
共聚焦荧光显微镜:确定小鼠纤维细胞中蛋白质定位的技术
Overview
资料来源:多米尼克·博利诺1,埃里克·勒贡佐夫2,托尼娅·韦伯1
1马里兰大学医学院微生物学和免疫学系,马里兰州巴尔的摩的马琳和斯图尔特·格林鲍姆综合癌症中心 21201
2马里兰大学医学院生物医学工程与技术中心,巴尔的摩,马里兰州 21201
与传统的"广域"显光显微镜相比,共聚焦荧光显微镜是一种成像技术,能够提高光学分辨率。共聚焦显微镜能够通过"激光扫描"实现更好的 x-y 光学分辨率, 通常是一组电压控制的反射镜(电流计或"galvo"反射镜), 一次将激光照明定向到试样的每个像素。更重要的是,共聚焦显微镜通过使用针孔去除来自未位于被扫描的 z 平面位置的聚焦光,从而达到卓越的 z 轴向分辨率,从而使探测器能够从指定的 z 平面收集数据。由于共聚焦显微镜可实现高 z 分辨率,因此可以从一系列 z 平面(也称为 z 堆栈)中收集图像,并通过软件构建 3D 图像。
在讨论共聚焦显微镜的机制之前,考虑样品如何与光相互作用是很重要的。光由光子、电磁能量包组成。冲击生物样品的光子可以通过四种方式之一与构成样品的分子相互作用:1)光子不相互作用,通过样品;2) 光子反射/散射;3) 光子被分子吸收,吸收的能量通过统称为非辐射衰变的过程以热量释放;4) 光子被吸收,然后能量通过荧光过程作为二级光子迅速重新发射。结构允许荧光发射的分子称为荧光。大多数生物样本含有可忽略不计的内源性荧光水;因此,必须使用外源性荧光草来突出样品中感兴趣的特征。在荧光显微镜期间,样品用适当波长的光照亮,供荧光吸收。吸收光子后,氟荧光剂被称为"兴奋",吸收过程称为"激发"。当荧光剂以光子的形式释放能量时,这个过程被称为"发射",发射光子称为荧光。
用于激发荧光的光束由显微镜的物镜聚焦,并汇聚在最大聚焦的"焦点"处。超越焦点,光线再次发散。进入和退出的光束可以可视化为一对锥体接触焦点(参见图1,左侧面板)。衍射现象限制了光束的聚焦程度——光束实际上聚焦到有限大小的点。决定焦点大小的两个因素:1)光的波长,2)物镜的光聚集能力,其特征是其数值光圈(NA)。焦"点"不仅在x-y平面上延伸,而且在z方向上延伸,实际上是一个焦点体积。此焦卷的尺寸定义了光学成像可实现的最大分辨率。虽然光子的数量在焦量内最大,但焦点上方和下方的锥形光路也包含较低的光子密度。因此,光道中的任何荧光光能激发。在传统的(广域)荧光显微镜中,从焦平面上方和下方发射的荧光导致荧光失焦("模糊背景"),从而降低图像分辨率和对比度,如图 1 所示,代表焦平面(红星)上方的荧光发射的红色立方体,导致失焦荧光(右上)。由于使用针孔,这个问题在共聚焦显微镜中得到改善。(图2,右下角)。如图 3 所示,针孔允许来自焦点位置的发射物到达探测器(左图),同时阻止失焦荧光(右)到达探测器,从而提高分辨率和对比度。
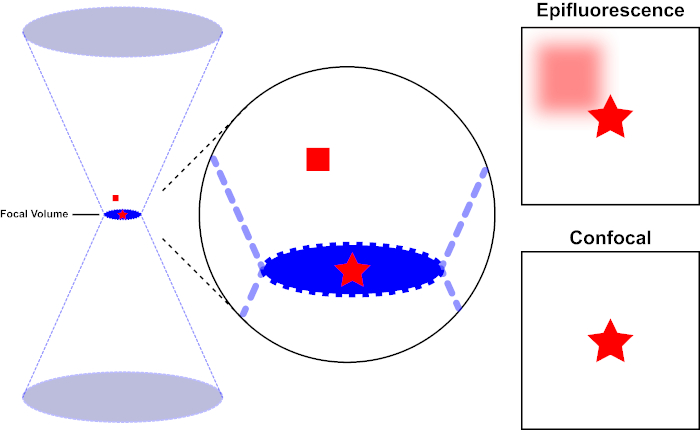
图 1.荧光与共聚焦显微镜的光学分辨率。请点击此处查看此图的较大版本。
用于激发荧光的光束由显微镜的物镜聚焦,并收敛在焦量上,然后分流(左)。红色星表示正在成像的样本的焦点平面,而红色方块表示焦平面上方的荧光发射。当使用表观显微镜拍摄此样本的图像时,从失焦的红色方块发射将可见,并造成"模糊背景"(右上)。共聚焦显微镜有一个针孔,可防止检测焦平面外发出的光,从而消除"模糊背景"(右下角)。
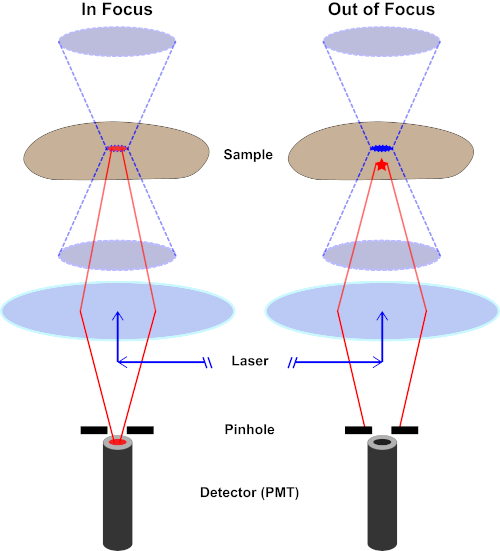
图 2.共聚焦显微镜中的针孔效应。请点击此处查看此图的较大版本。
虽然激发光的最高强度位于透镜的焦点(左,红色椭圆形),但样本的其他部分不在焦点(右,红星)将得到光和荧光。为了防止从这些失焦区域发出的光到达探测器,探测器前面有一个带针孔的屏幕。只有从焦平面发出的对焦中光(左)才能穿过针孔并到达探测器。探针孔挡住了失焦光(右),无法到达探测器。
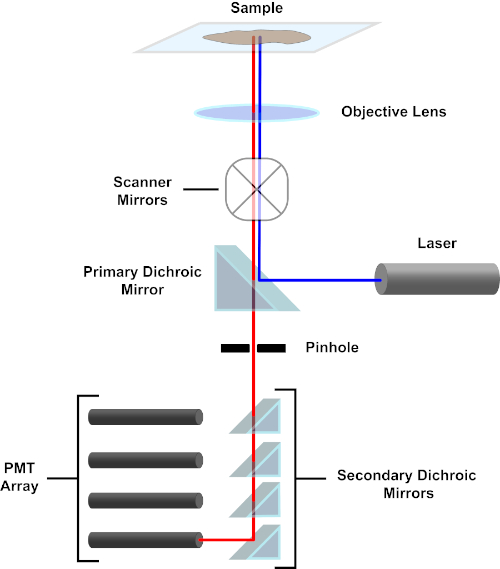
图 3.共聚焦激光扫描显微镜的主要部件。请点击此处查看此图的较大版本。
为了简单起见,共聚焦显微镜的机械描述将仅限于尼康Eclipse TI A1R的描述。虽然不同共聚焦显微镜之间可能存在细微的技术差异,但 A1R 还是很好地描述了共聚焦显微镜的功能。由一系列二极管激光器产生的激发光束由主二色镜反射到目标中,将光线聚焦到被成像的试样上。主二色镜选择性地反射激发光,同时允许其他波长的光通过。然后,光线会碰到扫描镜,以x-y的方式扫描整个试样上的光束,一次照亮一个(x,y)像素。光透镜收集光晕在发光像素处发出的荧光,并通过主二色镜到达一系列光电倍增管 (PMT)。二次二色镜将发射光定向到适当的PMT。样品散射到目标中的激励光由主二色镜反射回试样,从而阻止进入检测光路径并到达 PMT(参见图 3)。这使得相对微弱的荧光可以量化,而不会受到从激发光束散射的光污染,而荧光通常比荧光强度大一个数量级。由于针孔会阻挡焦量外部的光线,因此到达探测器的光线来自一个狭窄的选定z平面。因此,可以从一系列相邻的z平面中收集图像;这一系列图像通常称为"z 堆栈"。通过使用适当的软件,可以处理z堆栈以生成试样 3D 图像。共聚焦显微镜的一个特别优点是能够区分染色的亚细胞定位。例如,膜染色与细胞内染色的区分,这在传统的荧光显微镜(1,2,3)中极具挑战性。
样品制备是共聚焦成像的一个重要方面。光学显微镜技术的优势是成像活细胞或固定细胞的灵活性。当尝试生成 3D 图像时,由于 z 堆栈必须获取的图像数量、维持细胞健康的困难以及活细胞及其细胞器的运动,因此使用固定细胞是典型的。共聚焦荧光的固定和染色细胞的程序与免疫荧光中常用的程序类似。在腔室滑动或盖玻片上培养后,使用甲醛固定细胞以保存细胞形态。非特异性抗体结合被用牛血清白蛋白、牛奶或正常血清阻断。为了保持二次抗体的特异性,所使用的溶液不应来自产生主要抗体的同一物种。细胞用结合感兴趣的抗原的原抗体孵育。在标记多个细胞靶点时,主要抗体必须分别来自不同的物种。标记抗原的抗体然后由荧光酸结合的二级抗体结合。应选择荧光二级抗体,使其与共聚焦显微镜中可用的激光激发波长兼容。在可视化多个抗原时,荧光道的激发/发射光谱应足够不同,以便通过微观分析可以区分其信号。然后,将染色的试样安装在滑轨上进行成像。安装介质用于防止光漂白和样品脱水。如果需要,可以使用包含核反子(例如 DAPI 或 Hoechst)的安装介质 (4)。
在以下协议中,为表达CD1d(LCD1)而转染的小鼠成纤维细胞被识别CD1d和CD107a(LAMP-1)的抗体染色。CD1d 是一种主要的组织相容性复合体 1 (MHC 1) 样受体存在于抗原呈现细胞的表面,呈现脂质抗原。LAMP-1(脂质体相关膜蛋白-1)是一种横膜蛋白,主要存在于脂质膜中。为了获得适当的抗原表达,CD1d通过低pH的感其中体腔,因此LAMP-1被用作本协议的感其中体腔的标记。通过探测不同物种生产的具有抗CD1d和抗LAMP-1的LCD1细胞,可以使用具有独特荧光道的二次抗体来确定细胞中每种蛋白质的定位,以及LAMP-1阳性中是否存在CD1d利索体隔间。
Procedure
1. 材料
缓冲区
- 洗涤缓冲液:1 X 无钙或镁无菌磷酸盐缓冲盐 (PBS)
- 固定缓冲液:PBS中1%的甲醛
- 渗透缓冲器:PBS 中的 0.1% Triton X-100
- 阻断缓冲液:PBS中1%牛血清白蛋白
- 细胞生长培养基:DMEM辅以10%胎儿牛血清(FBS)、青霉素/链霉素和L-谷氨酰胺
设备
- 拉米纳尔流量罩
- 加湿培养箱 (37°C, 5% CO2)
- 共聚焦激光扫描显微镜;在这里,尼康Eclipse Ti激光
材料和试剂
- 室室培养幻灯片
Results
Application and Summary
References
- Claxton, N. S., Fellers, T. J. and Davidson, M. W. Laser scanning confocal microscopy. Department of Optical Microscopy and Digital Imaging, National High Magnetic Field Laboratory, Florida State University, 37 p., Unpublished (2010). Available at- http://www.vertilon.com/pdf/PP6207.pdf.
- Ojcius, D. M., Niedergang, F., Subtil, A., Hellio, R. and Dautry-Varsat, A. Immunology and the confocal microscope. Research in Immunology, 147 (3),175-88 (1996).
- Paddock, S. W. and Eliceiri K. W. Laser scanning confocal microscopy: history, applications, and related optical sectioning techniques. Methods in Molecular Biology, 1075, 9-47 (2014).
- Hoff. F. How to prepare your specimen for immunofluorescence microscopy. Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany. (2015) Available at- http://www.leica-microsystems.com.
Tags
跳至...
此集合中的视频:

Now Playing
共聚焦荧光显微镜:确定小鼠纤维细胞中蛋白质定位的技术
Immunology
43.2K Views

流细胞学和荧光活化细胞分拣(FACS):血性B淋巴细胞的分离
Immunology
93.0K Views

磁性活细胞分拣 (MACS): 胸腺 T 淋巴细胞的分离
Immunology
22.9K Views

ELISA Asas :间接、三明治和竞争
Immunology
238.4K Views

ELISPOT 分析:检测 IFN-- 分泌性细胞
Immunology
28.5K Views

免疫组织化学和免疫细胞化学:通过光显微镜进行组织成像
Immunology
78.9K Views

抗体生成:使用杂交瘤生产单克隆抗体
Immunology
43.6K Views

免疫荧光显微镜:石蜡内嵌组织部分的免疫荧光染色
Immunology
53.9K Views

基于免疫沉淀的技术:使用阿加玫瑰珠纯化内源性蛋白质
Immunology
87.7K Views

细胞周期分析:使用CFSE染色和流细胞测定法评估刺激后CD4和CD8 T细胞增殖
Immunology
24.2K Views

收养细胞转移:将供体小鼠孢子细胞引入宿主小鼠,并通过FACS评估成功
Immunology
22.3K Views

细胞死亡测定:细胞毒性能力的铬释放测定
Immunology
151.4K Views
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。