
Method Article
Einsatz von Drosophila S2-Zellen für Live Imaging of Cell Division
In diesem Artikel
Zusammenfassung
Zellteilungen können in Echtzeit mit fluoreszierend markierten Proteinen und Zeitraffermikroskopie visualisiert werden. Mithilfe des hier vorgestellten Protokolls können Benutzer die Timing-Dynamik der Zellteilung, die mitotische Spindelbaugruppe sowie den Chromosomenkongress und die Segregation analysieren. Defekte in diesen Ereignissen nach RNA-Interferenz (RNAi)-vermitteltem Genknockdown können bewertet und quantifiziert werden.
Zusammenfassung
Drosophila S2-Zellen sind ein wichtiges Werkzeug bei der Untersuchung der Mitose in der Gewebekultur und bieten molekulare Einblicke in diesen grundlegenden zellulären Prozess in einer schnellen und hochdurchsatzigen Weise. S2-Zellen haben sich sowohl für Fest- als auch für Live-Zell-Bildgebungsanwendungen bewährt. Insbesondere kann die Live-Zell-Bildgebung wertvolle Informationen darüber liefern, wie Verlust oder Knockdown eines Gens die Kinetik und Dynamik wichtiger Ereignisse während der Zellteilung beeinflussen kann, einschließlich mitotischer Spindelmontage, Chromosomenkongress und Segregation, sowie Gesamtzyklus-Timing. Hier verwenden wir S2-Zellen, die mit fluoreszierend mit dem Tag mCherry: -tubulin versehen sind, um die mitotische Spindel zu markieren, und GFP:CENP-A (in Drosophilaals "CID"-Gen bezeichnet), um das Zentromer zu markieren, um die Auswirkungen wichtiger mitotischer Gene auf das Timing der Zellteilungen, aus prophase (insbesondere bei Nuclear Envelope Breakdown; NEBD) zum Beginn der Anaphase. Dieses Bildgebungsprotokoll ermöglicht auch die Visualisierung der Spindelmikrotubuli- und Chromosomendynamik während der gesamten Mitose. Hierbei wollen wir ein einfaches, aber umfassendes Protokoll bereitstellen, das es den Lesern ermöglicht, S2-Zellen einfach für Live-Bildgebungsexperimente anzupassen. Die Ergebnisse solcher Experimente sollten unser Verständnis von Genen erweitern, die an der Zellteilung beteiligt sind, indem sie ihre Rolle in mehreren gleichzeitigen und dynamischen Ereignissen definieren. Beobachtungen in diesem Zellkultursystem können in vivo mit dem beeindruckenden Toolkit genetischer Ansätze bei Fliegen validiert und weiter untersucht werden.
Einleitung
Cell Division ist ein Prozess, der für alle mehrzelligen Organismen kritisch ist, sowohl in ihrer Entwicklung als auch in der Homöostase1. Drosophila wird seit langem als Modell für die Untersuchung der Zellteilung verwendet, mit Experimenten in verschiedenen Gewebetypen und genetischen Bedingungen, die wichtige Einblicke in den Prozess liefern. Während viele dieser Erkenntnisse aus festen Zellbedingungen stammen, ist die Zellteilung ein dynamisches Verfahren mit vielen beweglichen Teilen, das die Visualisierung von lebenden Zellen zur Beurteilung von RNAi- oder genetischen Knockout-Effekten auf zahlreichen Teilen der Zellteilung, einschließlich Spindelbildung, Chromosom kongressund Segregation und Zytokinese.
Viele Protokolle wurden im Laufe der Jahre entwickelt und genutzt, um Drosophila-Zellteilungen in vivo zu visualisieren. Verschiedene Gruppen haben Techniken kultiviert, um Abteilungen in larval imaginalen Scheiben und Larven Gehirne2,3,4,5,6,7,8 . Diese Techniken sind zwar nützlich für die Bildgebung in bestimmten Geweben, sind aber im Durchsatz begrenzt und erfordern häufig die Erzeugung und Pflege genetischer Bestände, um fluoreszierende Komponenten zu erzeugen und die Expression von Genen von Interesse zu verändern. Kultivierte S2-Zellen von Drosophila bieten eine Alternative mit höherem Durchsatz, um die Wirkung verschiedener Gene in der Zellteilung schnell zu testen. Darüber hinaus können S2-Zellen mit der Fähigkeit, verschiedene fluoreszierende Proteine zu transfekten, schnell modifiziert werden, um die Auswirkungen des RNAi-Knockdowns auf zahlreiche Komponenten der Zellteilung zu bestimmen. Fluoreszierend markierte Gene von Interesse können auch in der Zellteilung beobachtet werden, was eine dynamische Charakterisierung ihrer Funktion9ermöglicht.
Hier bieten wir ein detailliertes Protokoll für die Live-Bildgebung von mitotischen S2-Zellen mit Methoden, die wir vor kurzem beschrieben10. Unsere Methode verwendet stabil transfizierte Zellen mit fluoreszierenden Markern für Mikrotubuli und Zentromere, deren Expression unter der Kontrolle eines kupferinduzierbaren Motors (Metallothionein; pMT) steht. Diese Methode kann verwendet werden, um Spindel- und Chromosomendynamik in allen Phasen der Mitose unter Verwendung eines relativ einfachen Fluoreszenzmikroskops mit basisgrundlegender Bildgebungssoftware abzubilden. Es kann weiter an die individuellen Forschungsbedürfnisse angepasst werden, wobei transiente Transfektion und RNAi erweiterte Möglichkeiten zur Bestimmung der Rolle von Kandidatengenen bei der Mitose bieten. Aufgrund der relativen Einfachheit des Protokolls kann es für kleinräumige Funktionsverlust-Bildschirme verwendet werden, um Gene zu identifizieren, für die weitere Studien in vivo von Vorteil wären, was eine gezieltere Anstrengung und Effizienz mit anschließenden genetischen Manipulation in Fliegen.
Protokoll
1. Herstellung von S2-Zellen zur Behandlung mit RNAi
-
Aus einer Bestandskultur von konfluenten pMT:GFP:CID und pMT:mCherry:'-tubulin stabil transfizierte S2-Zellen (ca. 80% lebensfähig; bei 24-28 °C angebaut), Samenzellen in einer 6 gut sterilen Kulturplatte mit einer Dichte von 1 x 106 Zellen/ml in frischem, erwärmtem, 10% fetalem Rinderserum (FBS) Schneiders Insektenmedien (SIM).
HINWEIS: Stably transfizierte S2-Zellen können durch Befolgen eines Protokolls aus dem Drosophila RNAi Screening Center (DRSC) (https://dgrc.bio.indiana.edu/Protocols?tab=cells) erzeugt werden. Obwohl die hier verwendete spezifische stabile S2-Linie GFP und mCherry nutzt, gibt es sowohl innerhalb dieser fluoreszierenden Spektren als auch in anderen zahlreiche Alternativen. Eine ausführliche Diskussion dieser fluoreszierenden Proteine geht über den Rahmen dieses Protokolls hinaus, aber ihre Eigenschaften und potenziellen Vor- und Nachteile wurden an anderer Stelle fachmännisch überprüft 11.- Bestimmen Sie mit Hilfe eines Zellzählers oder Hämozytometers die Dichte des konfluenten Zellbestands.
- Bestimmen Sie das Volumen der konfluenten Zellsuspension, das erforderlich ist, um 1 x 106 Zellen/ml in einem Gesamtvolumen von 4 ml zu erreichen (z. B. 4 x 106 Zellen insgesamt). Fügen Sie dieses Volumen an Zellbeständen zu einem Volumen frischer SIM hinzu, um 4 ml/well Gesamtvolumen zu machen. Bestimmen Sie die Dichte des Zellbestands (Zellen/ml), indem Sie 100 l Zellen direkt aus dem Bestandskolben mit 100 L Medien verdünnen und diese 1:2-Verdünnung entweder manuell mit einem Hämozytometer oder mit einem automatischen Zellzähler, falls verfügbar, zählen.
HINWEIS: Bei Verwendung von S2-Zellen, die nicht stabil transfiziert sind, kann ein transienter Transfektionstest mit fluoreszierend markiertem (GFP, mCherry, etc.) -Tubulin und einem DNA-Marker (CENP-A, Histone H2B usw.) durchgeführt werden. Darüber hinaus sind stabil transfizierte Zelllinien mit verschiedenen mitotischen Spindeln und DNA-Markern im Drosophila Genetics Resource Center erhältlich, die besser auf die genauen experimentellen Bedürfnisse des Benutzers (https://dgrc.bio.indiana.edu/cells/Catalog) zugeschnitten sind. - Frisch gesäte Zellen für 36-48 h in 24-28 °C Inkubator geben.
2. Behandlung von Samenzellen mit dsRNA gegen Gene(s) von Interesse
HINWEIS: Im folgenden Beispiel und im Abschnitt Ergebnisse wird Shortstop (Shot), ein Actin-Mikrotubuli-Vernetzungsmittel, als Gen von Interesse verwendet. Verwenden Sie eine Scheinbehandlung ohne dsRNA oder mit dsRNA, die gegen ein irrelevantes Gen (z. B. Beta-Galactosidase, lacZ) als Negativkontrolle gerichtet ist.
- Warm Schneiders Insektenmedien nicht mit FBS (Serum free media; SFM) in 24-28 °C Inkubator.
-
Übertragen Sie Zellen aus dem zuvor gesäten Brunnen (ab Schritt 1.1.2) in ein 15 ml steriles Rohr, unter Berücksichtigung des Gesamtvolumens der übertragenen Zellen.
- Bewahren Sie ein kleines Volumen von Zellen (ca. 100 l) für die Zählung in einem Zellzähler oder Hämozytometer auf.
- Sanft Pelletzellen durch Zentrifugieren bei 1.000 x g für 3 min bei Raumtemperatur.
- Bestimmen Sie bei der Zentrifugieren von Zellen die Konzentration der Zellen pro ml mit einem Zellzähler oder Hämozytometer. Multiplizieren Sie diese Zahl mit dem Gesamtvolumen, das zentrifugiert wird (in 1.2.2.), um die Gesamtzahl der Zellen zu erhalten.
- Aspirieren Sie den Überstand aus den pelletierten Zellen.
-
Setzen Sie die Zellen in erwärmtem SFM wieder aus, um eine Konzentration von 3 x 106 Zellen/ml zu erhalten.
- 1 ml der resuspendierten Zellen in einen neuen Brunnen in einer 6-Well-Platte übertragen.
- Fügen Sie 10-50 g dsRNA (verdünnt in 100 l RNAase-freies Wasser) gegen Shot (oder das Zielgen von Interesse) direkt in die Zellen und wirbeln Sie die Platte zu mischen. Inkubieren Sie die Platte für 1 h im 24-28 °C Inkubator, um eine direkte dsRNA-Aufnahme in zellenzulausen.
- Während der 1 h Inkubation die 10% FBS-ergänzte SIM im 24-28 °C Inkubator erwärmen.
- Nach der Inkubation von Zellen mit dsRNA für 1 h, fügen Sie 2 ml von 10 % FBS ergänzte SIM direkt in den Brunnen, ohne die 1 ml des Mediums zu entfernen.
- Die Zellen 3-7 Tage lang in den Inkubator 24-28 °C geben.
HINWEIS: Wie bei der dsRNA-Konzentration muss die Gesamtbehandlungszeit möglicherweise für das gewünschte Zielgen optimiert werden. Um die RNAi-Wirksamkeit zu bewerten, führen Sie einen westlichen Fleck auf der ganzen Zelle Lysate mit einem Antikörper gegen das Zielgen. Wenn sich die anfängliche dsRNA-Zielsequenz als ineffektiv erweist, wird alternative dsRNAs entwickelt, die auf eindeutige Sequenzen innerhalb des Zielgens ausgerichtet sind.
3. Induktion der fluoreszierenden Proteinexpression und Vorbereitung von Zellen für die Bildgebung
- Wenn stabil transfizierte Zellen mit dem pMT-Plasmid (das einen induzierbaren Metallothionein-Promotor enthält) wie hier beschrieben erzeugt wurden, induzieren Sie die Expression fluoreszierender Proteine durch Behandlung von Zellen mit Kupfersulfat (CuSO4) in einer Endkonzentration von 500 m für 24-36 h vor der Bildgebung und 4 Tage nach der RNAi-Behandlung. Die Verwendung von konstitutiven Expressionsplasmiden wie pAct erfordert keine Kupferinduktion.
-
Vorbereiten von Zellen für die Bildgebung
- Erwärmen Sie die 10% FBS-ergänzte SIM im 24-28 °C Inkubator für 1 h.
- Übertragen Sie Zellen in ein 15 ml steriles Rohr, unter Berücksichtigung des Gesamtvolumens der übertragenen Zellen.
- Bewahren Sie ein kleines Volumen von Zellen (ca. 100 l) für die Zählung in einem Zellzähler oder Hämozytometer auf.
- Sanft Pelletzellen durch Zentrifugieren bei 1.000 x g für 3 min bei Raumtemperatur.
- Bestimmen Sie bei zentrifugierenden Zellen die Konzentration (Zellen/ml) mit einem Zellzähler oder Hämozytometer. Multiplizieren Sie diese Zahl mit dem Gesamtvolumen, das zentrifugiert wird (in 2.2.2.), um die Gesamtzahl der Zellen zu erhalten.
- Aspirieren Sie den Überstand aus den pelletierten Zellen.
- Resuspend ieren die Zellen in frischen, erwärmten 10% FBS ergänztS SIM, um eine Konzentration von 2 x 106 Zellen/ml zu erhalten. Fügen Sie eine angemessene Menge cuSO4 hinzu, um die Konzentration von 500 m beizubehalten.
- Übertragen Sie 200-500 l resuspendierte Zellen in einen Brunnen einer Multi-Well-Live-Zellkammer und legen Sie sie auf das invertierte Fluoreszenzmikroskop. Lassen Sie die Zellen in der Kammer für 15-30 min vor bildgebenden Experimenten absetzen.
HINWEIS: Lebende Zellkammerbrunnen können zur zusätzlichen Haftung mit Poly-L-Lysin vorbeschichtet werden.
4. Einrichten eines Live Cell Imaging-Programms
HINWEIS: Live Cell Imaging in diesem Experiment wurde mit einem invertierten Bildgebungssystem und der zugehörigen Software (z. B. Olympus IX83 mit dem Softwarepaket cellSens Dimensions) durchgeführt. Die Angaben unterscheiden sich je nach Mikroskophersteller und Softwarepaket; daher sind die allgemeinen Leitlinien und Operationen unten aufgeführt.
-
Bereiten Sie mit der Software, die das invertierte Fluoreszenzmikroskop betreibt, ein Programm für die Bildgebung einer Zelle (oder Zellen) im Laufe der Zeit vor.
- Öffnen Sie die Software, indem Sie auf das Software-Desktop-Symbol doppelklicken.
- Erstellen Sie eine neue experimentelle Datei, indem Sie auf Dateiund dann auf Neue experimentelle Dateiklicken.
- Fügen Sie zunächst eine Zeitrafferschleife ein, über der Bilder aufgenommen werden. Klicken Sie dazu in der Symbolleiste auf das Stoppuhrsymbol (Zeitrafferschleife). Legen Sie das Intervall in s auf der Registerkarte Experiment-Manager fest, und legen Sie dann die Anzahl der Zyklen auf derselben Registerkarte fest, indem Sie die gewünschte Gesamtversuchslänge (in s) durch das Intervall dividieren. Lassen Sie die Schleife über den gewünschten Gesamtzeitraum wiederholen.
HINWEIS: In der Regel werden Bilder alle 30-60 s über einen Zeitraum von 3-4 h aufgenommen. - Fügen Sie innerhalb der Zeitrafferschleifenschicht eine Infrarotfokusprüfung ein, um den Fokus des Ziels beizubehalten (sofern verfügbar). Fügen Sie dazu einen Z-Drift Compensation (ZDC)-Schritt innerhalb des Layers "Zeitrafferschleife" hinzu, indem Sie auf das Symbol Quadrat mit zwei Pfeilen (XY-Symbol verschieben) klicken und im Dropdown-Menü Z-Drift Compensation auswählen.
HINWEIS: Der Begriff Infrarotfokusprüfung bezieht sich auf ein System, mit dem ein Infrarotimpuls verwendet wird, um einen konstanten Abstand zwischen dem Objektiv und der Dia-/Bildkammer zu halten. Viele Mikroskope haben ein solches System, jedes mit seiner eigenen proprietären Nomenklatur. Leser sollten ihr Betriebshandbuch oder ihren Vertreter für spezifische Namensangaben konsultieren. - Fügen Sie nach der Infrarotfokusprüfung einen Schritt im Programm hinzu, um ein Mehrkanalbild (z. B. FITC für GFP und TRITC für mCherry) über einen 3-5 Z-Stack zu erstellen, indem Sie zuerst einen Z-Stack-Schritt einfügen und dann den Kanal angeben. Fügen Sie dazu einen Multichannel Group-Layer innerhalb des Zeitrafferschleifen-Layers hinzu, indem Sie auf das Symbol "Farbrad" (Symbolfür Multichannel-Gruppen) klicken. Fügen Sie als Nächstes einen Z-Stack Loop-Layer innerhalb des Layers Multichannel Group hinzu, indem Sie auf das 3-schichtige Symbol (Z-Stack-Loop-Symbol) klicken. Legen Sie die gewünschte Schrittgröße und Anzahl der Slices auf der Registerkarte Experiment Manager fest, und legen Sie die Belichtung jedes Kanals so niedrig wie möglich fest, um die Photobleichung zu minimieren.
HINWEIS: Das Z-Stack-Intervall, die Belichtungszeit und die prozentuale Durchlässigkeit für LEDs können variieren. Typisch in diesen Experimenten, verwenden Sie drei Z-Stacks, die über einen Bereich von 3 m übernommen werden, was ein Intervall von 1 m zwischen jedem Stapel ergibt. Die Definition der Mitte der Zelle anstelle der oberen und unteren Tendenz führt zu den besten Ergebnissen. Bilder werden dann für jeden Kanal(n) bei einer Belichtungszeit von 50 ms und einer prozentualen Transmission von 50 % ohne neutralen Dichtefilter (ND) gesammelt.
5. Bild-Teilen von Zellen mit Live Cell Imaging-Programm
HINWEIS: S2-Zellen benötigen kein CO2 und wachsen optimal bei 23-27 °C. Die gesamte Bildgebung wurde bei Umgebungstemperatur in einem gut kontrollierten Raum durchgeführt.
- Mit dem Okular finden Sie die obere (oder untere) Ecke des Brunnens und fokussieren das Objektiv (40-60x Öleintauchen) auf die Zellen mit dem mCherry-Kanal.
- Scannen Sie entlang der Oberseite (oder Unterseite) des Brunnens, um sich vom vertikalen Brunnenteiler zu entfernen.
HINWEIS: Die Zu nah an den Brunnenteilern zu nahestehende Bildgebung kann die Infrarotfokusprüfungen stören. - Suchen Sie eine Zelle (oder Zellen), die sich in der späten G2- oder frühen M-Phase (Prophase) befinden.
HINWEIS: Diese Zellen lassen sich am besten durch das Vorhandensein von genau zwei "sternartigen" Mikrotubulistrukturen (Zentrom) und einem intakten Kern (angezeigt durch einen kreisförmigen Bereich von gebrochenem Licht innerhalb der Zelle) identifizieren, die beide leicht mit dem Anregungsfilter. Die Auswahl von Zellen in früheren Interphasen, die durch ein oder null leicht sichtbare Zentrosomen bemerkenswert sind, kann aufgrund einer Verzögerung in der G2/M-Progression zu Zeitverschwendung führen. Umgekehrt verhindert die Auswahl von Zellen nach NEBD eine genaue Berechnung des mitotischen Timings und der Bildgebung der frühen Spindelbaugruppe und Chromosomendynamik. Außerdem enthalten S2-Zellen häufig >2 Zentrosomen. Obwohl diese in der Regel Cluster in eine bipolare Spindel12, Es wird empfohlen, dass Benutzer vermeiden diese Zellen, es sei denn, anders für den experimentellen Entwurf gewünscht. Bilder und die Diskussion der geeigneten Zellen zur Auswahl finden Sie im Abschnitt Repräsentative Ergebnisse. - Klicken Sie auf die Schaltfläche Live-Ansicht, um mit der Anzeige von Zellen im Softwarebildschirm zu beginnen. Mit dem fein fokussierten Knopf des Mikroskops fokussieren Sie die Zelle (oder Zellen) von Interesse. Legen Sie die Infrarotfokusprüfung fest, indem Sie auf die Schaltfläche Fokus festlegen klicken.
- Initiieren Sie das Zeitraffer-Imaging-Programm, indem Sie auf die Schaltfläche Start klicken. Passen Sie die Histogramme nach Bedarf an, indem Sie den gewünschten Kanal auswählen und die mittleren Pixelintensitäten anpassen, um die Zelle (oder Zellen) klar zu sehen.
- Lassen Sie das Programm laufen, überprüfen Sie die Zelle nach 15-20 min, um sicherzustellen, dass der Kernhüllenzusammenbruch (NEBD) aufgetreten ist, was durch das Verschwinden der runden Dunkelheit, einem gebrochenen Punkt in der Nähe des Zellzentrums, bestimmt wird.
- Lassen Sie das Programm weiterhin ausführen, und überprüfen Sie zeitweise (alle 15-20 min), um festzustellen, sobald ein Anaphasenbeginn aufgetreten ist. Beenden Sie das Programm an diesem Punkt, wenn im gesamten Zeitraum mehr Zeit verbleibt, und speichern Sie die Datei. Wenn Ereignisse während der Telophase und Zytokinese von Interesse sind, können Sie dem Programm eine ausreichende Zeit einplanen, um auch diese Prozesse abzubilden.
-
Führen Sie die Analyse von NEBD bis zum Anaphasenbeginn timing durch, indem Sie die Zeit von NEBD (tNEBD) in min und den Zeitpunkt der anfänglichen Chromosomentrennung (tAnaphase-Beginn)in min und subtrahieren tNEBD von tAnaphase-Beginn, um die NEBD zur Anaphase-Einbesetzeit für eine bestimmte Zelle.
- Klicken Sie hierdurch auf die Schaltfläche "Frame up" und notieren Sie die Frames, in denen NEBD und Anaphase-Beginn auftreten, subtrahieren Sie diese, um die Gesamtzahl der verstrichenen Frames zu bestimmen, und multiplizieren Sie dies mit dem Zeitintervall zwischen bildgebenden Frames.
- Suchen Sie weiterhin nach vorteilenden Zellen, um mehrere n für eine bestimmte Bedingung zu erhalten. Zellen können in der lebenden Zellkammer gut für bis zu 12 h nach ihrer anfänglichen Setzung abgebildet werden.
Ergebnisse
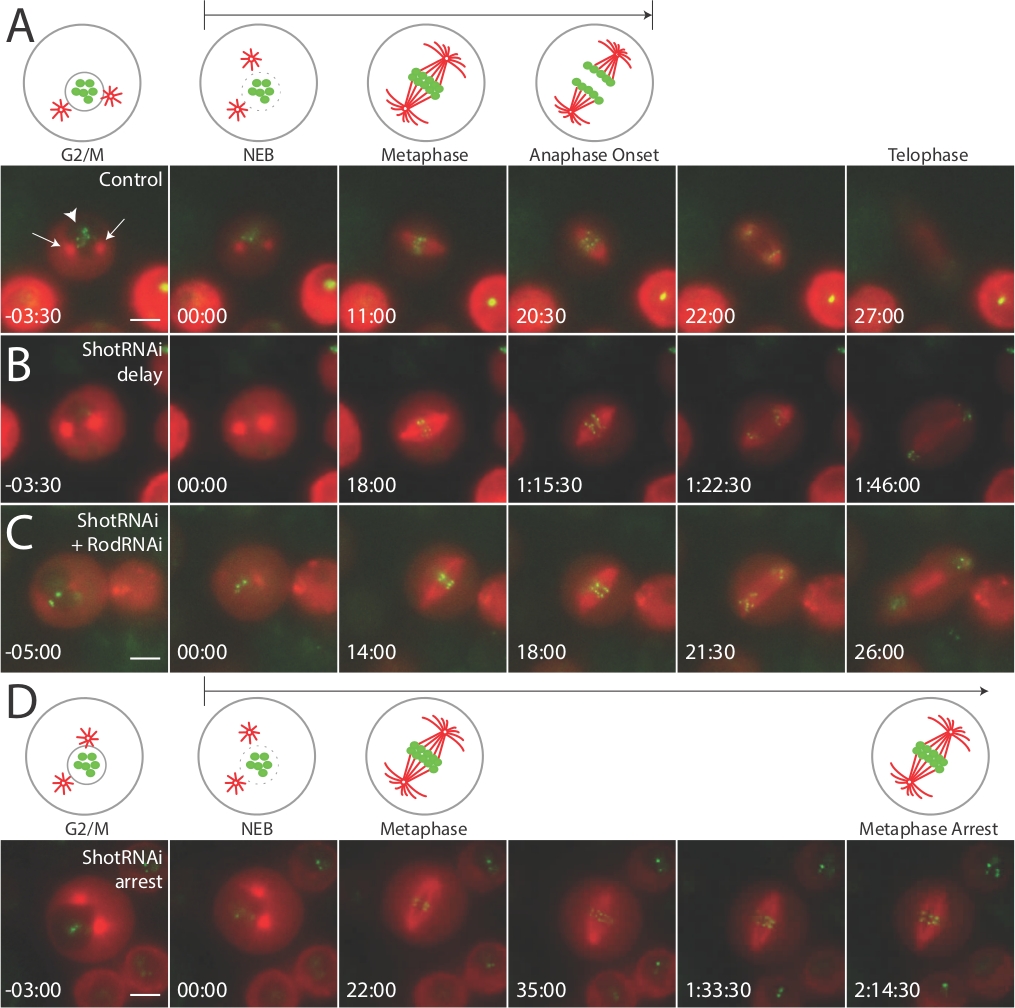
Die oben beschriebenen Methoden führen zur Identifizierung und Abbildung von Drosophila S2-Zellen, die sich einer Zellteilung unterziehen. Zellen, die kurz vor dem Eintritt in die M-Phase zu teilen sind, können durch das Vorhandensein von zwei Zentrosomen und einem intakten Kern, der durch gebrochenes Licht und einen dunkleren Fleck innerhalb der Zelle angezeigt wird, wenn sie im Kanal von Tubulin betrachtet werden (Abbildung 1, linke Panels, roter Kanal; Pfeilspitze in Abbildung 1A). Wir schätzen, dass etwa 2-5% der Zellen in diese Kategorie fallen, und zwischen bildgebenden Experimenten verbringen wir in der Regel maximal 2-3 Minuten damit, nach einer anderen qualifizierten Zelle zu suchen. NEBD kann durch das Verschwinden dieses dunklen Flecks visualisiert werden, was zu einer gleichmäßigen Färbung des Zytoplasmas führt (Abbildung 1, Tafeln mit der Aufschrift "00:00", rot). Nach NEBD kann die Zeit, die jede Zelle benötigt, um die Spindel, Kongresschromosomen und Segregate-Chromosomen zu bilden, einfach gemessen werden, indem die Zeitpunkte dieser Ereignisse relativ zu NEBD notiert werden.
Wir nutzen diese Divisionen, um das mitotische Timing, insbesondere von NEBD bis zum Anaphasenbeginn, zu bewerten und dabei den Punkt zu beachten, an dem Chromosomen zu trennen beginnen. Mehrheit (90 %) kupferinduzierten Zellen, exprimiert sowohl mCherry:-Tubulin als auch GFP:CID. Ein kleiner Prozentsatz der Zellen (ca. 10%) nur einen Marker oder keines der beiden marker ausgedrückt. Diese Zellen wurden bei der Zellauswahl vermieden. In der Regel zeigen S2-Zellen NEBD zum Anaphasenbeginn von 20-30 Minuten an (Abbildung1A, Steuerung). Als nächstes behandelten wir Zellen mit dsRNA gegen Shortstop (Shot), ein Actin-Mikrotubuli-Vernetzungsprotein, von dem vermutet wurde, dass es die Zellzyklusdynamik beeinflussen könnte. Tatsächlich zeigten die nach Demshot-Knockdown-Zellen eine signifikante mitotische Verzögerung (Abbildung1B, ShotRNAi-Verzögerung), wobei viele Zellen während des gesamten 2-3-stündigen Bildgebungsexperiments in eine Anaphase übergehen (Abbildung1D, ShotRNAi Verhaftung). Wir haben argumentiert, dass dieser Verzögerungs-/Verhaftungsphänotyp wahrscheinlich auf die Aktivierung des Spindelmontage-Checkpoints (d. h. des M-Phasen-Checkpoints) zurückzuführen sein könnte. Um diese Hypothese direkt zu testen, haben wir Zellen mit dsRNA gegen Shot and Rough Deal (Rod) kobehandelt, ein wichtiger Bestandteil dieses Checkpoints13. Dies führte zu einer Unterdrückung des Verhaftungsphänotyps, was zu NEBD zu Anaphasenzeiten ähnlich wie Control führte (Abbildung 1C, ShotRNAi+RodRNAi). So konnte dieses Live-Bildgebungsprotokoll zu dem Schluss kommen, dass Shot für eine rechtzeitige M-Phasen-Progression erforderlich ist und dass sein Verlust zu einer Aktivierung des Checkpoints führt, die mitotische Zellen verzögert oder festhält.

Abbildung 1: Live-Bildgebung zeigt Zellzyklusdefekte aufgrund der Aktivierung des mitotischen Checkpoints in S2-Zellen.
Unter allen Bedingungen wurde die Live-Zell-Bildgebung an S2-Zellen durchgeführt, die stabil mit induzierbarem GFP:CID und mCherry kotransfiziert wurden: "Tubulin, wie hier beschrieben. (A) Karikaturen veranschaulichen wichtige Sehenswürdigkeiten während der Mitose, und entsprechende Bilder werden aus repräsentativen Filmen von angegebenen Genotypen mit angegebenen Zeitpunkten relativ zu NEBD (t=00:00) gezeigt. Kontrollzellen erreichen in der Regel innerhalb von 20-30 min eine Anaphase. ShotRNAi-behandelte Zellen zeigen NEBD-Anaphasenverzögerung (B) und werden häufig einer Metaphasenverhaftung unterzogen, ohne im Bildgebungsexperiment (D) in eine Anaphase einzutreten. Die Ko-Behandlung von Zellen mit ShotRNAi und RodRNAi, einer Komponente des Spindelmontage-Checkpoints, unterdrückt den Shot-Phänotyp, was zu einer Anaphase-Onset-Kinetik ähnlich wie Kontrollzellen (C) führt. Die Pfeile in (A) zeigen die "sternartigen" Zentromsomenstrukturen und die große Pfeilspitze den Kern an. Jeder Maßstabsbalken stellt 5 Mikrometer dar. Diese Zahl wird mit Genehmigung von10 (https://www.molbiolcell.org/info-for-authors) angepasst und wiederveröffentlicht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Video 1: Zeitrafferfilm der S2-Zellenteilung "Control". Video zeigt einen repräsentativen Film der S2-Zellteilung im Genotyp "Kontrolle". Dieser Film entspricht Abbildung 1A. Dieses Video wird mit Genehmigung von10 (https://www.molbiolcell.org/info-for-authors) angepasst und wiederveröffentlicht. Bitte klicken Sie hier, um dieses Video herunterzuladen.
Video 2: Zeitrafferfilm der verzögerten 'ShotRNAi' S2-Zellteilung. Video zeigt einen repräsentativen Film der S2-Zellteilung im Genotyp "ShotRNAi", der zu einer phänotypischen mitotischen Verzögerung führt. Dieser Film entspricht Abbildung 1B. Bitte klicken Sie hier, um dieses Video herunterzuladen.
Video 3: Zeitrafferfilm der S2-Zellteilung 'ShotRNAi+RodRNAi'. Video zeigt einen repräsentativen Film der S2-Zellteilung im Genotyp "ShotRNAi+RodRNAi". Dieser Film entspricht Abbildung 1C. Dieses Video wird mit Genehmigung von10 (https://www.molbiolcell.org/info-for-authors) angepasst und wiederveröffentlicht. Bitte klicken Sie hier, um dieses Video herunterzuladen.
Video 4: Zeitrafferfilm der verhafteten S2-Zellenabteilung "ShotRNAi". Video zeigt einen repräsentativen Film der S2-Zellteilung im Genotyp "ShotRNAi", der zu einem phänotypischen mitotischen Arrest führt. Dieser Film entspricht Abbildung 1D. Bitte klicken Sie hier, um dieses Video herunterzuladen.
Diskussion
Identifizieren einer geeigneten Zelle
Der Schlüssel zur Abbildung, die S2-Zellen teilt, ist zunächst die richtige Zelle zu lokalisieren. Zeit kann verschwendet werden Bildzellen, die fälschlicherweise angenommen werden, bereit zu teilen, aber nicht in einem angemessenen Zeitrahmen zu tun. Es müssen Zellen identifiziert werden, die zwei unterschiedliche und sichtbare Zentrosomen und einen intakten Kern haben. Centrosomen müssen Mikrotubulifasern haben, die von ihnen austreten, was ihnen ein "sternartiges" Aussehen verleiht. Ein intakter Kern bricht Licht beim Einstellen des Fokus und macht auch die ungefähre Mitte der Zelle dunkler im Aussehen. Der Kern wird auch die GFP:CID "Punkte" enthalten, ein weiteres Unterscheidungsmerkmal, das bei der Identifizierung hilft. Zellen mit >2 Zentrosomen, Tubulinpunkta ohne Tubulinfasern, die von ihnen austreten, oder Zellen mit nur einem Zentromom sollten vermieden werden. Wenn zwei Zentrosomen sichtbar sind, ein Kern jedoch nicht, ist NEBD bereits aufgetreten und die Zelle ist in ihrer Division zu weit fortgeschritten, um abgebildet zu werden, wenn eine vollständige M-Phasen-Analyse gewünscht wird. Sobald eine geeignete Zelle gefunden wurde, ist die Geschwindigkeit beim Starten der Bildgebung entscheidend, da NEBD schnell auftritt (in der Regel innerhalb von 3-5 min) und die Verzögerung des Beginns der Bildgebung zu verpassten Gelegenheiten führen kann. Fokussieren Sie die Zelle in der Bildgebungssoftware schnell so, dass beide Zentrosomen sichtbar sind, oder wenn Zentrosomen a-ebenerweise miteinander liegen, stellen Sie den Brennpunkt zwischen den beiden ein, so dass jeder erfasst werden kann, wenn Z-Stacks über und unter dem definierten Zentrum gesammelt werden spitze. Sobald sie fokussiert sind, stellen Sie sofort den Offset zwischen der Bühne und der Live-Zellkammeroberfläche mithilfe der Infrarotfokusprüfung (falls verfügbar) ein und starten Sie das Bildgebungsprogramm. Obwohl das hier vorgestellte Protokoll speziell auf Experimente zur Bewertung der NEBD-zu-Anaphase-Dynamik zugeschnitten ist, könnten einfache Modifikationen für Leser geeignet sein, die alternative mitotische Ereignisse untersuchen. Für die NEBD-zu-Metaphase und anaphase-to-telophase-Bildgebung schlagen wir 10 s Bildaufnahmeintervalle vor, da diese Prozesse hochdynamisch sind und schnell in S2-Zellen (5-10 min) auftreten. Metaphase-zu-Anaphase-Übergang (unser typischer experimenteller Fokus) ist ein längerer Prozess (20-30 min) in S2-Zellen, und wir sammeln Bilder in 30 s Intervallen. Schließlich tritt die Telophase-durch-Zytokinese in S2-Zellen recht langsam auf, und wir schlagen 60 s-Intervalle vor, die genügend Auflösung für diesen ungefähren einstündigen Prozess bieten.
Aufrechterhaltung des Fokus während des gesamten Imaging-Programms
Wenn ein Infrarotfokus-Prüfgerät nicht verfügbar ist, achten Sie darauf, das Mikroskop oder den Tisch, auf dem es sitzt, nicht zu stoßen, da dies dazu führen kann, dass sich die Zelle während des Bildgebungsprogramms aus dem Fokus bewegt, was zu mehrdeutigen, nicht interpretierbaren Ergebnissen führt. Die Vorbeschichtung der Live-Zellkammerbrunnen mit Poly-L-Lysin kann bei der Zellhaftung helfen, was zellweise bewegungen vermeiden kann und besonders für Setups ohne Infrarotfokusprüfungen nützlich ist. Darüber hinaus können Setups, einschließlich stoßdämpfender Plattformen oder Lufttische, dazu beitragen, dass Zellen nicht mehr aus dem Fokus geraten. Schließlich haben einige Softwareprogramme Pause-Funktionen, die es dem Benutzer ermöglichen, das Programm neu zu fokussieren und fortzusetzen.
Bildgebung mehrerer Zellen
Hilfreich für die Akkumulation von Daten ist, dass oft mehrere Zellen, die zur Teilung bereit sind, innerhalb desselben Sichtfeldes für die Bildgebung platziert werden können. Dies kann besonders nützlich sein für Experimente, bei denen Zellen eine lange M-Phasen-Dauer oder eine längere Verkleehrung haben (z. B. Bedingungen, die eine Aktivierung des SAC induzieren). Für diese Zellen bilden wir in der Regel, bis die Zellen photobleached (ca. 2-3 h), aber das volle Timing der verhafteten Zellen sollte im Ermessen des Forschers sein. Manchmal können sich die mehrfachen vorteilenden Zellen in verschiedenen Fokusebenen befinden, in diesem Fall kann es nützlich sein, Z-Stacks hinzuzufügen, um alle Zellen aufzunehmen. Das Hinzufügen weiterer z-Stacks kann auch zur Verbesserung der Auflösung beitragen. Dabei ist jedoch Vorsicht geboten, da zellenstärker erstrahlen und zu schnellerer Photobleichung führen und zu großen Dateigrößen auf Festplattenspeichern führen.
Vermeidung von Photobleichungen und Phototoxizität
Unsere Methode beschreibt spezifische Einstellungen für Belichtungszeiten, Bildgebungsintervalle, Z-Stacks und prozentuale Durchlässigkeit für unsere LED-Lichtquelle, die in der Regel für unseren experimentellen Aufbau und die gewünschten Ergebnisse funktionieren. Da Mikroskope und experimentelle Ziele variieren können, kann das, was für unser System gut funktionieren könnte, zu vorzeitiger Photobleiche oder Phototoxizität in anderen führen. Photodamage kann einen Mangel an Spindelbewegung, Mikrotubuli-Fragmentierung, defekte Mikrotubuli-Dynamik und verlängerte Spindel-Checkpoint-Aktivierung darstellen. Eine mögliche Methode, um solche Fallstricke zu vermeiden, besteht darin, die Expositionszeit zu begrenzen. Die meisten modernen Kameras verfügen heute über große Dynamikbereiche, und Histogramme können angepasst werden, um Strukturen auch bei sehr geringen Belichtungen zu visualisieren. Eine andere Technik besteht darin, das Bildgebungsintervall (z. B. auf mehrere Minuten zwischen den Bildern) so zu erhöhen, dass die Anzahl der Personen, in denen eine Zelle über ein gesamtes Erfassungsintervall dem Licht ausgesetzt wird, reduziert wird. Dies ist insbesondere bei der Bildgebung über längere Zeiträume (viele Stunden) von Vorteil und kann zusätzlich zur Verringerung der Dateigröße solcher Experimente beitragen. Die Begrenzung der Anzahl der z-Stacks kann auch dazu beitragen, die Lichtexposition zu reduzieren, indem man 2 oder sogar nur 1 anstelle von 3 verwendet. Darüber hinaus verringert die Anpassung der prozentualen Lichtdurchlässigkeit (für LED-Lichtquellen) oder die Verwendung von Neutraldichtefiltern (nd) (für Halogenlampen- und LED-Lichtquellen) die Lichtintensität und gekoppelt mit längeren Belichtungszeiten kann die Sichtbarkeit erhalten. . Durch die Auswahl von Zellen mit bereits getrennten Zentrosomen kann die Gesamtexposition eingeschränkt werden, da diese Zellen nach Beginn der Bildgebung in der Regel schnell (innerhalb von ein oder zwei Minuten) in mitoseeintreten. Sie können auch die Zeit begrenzen, die Zellen vor NEBD abgebildet werden dürfen. Unser Labor legt diese Zeit in der Regel auf 10 min fest, aber ein konservativeres Limit kann leicht vom Benutzer auferlegt werden. Darüber hinaus empfehlen wir eine "invasivere" Maßnahme, die die Behandlung mit dsRNA gegen eine Komponente des SAC (wie Rod oder Mad2) abzielt. Wenn ein Arrest-Phänotyp auf unspezifische Zellschäden zurückzuführen ist (z. B. defekte Mikrotubuli-Dynamik), ist eine solche Behandlung weniger wahrscheinlich, um die Verhaftung zu unterdrücken, als eine gutgläubige Wirkung des Gens, das für das ursprüngliche Experiment von Interesse ist.
Zukünftige Richtungen
Die hier beschriebene Methode kann auf einem relativ einfachen Epifluoreszenzmikroskop eingesetzt werden, um lebende Zellteilungen schnell abzubilden und kann leicht an das besondere experimentelle Design und die Ziele eines Forschers angepasst werden. Für S2 und andere Drosophila-Zellen 9,14,15, wurden mehrere ausgezeichnete Methoden zur Kultivierung, RNAi-Knockdown-Ansätze, transiente Transfektion und Fluoreszenzmikroskopie veröffentlicht. 16 , 17. Unser Protokoll bietet ein paar Vorteile. (1) Die Verwendung einer doppelten stabilen Zelllinie (GFP:CID, mCherry:-Tubulin) markiert gleichzeitig zwei wichtige mitotische Strukturen, vermeidet den Ärger und mögliche Komplikationen der transienten Transfektion und stellt sicher, dass fast alle Zellen fluoreszierende Marker als potenzielle Kandidaten für die Bildgebung. (2) Die Anpassung an die Mitose ergänzt die veröffentlichten Protokolle zur Untersuchung der zytoskelettalen Dynamik in motilen, nicht teilenden Zellen und erstreckt sich auf die Untersuchung von mitotischen Ereignissen in Echtzeit. Während wir glauben, dass unsere eine leistungsstarke Technik ist, die das Repertoire anderer erweitert, kann es immer Verbesserungen geben. Ein besonderer Bereich der Zellteilung, den wir schwer abzubilden gefunden haben, ist die zentromische Teilung. Dies geschieht vor NEBD und es ist schwierig zu bestimmen, wann ein einzelnes Zentromer bereit ist, sich in zwei zu trennen. Die Verwendung von fluoreszierenden Zellzyklusmarkern (Cyclin A und Cyclin B) könnte helfen, dieses Problem zu lösen, geht aber auf Kosten von Kanälen, die zur Visualisierung von Zellteilungskomponenten verwendet werden könnten. Unsere beste Strategie für den Versuch, die Zentrosome-Division zu visualisieren, besteht darin, dem Bildgebungsprogramm eine Multipoint-Erfassung hinzuzufügen (verschiedene Punkte in der XY-Ebene zu bilden), aber dies kann zu großen Dateien führen und kann auch erfordern (abhängig von der Anzahl der Punkte) Das Intervall zwischen der Bildsammlung zu erhöhen, die Zeitauflösung des resultierenden Films zu verringern. Eine andere Lösung könnte die Synchronisation von Zellen in einem gewünschten Zellzyklusstadium mit Medikamenten sein, die zellzyklusregulatore zielen, gefolgt von anschließendem Auswaschen vor der Bildgebung, obwohl diese Methoden in S2-Zellen möglicherweise nicht zuverlässig sind.
Das hier vorgestellte Protokoll bildet auch eine Grundlage für zukünftige Anwendungen in der Live-Zell-Bildgebung. Mit verbesserter Bildgebungs- und Softwaretechnologie könnten wirklich hohe Durchsatzanpassungen dieses Protokolls mit höherer Kapazität möglich sein, Multikammerplatten. Solche Innovationen würden großflächige RNAi-Bildschirme, wie sie bereits in festen Präparaten erreicht wurden,12,18, in einem Live-Zell-Format beweglicher machen. Kleine Molekül-Wirkstoff-Screens könnten auch als mittels zur Identifizierung neuartiger Verbindungen gedacht werden, die auf Zellteilungsprozesse abzielen. Die Verbesserung von Fluorophoren und optischen Filtern könnte auch zur Abbildung mehrerer mitotischer Komponenten (nicht nur der beiden, die wir beschreiben) führen und die Abbildung bestimmter mitotischer Regulatoren und ihrer Wechselwirkungen mit DNA und/oder Spindel ermöglichen. Beispielsweise wäre die Generierung von Zellen mit fluoreszierenden Reportern, die nach dem DNA-Schaden oder während der Apoptose ausdrücken, nützliche Werkzeuge, um neuartige Gene zu identifizieren, die an diesen Prozessen beteiligt sind. Ähnliche Ansätze könnten verwendet werden, um die Zellzyklusdynamik mit der Expression von fluoreszierend markierten Cyclinen19zu untersuchen.
Offenlegungen
Die Autoren haben nichts zu erklären.
Danksagungen
Diese Arbeit wurde von den National Institutes of Health (R01 GM108756) finanziert. Wir danken Gary Karpen (University of California, Berkeley) dafür, dass er uns großzügig einen GFP:CID S2-Zelllinienbestand zur Verfügung gestellt hat, aus dem unsere GFP:CID/mCherry:'Tubulin-Linie generiert wurde10.
Materialien
| Name | Company | Catalog Number | Comments |
| Bright-Line Hemacytometer | Sigma-Aldrich | Z359629-1EA | for cell counting |
| cellSens imaging software | Olympus | ||
| CELLSTAR Cell Culture Flask, 50 mL, 25 CM2, PS, Red Filter Screw Cap, Clear, Sterile, 10 PCS/BAG | Greiner Bio-One | 690175 | |
| CELLSTAR Cell Culture Multiwell Plate, 6 well, PS, Clear, TC, Lid with condensation rings, sterile, single packed | Greiner Bio-One | 657160 | |
| Centrifuge 5804 R | eppendorf | Cat. 022623508 | |
| Copper(II) sulfate pentahydrate, minimum 98% | Sigma-Aldrich | C3036-250G | |
| Corning Fetal Bovine Serum | Fisher Scientific | MT35015CV | |
| Effectene Transfection Reagent 1 mL | Qiagen | 301425 | for transient transfection |
| IX-83 Inverted Epifluorescent Microscope | Olympus | ||
| LabTek Chambered Slide Insert | Applied Scientific Instruments | I-3016 | |
| MEGAscript T7 Transcription Kit | Thermo Fisher Scientific | AM1334 | For dsRNA production |
| MOXI Z Mini Automated Cell Counter Kit | ORFLO | MXZ001 | for cell counting |
| MS-2000 XY Flat-Top Automated Stage and Controller | Applied Scientific Instruments | ||
| Nunc Lab-Tek II Chambered Coverglass (no 1.5 borosilicate glass) 8-well | Thermo Fisher Scientific | 155409 | |
| Orca-Flash 4.0 LT Camera | Hammamatsu Photonics K.K. | C11440-42U | |
| pMT/V5-His A Drosophila Expression Vector | Thermo Fisher Scientific | V412020 | |
| Poly-L-Lysine | Cultrex | 3438-100-01 | |
| Purifier Logic+ Class II, Type A2 Biosafety Cabinets | Labconco | 302310000 | |
| Schneider's insect medium | Sigma-Aldrich | S0146-100ML | |
| Spectra Tub Centrifuge Tubes | VWR | 470224-998 | |
| Uner Counter BOD Incubator | Sheldon manufacturing (VWR) | 89409-346 | |
| X-CITE 120 LED | ExcelitasTechnologies | Led light source | |
| Z -Drift Compensator (ZDC) | Olympus | infrared focus check |
Referenzen
- Ragkousi, K., Gibson, M. C. Cell division and the maintenance of epithelial order. Journal of Cell Biology. 207 (2), 181-188 (2014).
- Aldaz, S., Escudero, L. M., Freeman, M. Live imaging of Drosophila imaginal disc development. Proceedings of the National Academy of Science U. S. A. 107 (32), 14217-14222 (2010).
- Cabernard, C., Doe, C. Q. Live imaging of neuroblast lineages within intact larval brains in Drosophila. Cold Spring Harbor Protocols. 2013 (10), 970-977 (2013).
- Lerit, D. A., Plevock, K. M., Rusan, N. M. Live imaging of Drosophila larval neuroblasts. Journal of Visualized Experiments. (89), (2014).
- Morris, L. X., Spradling, A. C. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 138 (11), 2207-2215 (2011).
- Prasad, M., Jang, A. C., Starz-Gaiano, M., Melani, M., Montell, D. J. A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nature Protocols. 2 (10), 2467-2473 (2007).
- Restrepo, S., Zartman, J. J., Basler, K. Cultivation and Live Imaging of Drosophila Imaginal Discs. Methods in Molecular Biology. 1478, 203-213 (2016).
- Tsao, C. K., Ku, H. Y., Lee, Y. M., Huang, Y. F., Sun, Y. H. Long Term Ex Vivo Culture and Live Imaging of Drosophila Larval Imaginal Discs. PLoS One. 11 (9), e0163744 (2016).
- Rogers, S. L., Rogers, G. C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nature Protocols. 3 (4), 606-611 (2008).
- Dewey, E. B., Johnston, C. A. Diverse mitotic functions of the cytoskeletal cross-linking protein Shortstop suggest a role in Dynein/Dynactin activity. Molecular Biology of the Cell. 28 (19), 2555-2568 (2017).
- Rodriguez, E. A., et al. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends in Biochemical Sciences. 42 (2), 111-129 (2017).
- Kwon, M., et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes and Development. 22 (16), 2189-2203 (2008).
- Basto, R., Gomes, R., Karess, R. E. Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nature Cell Biology. 2 (12), 939-943 (2000).
- Currie, J. D., Rogers, S. L. Using the Drosophila melanogaster D17-c3 cell culture system to study cell motility. Nature Protocols. 6 (10), 1632-1641 (2011).
- Lu, W., Del Castillo, U., Gelfand, I. V. Organelle transport in cultured Drosophila cells: S2 cell line and primary neurons. Journal of Visualized Experiments. (81), e50838 (2013).
- Yang, J., Reth, M. Drosophila S2 Schneider cells: a useful tool for rebuilding and redesigning approaches in synthetic biology. Methods in Molecular Biology. 813, 331-341 (2012).
- Zhou, R., Mohr, S., Hannon, G. J., Perrimon, N. Inducing RNAi in Drosophila cells by soaking with dsRNA. Cold Spring Harbor Protocols. 2014 (5), (2014).
- Goshima, G., et al. Genes required for mitotic spindle assembly in Drosophila S2 cells. Science. 316 (5823), 417-421 (2007).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten