison du nombre d'images qui doivent être acquises pour une z-pile, la difficulté de maintenir la santé cellulaire, et le mouvement des cellules vivantes et de leurs organites, l'utilisation de cellules fixes est typique. La procédure pour fixer et tacher des cellules pour la fluorescence confocale est semblable à celle conventionnellement employée dans l'immunofluorescence. Après la culture dans les diapositives de chambre ou sur les couvertures, les cellules sont fixées à l'aide de paraformaldéhyde pour préserver la morphologie cellulaire. La liaison non spécifique d'anticorps est bloquée utilisant l'albumine bovine de sérum, le lait, ou le sérum normal. Afin de maintenir la spécificité des anticorps secondaires, la solution utilisée ne doit pas provenir de la même espèce dans laquelle les anticorps primaires ont été générés. Les cellules sont incubées avec des anticorps primaires qui lient l'antigène d'intérêt. Lors de l'étiquetage de plusieurs cibles cellulaires, les anticorps primaires doivent être dérivés d'une espèce différente. Les anticorps taguant un antigène sont alors liés par des anticorps secondaires fluorophore-conjugués. Les anticorps secondaires conjugués au fluorophore doivent être sélectionnés afin qu'ils soient compatibles avec les longueurs d'onde de l'excitation laser disponibles dans le microscope confocal. Lors de la visualisation de plusieurs antigènes, les spectres d'excitation/émission des fluorophores devraient différer suffisamment pour que leurs signaux puissent être discriminés par l'analyse microscopique. Le spécimen taché est ensuite monté sur une glissière pour l'imagerie. Un support de montage est utilisé pour prévenir le photoblanchiment et la déshydratation des spécimens. Si vous le souhaitez, un support de montage contenant une contre-tache nucléaire (p. ex. DAPI ou Hoechst) peut être utilisé (4).
Dans le protocole suivant, les fibroblastes de souris transfectés pour exprimer cD1d (LCD1) ont été souillés avec des anticorps reconnaissant CD1d et CD107a (LAMP-1). CD1d est un complexe d'histocompatibilité majeur 1 (MHC 1)-comme le récepteur présent sur la surface des cellules présentant d'antigène qui présente des antigènes. LAMP-1 (protéine de membrane associée lysosomal-1) est une protéine transmembranaire principalement présente dans les membranes lysosomal. Pour la présentation appropriée d'antigène, CD1d est trafiqué par le compartiment lysosomal de pH bas, ainsi LAMP-1 est employé comme marqueur du compartiment lysosomal pour ce protocole. En sondant les cellules LCD1 avec anti-CD1d et anti- LAMP-1 qui ont été produites dans différentes espèces, les anticorps secondaires avec des fluorophores uniques peuvent être utilisés pour déterminer la localisation de chaque protéine dans la cellule et si CD1d est présent dans le LAMP-1 positif compartiments lysosomal.
1. Matériaux
Tampons
- Tampon de lavage : 1 X saline stérile tamponnée de phosphate (PBS) sans calcium ni magnésium
- Tampon de fixation : 1% de paraformaldehyde en PBS
- Tampon de perméabilisation : 0,1 % Triton X-100 en PBS
- Tampon de blocage : 1% d'albumine de sérum bovin dans PBS
- Milieu de croissance cellulaire : DMEM complété par 10% de sérum bovin fœtal (FBS), pénicilline/streptomycine, et L-glutamine...
Dans cette expérience, les fibroblastes de souris exprimant le gène de glycoprotéine de surface CD1d ont été fixés, immunostained et imaged sur un microscope confocal. Une image représentative obtenue à l'aide du protocole ci-dessus est indiquée à la figure 4. Dans le panneau supérieur de A, des images monocanaux montrant le motif de coloration de chaque cible individuelle sont présentées. Ces images comprennent une seule section (tranche) de la z-stack capturée. Le panneau ...
La coloration fluorescente confocale est une procédure relativement simple qui donne lieu à des images de très haute qualité de spécimens qui sont préparés de la même manière que pour la microscopie à fluorescence conventionnelle. En bref, les échantillons sont fixes, perméabilisés, puis bloqués. Les anticorps primaires contre une protéine ou des protéines d'intérêt sont autorisés à se lier, puis des anticorps secondaires conjugués au fluorophore sont utilisés pour visualiser la coloration. La micro...
- Claxton, N. S., Fellers, T. J. and Davidson, M. W. Laser scanning confocal microscopy. Department of Optical Microscopy and Digital Imaging, National High Magnetic Field Laboratory, Florida State University, 37 p., Unpublished (2010). Available at- http://www.vertilon.com/pdf/PP6207.pdf.
- Ojcius, D. M., Niedergang, F., Subtil, A., Hellio, R. and Dautry-Varsat, A. Immunology and the confocal microscope. Research in Immunology, 147 (3),175-88 (1996).
- Paddock, S. W. and Eliceiri K. W. Laser scanning confocal microscopy: history, applications, and related optical sectioning techniques. Methods in Molecular Biology, 1075, 9-47 (2014).
- Hoff. F. How to prepare your specimen for immunofluorescence microscopy. Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany. (2015) Available at- http://www.leica-microsystems.com.
Tags
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved