
Method Article
酶的结晶和结构确定:多功能微流体芯片中串行晶体学的基板复合物
摘要
描述了一种多功能的微流体装置,它利用反扩散方法使酶结晶,通过浸泡在晶体中引入基材,以及酶的3D结构测定:通过在室温下对芯片内部晶体进行连续分析,使酶结晶复杂。
摘要
在X射线分析之前,制备井喷晶体及其处理是生物晶体学研究的两个关键步骤。我们描述了一种多功能的微流体芯片,它通过有效的反扩散方法使晶体得以生产。微流体通道提供的无对流环境是晶体生长的理想之选,有助于将基板扩散到晶体酶的活性部位。在这里,我们应用这种方法的CCA添加酶的精神病细菌 普莱诺克氏杆菌 在介绍的例子。经过结晶和基底扩散/浸泡后,通过连续晶体学和芯片内多个晶体的分析,在室温下确定了酶的晶体结构:基底复合物。整个过程保留了样品的真正衍射特性,因为它不需要水晶处理。
引言
晶体学是破译生物大分子3D结构的方法。后者对于了解酶如何选择和处理其基质非常重要。晶体结构的确定需要目标大分子的结晶和晶体的调理,以进行X射线衍射1的分析。晶体制备和处理都是关键但微妙的步骤,可以影响晶体质量和衍射特性,从而影响由此产生的 3D 结构的分辨率(即精度)。为了方便制备高品质的晶体,并消除不必要的处理,以保持其衍射性能,我们设计了一个用户友好和多功能的微流体设备称为ChipX2,3,4。
在本文中,我们将演示如何使用传统的实验室材料将蛋白质溶液加载到 ChipX 通道中,通过反扩散来制备晶体。由于结晶剂5、6的扩散产生的浓度梯度,这种结晶方法可有效筛选含有酶溶液的微流道沿线的超饱和度和潜在核化条件。
芯片设置简单,仅使用标准实验室管道,不需要任何昂贵的设备。当晶体在 ChipX 中生长时,酶的配体可以通过扩散引入。然后,利用同步加速器 X 射线源在芯片通道中包含的一系列晶体的室温下收集衍射数据。这里描述的结构研究导致确定一个tRNA成熟酶的结构在其apo形式和复杂与模拟其CTP基板引入浸泡。这种称为CCA添加酶的蛋白质在tRNA的3端聚合CCA三核苷酸尾部。通过串行晶体学获得的两个3D图像的比较揭示了与配体结合相关的局部构象变化,这些变化在比低温晶体学中使用的条件下更具生理性。本视频中描述的协议通常适用于任何生物分子,无论是蛋白质、核酸还是多成分复合物。
研究方案
1. 在芯片X中设置结晶测定
注:芯片X微流体设备可以从作者那里获得。芯片的描述在 图1中给出。含有用于触发结晶的结晶剂(或结晶剂)的溶液可能来自商业或由实验者准备。
- 加载生物分子样本
注:在 ChipX 每个通道的直段执行单独反扩散检测所需的样本量为 300 nL。然而,为了方便起见,我们建议加载 5 μL 以完全填充 8 个通道,同时考虑其弯曲部分和入口死卷的可变长度。- 使用标准 10 μL 移液管和尖端的 5 μL 酶溶液。
- 将尖端垂直引入样品入口,并注入溶液,直到八个通道填充到其另一端(晶体储层的入口)。
- 在样品入口中注入 1 μL 石蜡油,以便断开彼此之间的通道。
- 使用标准 10 μL 移液管在每个通道四肢的晶体储层中恢复额外的溶液。
- 用 1 厘米 x 1 厘米的胶带密封样品入口。
- 加载结晶解决方案
- 使用标准 10 μL 移液管和尖端的 5 μL 结晶溶液。储层体积为 10 μL,但仅装载一半,可避免用胶带密封时溢出,并有助于进一步添加用于浸泡实验的配体。如果通过蒸汽扩散获得初始结晶条件,则结晶体浓度增加 1.5 - 2 倍。解决方案可以在每个储层中有所不同(在所展示的情况下,1 M 磷酸氢钠、100 mM 醋酸钠、pH4.5 在整个储层中均使用)。
- 将管道尖端定向到储液管形状部分导管的入口,以避免在溶液沉积时形成气泡。它将防止两种解决方案之间的接触和晶体扩散到通道中。
- 将晶体溶液注入储层。
- 用 2.5 厘米 x 1 厘米的胶带密封储层。
- 在 20 °C 下孵化芯片(温度可根据目标进行调整,通常在 4 至 37 °C 4之间)。
2. 蛋白质标签与卡盒甲胺荧光检测
注意:此步骤是可选的。必须在样品装载前执行,以便于使用荧光检测芯片中的晶体。详细的微量荧光标签方法是由普西和同事7描述的。所有步骤均在室温下执行。
- 将 5 毫克的卡盒霉素酯粉溶解在 1 mL 无水性二甲基成型酰胺中,将溶液分解成 0.6 μL 方位,储存在 -20 °C。
- 准备一个1M纳-波拉特pH 8.75股票解决方案。
- 稀释股票,以准备反应缓冲在0.05 M纳-波拉特pH 8.75。
- 用 800μL 的反应缓冲冲洗脱盐柱(7 kDa MWCO,0.5 mL)。
- 在 1400 x g 时将柱子离心 1 分钟,取出过滤液。
- 重复此操作两次(步骤 2.4-2.5)以洗涤柱子。
- 将 80 μL 的蛋白质存放在柱子上的存储缓冲区(蛋白质可以稀释到 1 毫克/mL,以便在需要时增加体积)。
- 在1400 x g时将柱子离心1分钟。此步骤旨在将蛋白质从其存储缓冲区转移到反应缓冲区。
- 恢复流经(在反应缓冲区中含有蛋白质),并将其与 0.6 μL 的卡博瑟霍达明溶液混合。
- 在室温下孵化5分钟。
- 同时,用存储缓冲液冲洗列 3 x,在 1400 x g 处将柱子离心 1 分钟,然后丢弃过滤液。
- 将反应解决方案存放在柱子上。
- 在 1400 x g 下将柱子离心 1 分钟,并恢复流经(即存储缓冲区中标记的蛋白质溶液)。
- 补充股票蛋白溶液与0.1-1%(w/w)的标签蛋白质。
- 设置第 1 节中描述的 ChipX 结晶分析。
- 通过用520nm波长的光源刺激荧光探针,检查检测中蛋白质晶体的存在。
3. 晶体观测
注意:ChipX 设备无需特别小心处理,即使里面有晶体,除非温度需要控制。
- 使用任何立体显微镜检查 ChipX 中的结晶检测结果。其脚印具有显微镜滑梯的标准尺寸,与任何系统和滑梯支架兼容。
- 检查从晶体浓度最高的储层开始的微流体通道的含量,以检查晶体浓度最低的样品入口。ChipX材料对可见光是透明的,与极化器的使用兼容,以及紫外线照明,通过内在的尝试性磷烷荧光8进行蛋白质晶体识别。
- 使用沿通道浮雕的标签记录晶体位置,或在芯片表面绘制旁边的色点,用永久标记标记水晶位置。
4. 水晶浸泡与配体
注:此过程是可选的。它用于将配体、酶基材或重原子引入晶体中,在X射线分析前至少应进行24-48小时,以便化合物沿着通道扩散到晶体中。
- 轻轻地从储层中取出密封胶带。
- 使用 10 μL 微管在一个或几个储层中加起来多达 5 μL 的配体溶液(例如,添加了 3 μL 的 10 m 环二丁-5'-[α,β) - 甲基磷酸酯 (CMPcPP) 溶液,最终浓度为 3.75 mm)。CMPcPP 是 CTP 的不可水解模拟,CTP 是酶的天然基材。
- 用 2.5 厘米 x 1 厘米的胶带密封储层。
- 在受控温度下孵化芯片24-48小时,使配体沿着芯片的通道扩散。
5. 通过串行晶体学进行晶体分析
注:协议的这一部分需要根据光束线设置和晶体的衍射特性进行调整。根据X06DA光束线(瑞士维利根的SLS)进行的实验,只给出晶体分析的一般指示。
- 光束线日志仪上的芯片X安装
注:芯片X持有人的3D打印文件在参考4中提供。- 关闭光束线的低温喷射。这里的分析是在室温下进行的。
- 将 ChipX 安装在专用支架上,该支架的通道包含要分析的晶体,这些晶体位于支架的中心。ChipX 支架4不需要任何螺丝或附加部分,因为它旨在为 ChipX 提供完美的配合。
- 将支架连接到日光计。
- 数据采集
- 将 ChipX 最厚的层(顶层、 图 1)定向(在此方向标签中,沿通道使用光束线的中心摄像头直接可读),朝向直接光束和晶体后面最薄的脸,以最大限度地减少 ref3中描述的衍射信号的衰减。
- 为了避免 ChipX 与周围材料(光束停止、对准器)碰撞,限制范围±30° (0° 与 X 射线光束垂直的通道)的日光计运动。
- 在沿通道浮雕的标签的帮助下找到晶体位置。
- 选择晶体位置。
- 通过标准低剂量网格/栅格筛选或一键式程序(视频显示网格筛选示例)将晶体集中。
- 收集范围内的衍射数据 -30° / +30°。
- 在芯片转换后,在同一通道中的另一个晶体上以步骤 5.2.4-5.2.6 重新启动该过程。
- 手动重新调整位于支架中心的另一个 ChipX 通道,并在此通道中对存在的晶体进行数据收集。
- 使用标准的晶体学包和程序处理和合并数据,然后解决和细化结构。
结果
此处描述的微流体芯片设计用于在室温下轻松设置结晶测定和晶体分析。上述程序和视频中所述的程序应用于从冷适应的杆菌血友病中加入CCA酶的结构特征框架中。这种酶属于一个基本的聚合酶家族,利用CTP和ATP9,10促进tRNA上3'CCA序列的顺序添加。
该芯片首先用于通过反扩散的方法准备酶晶体进行结构分析。为此,酶溶液通过在芯片样品入口的单次注射加载到8个微流体通道(结晶室)中(见 图1)。该酶用于5.5毫克/mL的存储缓冲区,包含20 mM三叶草/HCl pH 7.5,200 mM NaCl和5 mM MgCl2。此步骤使用标准 10 μL 微管手动执行。结晶溶液(100 mM 醋酸钠 pH 4.5,1 M 磷酸氢)随后沉积在通道另一四肢的储层中。
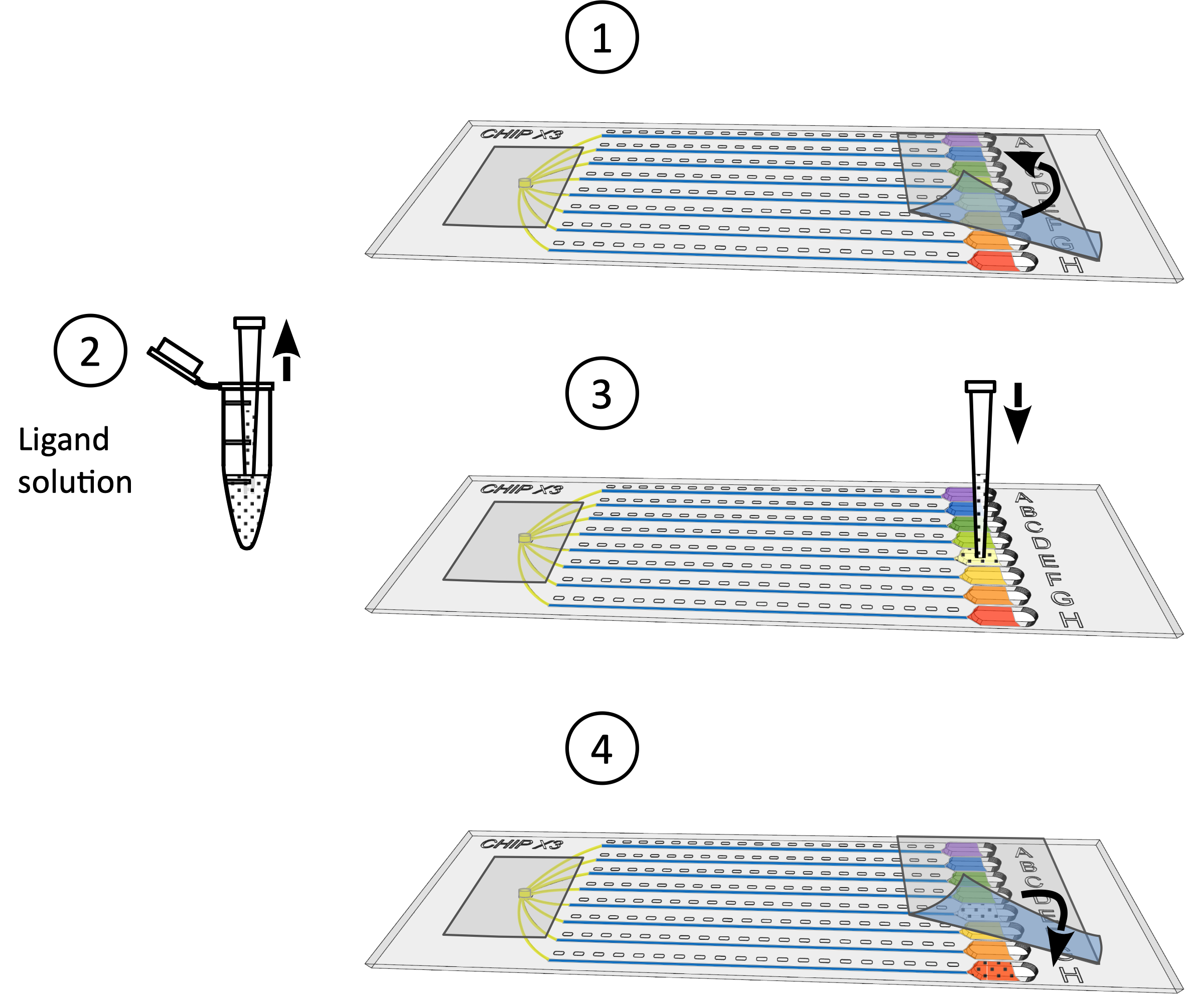
加载过程简单明了,不超过五分钟(图 2)。然后,晶体扩散到通道中,产生浓度梯度,触发晶体核化和生长。这种梯度动态演化,探索一个连续的超饱和状态5,6,直到达到通道和水库之间的晶体浓度平衡。结晶测定通常在 2 - 4 周内在 micoscope 下检查,以跟踪晶体的生长。在20°C(图3)的潜伏期后,CCA添加酶的双层晶体出现在整个通道中。蛋白质的可选荧光标签7大大方便了蛋白质晶体的识别及其对盐晶体的歧视(图4)。
我们利用芯片通道中的扩散环境,为建立晶体的酶提供基材。在本例中,CMPcPP(CTP 模拟)在最终浓度为 3.75 mM(图 5)时添加到水库解决方案中。这一补充是在晶体分析前两天进行的,以便CMPcPP能够到达并占据酶的催化位点,后来由晶体结构确认(见下文)。
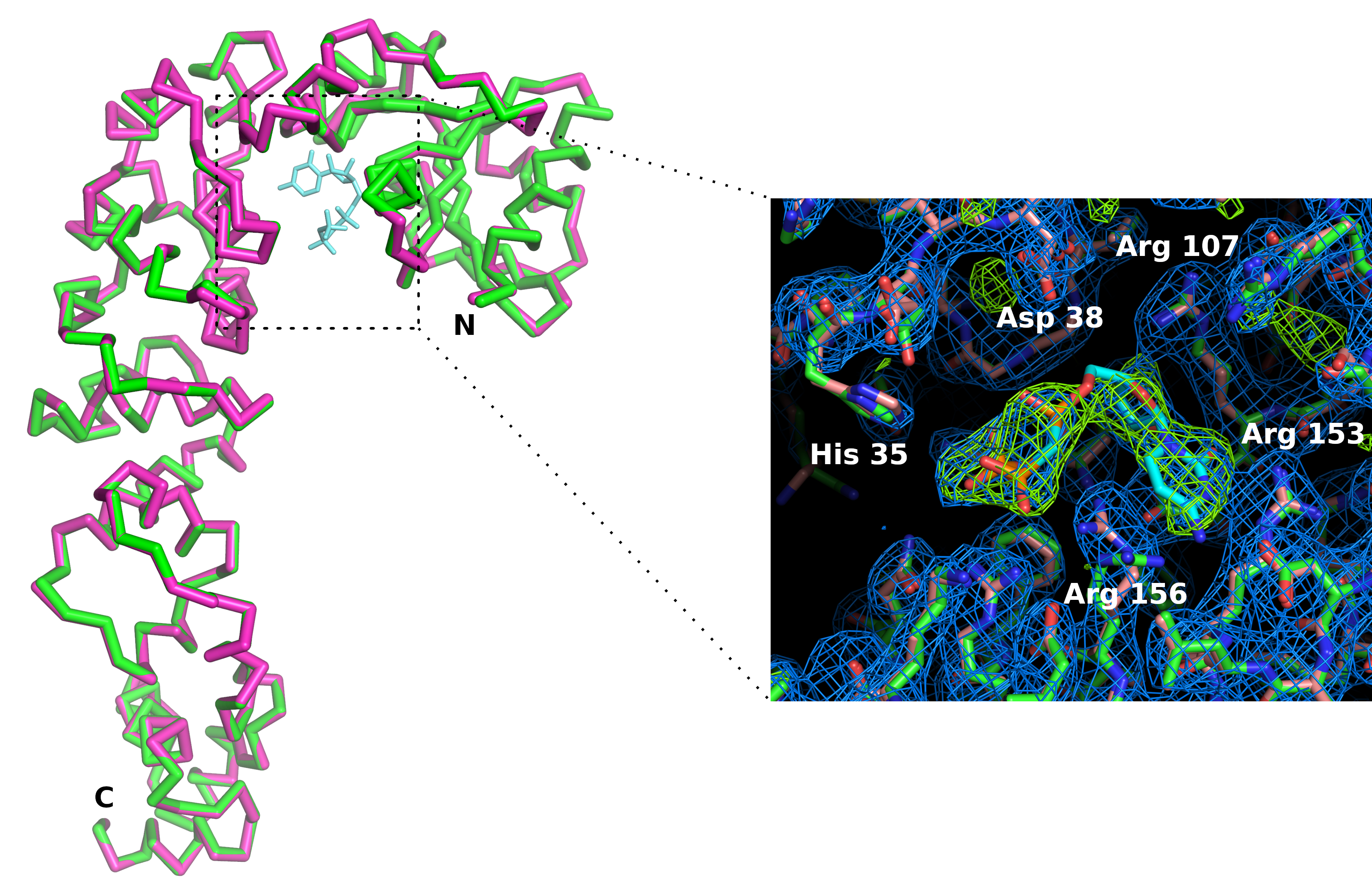
我们用3D打印机制造了多乳酸芯片支架(图6)。支架允许使用标准磁头将芯片安装在测高仪上。因此,芯片可以很容易地定位和翻译在X射线束中,使晶体处于衍射位置。数据收集策略需要根据光束线特性和晶体特性进行调整。以CCA添加酶为例,数据分别在瑞士光源(SLS)X06DA和X10SA光束线采集,X射线波长分别为1.0 é和Pilatus 2M-F和6M像素探测器。在室温下,每个晶体上采集30-60°的旋转,图像为0.1°或0.2°和0.1°(见表1)。当衍射模式的分辨率因辐射损伤而开始衰变时,部分数据集被单独处理和切割(通过信号噪声比  和CC1/2的降低以及高分辨率外壳中Rmeas的增加来检测)。通过合并来自 5 个晶体(表 1)的数据,重新构造了完整的数据集。晶体结构是利用标准晶体学封装和数据处理程序11和细化12的分子置换而成的。酶及其复合物的结构与CMPcPP的比较揭示了伴随CA添加酶活性位点的基板结合的局部构象适应(图7)。
和CC1/2的降低以及高分辨率外壳中Rmeas的增加来检测)。通过合并来自 5 个晶体(表 1)的数据,重新构造了完整的数据集。晶体结构是利用标准晶体学封装和数据处理程序11和细化12的分子置换而成的。酶及其复合物的结构与CMPcPP的比较揭示了伴随CA添加酶活性位点的基板结合的局部构象适应(图7)。

图1:芯片设计。 芯片由COC(厚度:1毫米)制成的顶层组成,其中印有8个微流体通道和储层。整个芯片用一层 COC 密封(厚度:0.1 mm)。所有通道都连接到左侧的单个入口,用于同时进行样品注射,并连接到右侧的单个储层,其中结晶溶液沉积。这些通道构成芯片的实际结晶室,长4厘米,横截面为80微米×80微米。 沿通道浮雕的标签(A1、A2、A3等)便于显微镜下的晶体定位,并准备一份样本清单供数据收集。ChipX 具有标准显微镜滑梯的大小(7.5 厘米 x 2.5 厘米)。 请点击这里查看此数字的较大版本。
{kind=link}

图2:在ChipX中设置结晶测定。 1) 使用标准 10 μL 移液器和尖端沉积 5-6 μL 的酶溶液。2) 在样品入口中垂直引入尖端,并在八个通道中注入溶液。3) 派普特 1μL 石蜡油。4) 将尖端垂直引入样品入口,然后注入油,以便断开彼此之间的通道。5) 用胶带密封入口。6) 使用标准 10μL 移液和尖端的 5 μL 结晶溶液。每个储层的解决方案可能有所不同(例如,从筛选套件中)。7) 将管道尖端定向到水库漏斗形状部分的通道入口(以避免溶液沉积时形成气泡),并将晶体溶液注入储层中。8) 用胶带密封储层,并在受控温度下孵育芯片。 请点击这里查看此数字的较大版本。
{kind=link}

图3:CCA添加酶的晶体,通过在ChipX的微流体通道中反扩散而生长。比例尺是 0.1 毫米。请点击这里查看此数字的较大版本。
{kind=link}

图4:晶体浸泡程序。1)轻轻地从储层中取出胶带。2) 使用 10μL 微管存放高达 5 μL 的配体溶液。3) 将配体添加到一个或几个水库。4) 再次用胶带密封储层,并在数据收集前在受控温度下孵育芯片 24-48 小时。 请点击这里查看此数字的较大版本。
{kind=link}

图5:微量荧光标签区分蛋白质(左)与盐(右)晶体。 CCA添加的酶溶液中含有0.4%(w/w)的蛋白质,标签为卡博奇霍达明。在右侧,晶体用 520 nm 波长的光源照明,图像使用 550 nm (LP550) 的低通滤镜拍摄:(插图)卡博奇罗达明-苏奇尼米迪尔酯的结构。 请点击这里查看此数字的较大版本。
{kind=link}

图6:(左)ChipX支架的绘图和安装在SLS(瑞士维利根)光束线X06DA的测序仪上的芯片X图,以便进行串行晶体分析。请点击这里查看此图的较大版本。
{kind=link}

图7:比较AC添加酶活性位点在appo形式(粉红色)和复杂与CTP模拟(绿色)。 虽然酶的整体构象不受影响,但CMPcPP配体的结合伴随着活动部位侧链的轻微重组。2Fo-Fc 电子密度图(蓝色)轮廓为 1.2 西格玛。以 4 西格玛(绿色)轮廓的电子密度图的差异确认了活性站点中配体的存在。 请点击这里查看此数字的较大版本。
{kind=link}
| 结晶样品 | CCA添加酶 | 加CA酶+CMPcPP |
| 晶体分析 | ||
| X射线光束线 | SLS-X06DA | 单反-X10SA |
| 波长(=) | 1.000 | 1.000 |
| 温度 (K) | 293 | 293 |
| 探测器 | 皮拉图斯 2M-F | 皮拉图斯 6M |
| 水晶探测器距离(毫米) | 300 | 400 |
| 收集的晶体 | 6 | 14 |
| 选择水晶 | 5 | 5 |
| 每个图像的旋转范围(°) | 0.1 | 0.2 |
| 每张图片的曝光时间 | 0.1 | 0.1 |
| 不。选定的图像 | 1000 | 540 |
| 总旋转范围(°) | 100 | 108 |
| 空间组 | P43212 | P43212 |
| a, c (\) | 71.5, 293.8 | 71.4, 293.6 |
| 平均马赛克(°) | 0.04 | 0.04 |
| 分辨率范围(=) | 46 – 2.54 (2.6 – 2.54) | 48 – 2.3 (2.4 – 2.3) |
| 总计否。反射 | 176105 (9374) | 232642 (32937) |
| 不。独特的反射 | 23922 (1598) | 34862 (4066) |
| 完整性 (%) | 90.6 (84.6) | 99.5 (100.0) |
| 冗余 | 7.5 (6.0) | 6.7 (8.1) |
| 8.1 (1.3) | 6.9 (0.7) |
| 雷米亚斯 (%)• | 18.6 (126.0) | 18.0 (231.2) |
| CC1/2 (%) £ | 98.7 (55.0) | 98.7 (46.9) |
| 威尔逊情节的整体 B 因子(=2) | 57.4 | 60.6 |
| 晶体精炼 | ||
| 不。反射、工作集/测试集 | 23583 / 1180 | 34840 / 3405 |
| 最终克里斯特 (%) / R免费 (%) | 18.8 / 21.4 | 20.0 / 22.9 |
| 不。非H原子:整体/蛋白质/配体/溶剂 | 2998 / 2989 / 0 / 9 | 3057 / 2989 / 29 / 10 |
| 债券的 R.m.s. 偏差 (+) / 角度 (+) | 0.009 / 1.23 | 0.010 / 1.22 |
| 平均 B 因子(+2):整体 / 蛋白质 / 配体 / 溶剂 | 60.1 / 60.1 / 0 / 52.7 | 62.5 / 62.6 / 60.1 / 55.5 |
| 拉马尚德兰情节:最青睐(%)/允许(%) | 98.1 / 1.9 | 97.2 / 2.8 |
| PDB ID | 6IBP | 6Q52 |
表 1:数据收集和细化统计
• 冗余独立Rmeas = Σhkl(N/N-1)1/2Σi |i i(hkl)- - 我(hkl)= |/Σ hkl Σ ii i(hkl),其中N是数据多重性17。
£ 基于 CC1/2 标准(数据集的两个随机半部分之间的相关性)包含在外壳 (+lt;2.0) 中具有低 +lt;I/σ (I) 的数据正如卡加洛斯和迪德里希斯 18提出的。
讨论
目前生物晶体学中的协议包括使用蒸汽扩散或第13、14批等方法制备晶体,并在低温条件下在氮气喷射中进行扩散分析之前,将其转移到微循环中进行低温冷却15、16。 相比之下,在 ChipX3中不可能直接进行晶体低温冷却,也无法从其微流体通道中提取晶体,这可被视为此设置的局限性。然而,本文中描述的协议为在室温下(即在更生理条件下)确定晶体结构提供了一个完全集成的管道。尽管室温下的数据收集会导致辐射损伤增加19,但这种效果通过快速数据采集时间(每个晶体上最多收集 60° 旋转)和合并多个部分数据集来抵消。ChipX 的设计和材料都进行了优化,以减少背景散射和衍射信号衰减3,数据收集可以在晶体上进行,其尺寸相当于通道大小的一半 (40μm)4。
综上所述,协议的主要优点如下。这些晶体是在无对流环境中(微流体通道)生产的,非常有利于优质晶体的生长。ChipX中实施的反扩散方法在筛选超饱和景观方面非常有效:晶体扩散到芯片通道中,形成浓度和超饱和波,有助于确定适当的核化和生长条件5。晶体从不直接处理,而是在芯片内部进行原位分析,从而保留其真正的衍射特性(即不通过物理相互作用或低温吸附改变晶体马赛克)20。衍射分析对分布在低剂量暴露芯片通道上的一系列晶体进行,以最大限度地减少辐射损伤,并通过合并该系列的部分数据来组装完整的数据集。ChipX 的标准足迹和简单的设计将允许将来使用同步加速器或 XFEL 设施实现 现场 数据收集的完全自动化。协议的所有步骤均在 ChipX 中执行。从实验者的角度来看,芯片设置简单易用的标准移液器执行,不需要任何额外的设备。样品入口的树状通道连接可最大限度地减少系统中的死体积,这在处理难以纯化或仅限数量有限的样品时非常重要。
总之,ChipX 中实施的片上实验室方法通过反扩散和晶体结构测定简化并有效地小型化结晶过程,从而允许在单个设备中从样品到其 3D 结构。它广泛适用,为室温下的常规连续生物晶体扫描研究提供了用户友好、经济高效的解决方案。
披露声明
作者没有什么可透露的。
致谢
作者承认瑞士光源(瑞士维利根)在光束线 X10SA (PXII) 和 X06DA (PXIII) 上的光束时间分配,亚历山德拉·布卢姆对结构优化的贡献,克拉丽莎·沃斯代尔对画外音的录制,弗朗索瓦·施内尔(斯特拉斯堡大学)协助视频编辑和 SFX。这项工作得到了斯特拉斯堡大学法国国家科学研究中心(CNRS)的支持, LabEx财团"NetRNA"(ANR-10-LABX-0036_NETRNA),斯特拉斯堡大学卓越计划(IdEx)的博士基金,在法国国家项目"Avenir投资"的框架下,由法国-德国大学(UFA-DFH)向K.R.提供博士学位,授予第1名。CT-30-19),德国福松斯格梅因沙夫特(授予号)莫 634/10-1)。作者受益于普罗科佩·休伯特·库里安合作项目(法国外交部和德国外交部长阿卡德米舍尔·奥斯塔施蒂安斯特)。
材料
| Name | Company | Catalog Number | Comments |
| Axioscope A1 stereomicroscope | Zeiss | Crystal observation (step 3) | |
| Carboxyrhodamine succinimidyl ester | Invitrogen | C-6157 | Protein labeling (step 2) |
| CMPcPP | Jena Bioscience | NU-438 | Crystal soaking (step 4) |
| Crystal clear sealing tape | Hampton research | HR3-511 | ChipX sealing (step 1) |
| Parafin oil | Hampton research | HR3-411 | ChipX loading (step 1) |
| Ultimaker 2 extended+ | Ultimaker | 3D printer - Representative results | |
| UV light source | Xtal Concepts Gmbh | XtalLight100c | Crystal observation (step 3) |
| Zeba spin desalting column 7K MWCO | ThermoFisher Scientific | 89882 | Protein labeling (step 2) |
参考文献
- Giegé, R., Sauter, C. Biocrystallography: past, present, future. HFSP Journal. 4 (3-4), 109-121 (2010).
- Dhouib, K., et al. Microfluidic chips for the crystallization of biomacromolecules by counter-diffusion and on-chip crystal X-ray analysis. Lab on a Chip. 9 (10), 1412-1421 (2009).
- Pinker, F., et al. ChipX: A Novel Microfluidic Chip for Counter-Diffusion Crystallization of Biomolecules and in Situ Crystal Analysis at Room Temperature. Crystal Growth & Design. 13 (8), 3333-3340 (2013).
- de Wijn, R., et al. A simple and versatile microfluidic device for efficient biomacromolecule crystallization and structural analysis by serial crystallography. IUCrJ. 6 (3), 454-464 (2019).
- García-Ruiz, J. M. A supersaturation wave of protein crystallization. Journal of Crystal Growth. 232 (1-4), 149-155 (2001).
- Otálora, F., Gavira, J. A., Ng, J. D., García-Ruiz, J. M. Counterdiffusion methods applied to protein crystallization. Progress in Biophysics and Molecular Biology. 101 (1-3), 26-37 (2009).
- Pusey, M., Barcena, J., Morris, M., Singhal, A., Yuan, Q., Ng, J. Trace fluorescent labeling for protein crystallization. Acta Crystallographica Section F Structural Biology Communications. 71 (7), 806-814 (2015).
- Meyer, A., Betzel, C., Pusey, M. Latest methods of fluorescence-based protein crystal identification. Acta Crystallographica Section F Structural Biology Communications. 71 (2), 121-131 (2015).
- Betat, H., Rammelt, C., Mörl, M. tRNA nucleotidyltransferases: ancient catalysts with an unusual mechanism of polymerization. Cellular and Molecular Life Sciences. 67 (9), 1447-1463 (2010).
- Ernst, F. G. M., Erber, L., Sammler, J., Jühling, F., Betat, H., Mörl, M. Cold adaptation of tRNA nucleotidyltransferases: A tradeoff in activity, stability and fidelity. RNA Biology. 15 (1), 144-155 (2018).
- Kabsch, W. XDS. Acta Crystallographica. Section D, Biological Crystallography. 66 (2), 125-132 (2010).
- Adams, P. D., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica. Section D, Biological Crystallography. 66 (2), 213-221 (2010).
- Dessau, M. A., Modis, Y. Protein Crystallization for X-ray Crystallography. Journal of Visualized Experiments. (47), e2285 (2011).
- Sauter, C., Lorber, B., McPherson, A., Giegé, R., Arnold, E., Himmel, D. M., Rossmann, M. G. Crystallization - General Methods. International Tables of Crystallography, Vol. F, Crystallography of Biological Macromolecules. , 99-120 (2012).
- Garman, E. "Cool" crystals: macromolecular cryocrystallography and radiation damage. Current Opinion in Structural Biology. 13 (5), 545-551 (2003).
- Li, D., Boland, C., Aragao, D., Walsh, K., Caffrey, M. Harvesting and Cryo-cooling Crystals of Membrane Proteins Grown in Lipidic Mesophases for Structure Determination by Macromolecular Crystallography. Journal of Visualized Experiments. (67), e4001 (2012).
- Diederichs, K., Karplus, P. A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nature Structural Biology. 4 (4), 269-275 (1997).
- Karplus, P. A., Diederichs, K. Linking Crystallographic Model and Data Quality. Science. 336 (6084), 1030-1033 (2012).
- de la Mora, E., Coquelle, N., Bury, C. S., Rosenthal, M., Holton, J. M., Carmichael, I., Garman, E. F., Burghammer, M., Colletier, J. -. P., Weik, M. Radiation damage and dose limits in serial synchrotron crystallography at cryo- and room temperatures. Proceedings of the National Academy of Sciences of the United States of America. 117 (8), 4142-4151 (2020).
- Nave, C. A. Description of Imperfections in Protein Crystals. Acta Crystallographica. Section D, Biological Crystallography. 54 (5), 848-853 (1998).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。