
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Análisis de transcriptómica
En este artículo
Resumen
Galaxy y David se han convertido en herramientas populares que permiten a los investigadores sin formación bioinformática para analizar e interpretar los datos de RNA-Seq. Se describe un protocolo para C. elegans los investigadores realizar RNA-Seq experimentos, acceder y procesar el conjunto de datos usando Galaxy y obtener información biológica significativa de las listas de genes usando DAVID.
Resumen
generación de secuenciación de próxima (NGS) tecnologías han revolucionado la naturaleza de la investigación biológica. De estos, RNA Sequencing (RNA-Seq) ha surgido como una poderosa herramienta para el análisis de expresión génica y mapeo transcriptoma. Sin embargo, el manejo de bases de datos de RNA-Seq requiere experiencia computacional sofisticado y plantea retos inherentes para investigadores de la biología. Este cuello de botella ha sido mitigada por el proyecto de acceso abierto Galaxy que permite a los usuarios sin conocimientos de bioinformática para analizar los datos de RNA-Seq, y la base de datos para la anotación, visualización, y Integrado de Discovery (DAVID), una ontología de genes (GO) Suite término análisis que ayuda entender el significado biológico de grandes conjuntos de datos. Sin embargo, para los usuarios de primera vez y los aficionados bioinformática, el autoaprendizaje y la familiarización con estas plataformas puede llevar mucho tiempo y difícil. Se describe un flujo de trabajo sencillo que ayudará a C. elegans investigadores aislar ARN de gusano, llevar a cabo un experimento de RNA-Seqy analizar los datos utilizando plataformas Galaxy y David. Este protocolo proporciona instrucciones paso a paso para el uso de los diversos módulos Galaxy para acceder a los datos en bruto NGS, controles de calidad, la alineación y análisis de la expresión génica diferencial, guiando al usuario con los parámetros en cada paso para generar una lista de genes que se pueden cribar para el enriquecimiento de clases de genes o procesos biológicos utilizando DAVID. En general, esperamos que este artículo se proporcionará información a los investigadores que realizan C. elegans experimentos de RNA-Seq, por primera vez, así como usuarios frecuentes que ejecutan un pequeño número de muestras.
Introducción
La primera secuenciación del genoma humano, lleva a cabo mediante el método de secuenciación de didesoxinucleótidos-Fred Sanger, tomó 10 años, y un costo estimado de US $ 3 billón 1, 2. Sin embargo, en poco más de una década desde su creación, Next-Generation Sequencing Technology (NGS) ha hecho posible secuenciar el genoma humano completo en dos semanas y por US $ 1,000. NGS nuevos instrumentos que permiten velocidades cada vez mayores de la colección de secuenciación de datos con una eficiencia increíble, junto con fuertes reducciones en el costo, están revolucionando la biología moderna en formas inimaginables como proyectos de secuenciación del genoma están convirtiendo rápidamente en un lugar común. Además, estos desarrollos han galvanizado avances en muchas otras áreas tales como el análisis de expresión génica a través de RNA-Sequencing (RNA-Seq), el estudio de las modificaciones epigenéticas en todo el genoma, las interacciones ADN-proteína, y la detección de la diversidad microbiana en huéspedes humanos. NGS-basado RNA-Seq en particular, ha hecho que sea posible identificar y transcriptomes mapa integral con precisión y sensibilidad, y ha sustituido a la tecnología de microarrays como el método de elección para los perfiles de expresión. Mientras que la tecnología de microarrays se ha utilizado ampliamente, que está limitado por su dependencia de las matrices de pre-existentes con la información genómica conocida, y otros inconvenientes tales como hibridación cruzada y la gama restringida de los cambios de expresión que se puede medir de forma fiable. RNA-seq, por otra parte, puede ser utilizado para detectar tanto las transcripciones conocidos y desconocidos, mientras que la producción de bajo nivel de ruido de fondo debido a su naturaleza inequívoca asignación de ADN. RNA-Seq, junto con las numerosas herramientas genéticas ofrecidas por organismos modelo tales como levaduras, moscas, gusanos, peces y ratones, ha servido de base para muchos descubrimientos biomédicos recientes importantes. Sin embargo, sigue habiendo retos importantes que hacen NGS inaccesibles para la comunidad científica en general, incluidas las limitaciones de almacenamiento, procesamiento y, sobre todo, m análisis bioinformática eaningful de grandes volúmenes de datos de secuenciación.
Los rápidos avances en las tecnologías de secuenciación y la acumulación exponencial de los datos han creado una gran necesidad de plataformas computacionales que permitan a los investigadores acceder, analizar y comprender esta información. Los primeros sistemas dependían en gran medida de los conocimientos de programación informática, mientras que, genoma navegadores tales como NCBI que permitieron a los no programadores para acceder y visualizar datos no permitió análisis sofisticados. La plataforma, de acceso libre basada en la web, Galaxy ( https://galaxyproject.org/ ), ha llenado este vacío y ha demostrado ser una tubería valiosa que permite a los investigadores para procesar datos de NGS y llevar a cabo una variedad de fácil de complejo análisis de la bioinformática. Galaxy se estableció inicialmente, y se mantiene, por los laboratorios de Anton Nekrutenko (Penn State University) y James Taylor (Universidad Johns Hopkins)f "> 3. El Galaxy ofrece una amplia gama de tareas de cómputo por lo que es una 'ventanilla única' para las necesidades de la bioinformática innumerables, incluyendo todos los pasos involucrados en un estudio de RNA-Seq. Itallows usuarios para realizar el procesamiento de datos, ya sea en sus servidores o localmente en sus propias máquinas. los datos y flujos de trabajo pueden ser reproducidos y compartidos. los tutoriales en línea, sección de ayuda, y una página-wiki ( https://wiki.galaxyproject.org/Support ) dedicada al Proyecto Galaxy proporcionan un apoyo constante. Sin embargo, para los usuarios de primera vez, especialmente aquellos que no tienen la formación bioinformática, la tubería puede parecer desalentador y el proceso de auto-aprendizaje y familiarización puede llevar mucho tiempo. Además, el sistema biológico estudiado, y los detalles del experimento y los métodos utilizados, el impacto las decisiones analíticas en varios pasos, y estos pueden ser difíciles de navegar sin instrucción.
El RN general A-Seq Galaxy de flujo de trabajo consiste en la carga de datos y verificación de la calidad seguido por análisis utilizando el Tuxedo Suite 4, 5, 6, 7, 8, 9, que es un colectivo de varias herramientas requeridas para las diferentes etapas de análisis de datos RNA-Seq 10, 11, 12, 13, 14. Un experimento típico RNA-Seq consiste en la parte experimental (preparación de la muestra, el aislamiento de ARNm y ADNc de preparación de la biblioteca), la NGS y el análisis de la bioinformática datos. Una visión general de estas secciones, y los pasos involucrados en la tubería Galaxy, se muestran en la Figura 1.
3fig1.jpg"/>
Figura 1: Visión general de un RNA-Seq Workflow. Ilustración de los pasos experimentales y computacionales que participan en un experimento RNA-Seq para comparar los perfiles de expresión génica de dos cepas de gusanos (A y B, líneas de color naranja y verde y flechas, respectivamente). Los diferentes módulos de Galaxy utilizado se muestran en recuadros con el paso correspondiente en el protocolo indicado en rojo. Las salidas de diversas operaciones están escritos en gris con los formatos de archivo que se muestran en azul. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
La primera herramienta en el Tuxedo Suite es un programa de alineación llamada 'Tophat'. Se descompone la entrada NGS lee en fragmentos más pequeños y luego los asigna a un genoma de referencia. Este proceso de dos pasos asegura que lee abarca regiones intrónicas cuya alineación puede ser de otro modo disrupted o perdidas se contabilizan y se asigna. Esto aumenta la cobertura y facilita la identificación de nuevas uniones de empalme. Salida Tophat se informa como dos archivos, un archivo de BED (con información sobre las uniones de corte y empalme que incluyen localización genómica) y un archivo de BAM (con detalles de mapeo de cada lectura). A continuación, el archivo de BAM se alinea contra un genoma de referencia para estimar la abundancia de las transcripciones individuales dentro de cada muestra con la función posterior en la Suite Tuxedo llamada 'Gemelos'. Gemelos funciones mediante el escaneo de la alineación reportar fragmentos de transcripción de longitud completa o 'transfrags' que abarcan todas las posibles variantes de empalme en los datos de entrada para cada gen. Basado en esto, se genera un 'transcriptoma' (montaje de todas las transcripciones generadas por gen para cada gen) para cada muestra que se secuenciaron. Estos conjuntos de mancuernas se colapsaron luego o se fusionaron junto con la referencia genoma para producir un único archivo de anotación para el análisis diferencial de aguas abajo usando la siguiente herramienta, 'Cuffmerge'. Por último, la expresión génica herramienta medidas diferencial la 'Cuffdiff' entre las muestras mediante la comparación de las salidas el sombrero de copa de cada una de las muestras para el archivo de salida Cuffmerge final (Figura 1). Gemelos utiliza FPKM / RPKM (Fragmentos / Lee por kilobase de transcripción por millón asignada lecturas) los valores reportar transcripción abundancias. Estos valores reflejan la normalización de los datos NGS primas para la profundidad (número promedio de lecturas de una muestra que se alinean con el genoma de referencia) y la longitud de genes (genes tener longitudes diferentes, por lo recuentos tienen que ser normalizado para la longitud de un gen para comparar los niveles entre los genes). FPKM y RPKM son esencialmente los mismos con RPKM ser utilizados para un solo extremo RNA-Seq donde cada lectura corresponde a un solo fragmento, mientras que, FPKM se utiliza para-Extremo emparejado RNA-Seq, ya que representa el hecho de que dos lecturas pueden corresponder al mismo fragmento. En última instancia, el resultado de estos análisis es una lista de genes expresados diferencialmente entre las condiciones y / o las cepas ensayadas.
Una vez que una carrera exitosa Galaxy se ha completado y se genera una 'lista de genes', el siguiente paso lógico requiere más análisis de la bioinformática para deducir conocimiento significativo de los conjuntos de datos. Muchos paquetes de software han surgido para atender a esta necesidad, incluyendo paquetes computacionales basados en web disponibles públicamente como David (la base de datos para la anotación, y Visualización Integrada Discovery) 15. DAVID facilita la asignación de significado biológico a grandes listas de genes de alto rendimiento estudios comparando la lista de genes subido a su base de conocimiento biológico integrado y revelar las anotaciones biológicas asociadas con la lista de genes. Esto es seguido por análisis de enriquecimiento, es decir, las pruebas a identify si cualquier clase de proceso o gen biológico se excesivamente en la lista (s) de genes de una manera estadísticamente significativa. Se ha convertido en una opción popular debido a una combinación de una amplia, base de conocimiento integrado y algoritmos de análisis de gran alcance que permiten a los investigadores a detectar temas biológicos enriquecidos dentro de la genómica-deriva '' listas de genes 10, 16. Las ventajas adicionales incluyen su capacidad para procesar las listas de genes creado en cualquier plataforma de secuenciación y una interfaz muy fácil de usar.
El nematodo Caenorhabditis elegans es un sistema modelo genético, bien conocida por sus muchas ventajas, tales como tamaño pequeño, cuerpo transparente, plan de cuerpo simple, facilidad de la cultura y gran susceptibilidad a la disección genética y molecular. Worms tienen un pequeño, simple y bien anotado genoma que incluye hasta un 40% de genes conservados con homólogos humanos conocidos 17. De hecho, C. elegansfue la primera metazoan cuyo genoma fue secuenciado por completo 18, y una de las primeras especies que se utilizó RNA-Seq para mapear transcriptoma de un organismo 19, 20. Estudios gusano temprano involucrados experimentación con diferentes métodos para alto rendimiento de captura de RNA, la preparación de la biblioteca y la secuenciación, así como tuberías de bioinformática que contribuyeron al avance de la tecnología de 21, 22. En los últimos años, la experimentación basada en ARN-Seq en los gusanos se ha convertido en un lugar común. Pero, para los biólogos del gusano tradicionales los retos que plantea el análisis computacional de los datos de RNA-Seq siguen siendo un obstáculo para una mayor y mejor utilización de la técnica.
En este artículo, se describe un protocolo para el uso de la plataforma Galaxy para analizar los datos de RNA-Seq alto rendimiento generados a partir de C. elegans. Para muchos por primera vez y de pequeña scaLe usuarios, la forma más rentable y sencillo para llevar a cabo un experimento de RNA-Seq es aislar ARN en el laboratorio y utilizar una instalación comercial NGS (o en casa) para la preparación de bibliotecas de ADNc de secuenciación y el propio NGS. Por lo tanto, hemos detallado primero las etapas implicadas en el aislamiento, la cuantificación y evaluación de la calidad de C. elegans muestras de ARN para la ARN-Seq. A continuación, se proporcionan instrucciones paso a paso para el uso de la interfaz de Galaxy para el análisis de los datos de NGS, comenzando con las pruebas de los controles de calidad post-secuenciación seguido de alineación, el montaje, y la cuantificación diferencial de la expresión génica. Además, hemos incluido direcciones para escudriñar los listas de genes resultantes de Galaxy para estudios de enriquecimiento biológicos utilizando DAVID. Como paso final en el flujo de trabajo, se proporcionan instrucciones para cargar los datos de RNA-Seq a los servidores públicos, tales como la secuencia de lectura del archivo (SRA) en el NCBI ( http: // www.ncbi.nlm.nih.gov/sra) para que sea libremente accesible para la comunidad científica. En general, esperamos que este artículo se proporcionará información completa y suficiente para los biólogos del gusano que llevan a cabo experimentos de RNA-Seq, por primera vez, así como usuarios frecuentes que ejecutan un pequeño número de muestras.
Protocolo
1. Aislamiento de ARN
- Medidas de precaución
- Limpie toda la superficie, instrumentos de trabajo y pipetas utilizando un spray RNasa disponible comercialmente para eliminar cualquier RNasas presente.
- Use guantes en todo momento, que cambia regularmente por otras nuevas durante las diferentes etapas del protocolo.
- Utilice sólo las puntas de filtro y mantener todas las muestras en hielo tanto como sea posible para evitar la degradación del ARN.
NOTA: Para obtener los mejores datos desde plataformas NGS, es fundamental comenzar con ARN de alta calidad. el aislamiento de ARN y preparación de los métodos varían dependiendo del origen de la muestra, el método de secuenciación y investigador preferencia. Varios kits disponibles en el mercado pueden ser utilizados para este propósito o ARN también se pueden aislar utilizando un método de fenol-cloroformo estándar de extracción de RNA. Con cualquiera de los métodos, las medidas de precaución antes mencionados se deben seguir durante todo el proceso para minimizar la contaminación y OBTLas muestras de ARN prístina Ain.
- cosecha de Worms
- Sincronizar la población de gusanos por tratamiento de blanqueo de hipoclorito 23 para obtener 1.000-1.500 emparejados por edad C. elegans gusanos adultos por cepa.
- Lavar los gusanos de placas utilizando solución tampón M9 y centrifugado a 325 xg en una centrífuga de mesa durante 30 s. Aspirar a cabo el buffer M9 dejando tras de sí un pellet de gusanos. Repita este paso al menos tres veces para eliminar el arrastre de bacterias.
- Para el sedimento gusano, añadir ~ 500! L de tampón de lisis (si se utiliza un kit comercial) o Trizol (una solución de mono-fásica de fenol e isotiocianato de guanidina; si fenol: se lleva a cabo extracción con cloroformo se describe en 1.3.3) para interrumpir tejidos gusano , desactivar RNasas y estabilizar los ácidos nucleicos.
NOTA: El protocolo se puede pausar aquí por el flash congelación de las muestras en nitrógeno líquido seguido de almacenamiento a -80 ° C.
- Aislamiento de ARN
- muestras de gusano sonicado en amplitud 45% en ciclos de 20 s. 'ON' y 40 s. 'OFF' (8-12 ciclos por cepa). Mantener las muestras en hielo en todo momento.
NOTA: asegúrese de que la sonda de ultrasonidos se sumerge en el tampón y se mantiene a un nivel constante a lo largo. Evitar la formación de espuma de la muestra y limpiar la sonda a fondo en-entre las muestras. ciclos de sonicación pueden variar en función del tipo de aparato de ultrasonidos usado. Se recomienda que las condiciones de sonicación se optimizan primero en una muestra de prueba antes de comenzar un experimento. - Si se utiliza un kit disponible en el mercado, proceder con Aislamiento de ARN según el protocolo prescrito. Para el aislamiento de ARN usando un método de fenol-cloroformo, realice los siguientes pasos.
- Centrifugar sonicó muestras a 16.000 xg durante 10 min. a 4 ° C.
- Transferir el sobrenadante a un tubo de microcentrífuga de 1,5 ml RNasa libre y añadir 100 l de cloroformo (1/5 del volumen de reactivo de aislamiento de ARN / ADN).
Precaución: Cloroformo es tóxico. Para minimizar la exposición y evitar la inhalación, el trabajo en una campana química cuando el manejo de esta sustancia. - Vórtice las muestras a fondo durante 30 - 60 s. y dejar que las muestras se sientan a temperatura ambiente durante 3 min.
- Centrifugar a 11.750 xg durante 15 min. a 4 ° C. Transferir sólo la capa acuosa superior a un nuevo tubo de microcentrífuga libre de RNasa teniendo cuidado de no aspirar el interfaz blanco que contiene ADN. Repita los pasos 1.3.4 a través de 1.3.6.
- Añadir 250! L (70% de fase acuosa o media de ARN / ADN aislamiento volumen de reactivo) de 2-propanol y se invierte el tubo para mezclar. Deje tubos se sientan a temperatura ambiente durante 10 min o dejan durante la noche a -80 ° C.
- Centrifugar las muestras a 11.750 xg durante 10 min. a 4 ° C. Se decanta el sobrenadante con mucho cuidado, dejando tras de sí unos cuantos l en la parte inferior del tubo de manera que la pastilla no se ve perturbado.
- Lavar pellet con 500 l de etanol al 75% (fabricado con agua libre de ARNasa) y centrifugar a 16.000 xg durante 5 min. unt 4 ° C.
- Eliminar la mayor cantidad sobrenadante como sea posible sin perturbar el sedimento. Deje secar al aire el sedimento en una campana durante unos minutos.
- Añadir 30 l de agua libre de RNasa y ayudar a disolver el sedimento de ARN mediante calentamiento durante 10 min. a 60 ° C.
- Comprobar la calidad y la cantidad de ARN usando un Bioanalyzer.
NOTA: Bioanalyzer genera un umber ntegridad N R NA I (RIN) como una medida de la calidad del ARN. Un RIN de al menos 8 es el umbral recomendado para muestras de ARN-Seq (mayor es mejor). cantidad y calidad del ARN también se pueden verificar por espectrofotometría sino que también debe ser seguido por la evaluación visual de la integridad del ARN. Para ello, ejecute las muestras en un gel de agarosa al 1,2% el tiempo suficiente para obtener la separación adecuada de los 28s y 18s bandas de ARN ribosomal. La presencia de dos bandas distintas (1,75 kb para ARNr 18S y 3,5 kb para ARNr 28S en el caso de C. elegans) es una medida aceptable de la calidad del ARN. - Usar ~ 100 ng /! L de ARN a ship para la instalación / NGS proveedor para la preparación de bibliotecas de secuenciación.
NOTA: Las muestras de ARN se deben enviar en hielo seco al proveedor de servicios de secuenciación. La mayoría de los proveedores de llevar a cabo una prueba de control de calidad independiente antes de la preparación de ARN biblioteca.
- muestras de gusano sonicado en amplitud 45% en ciclos de 20 s. 'ON' y 40 s. 'OFF' (8-12 ciclos por cepa). Mantener las muestras en hielo en todo momento.
2. ARN-Sec Análisis de Datos
- Descarga de datos sin procesar de secuenciación
- Descargar los datos de secuenciación FASTQ prima comprimido codificados en el formato fastq.gz del proveedor de NGS utilizando un "protocolo de transferencia de archivos" (FTP).

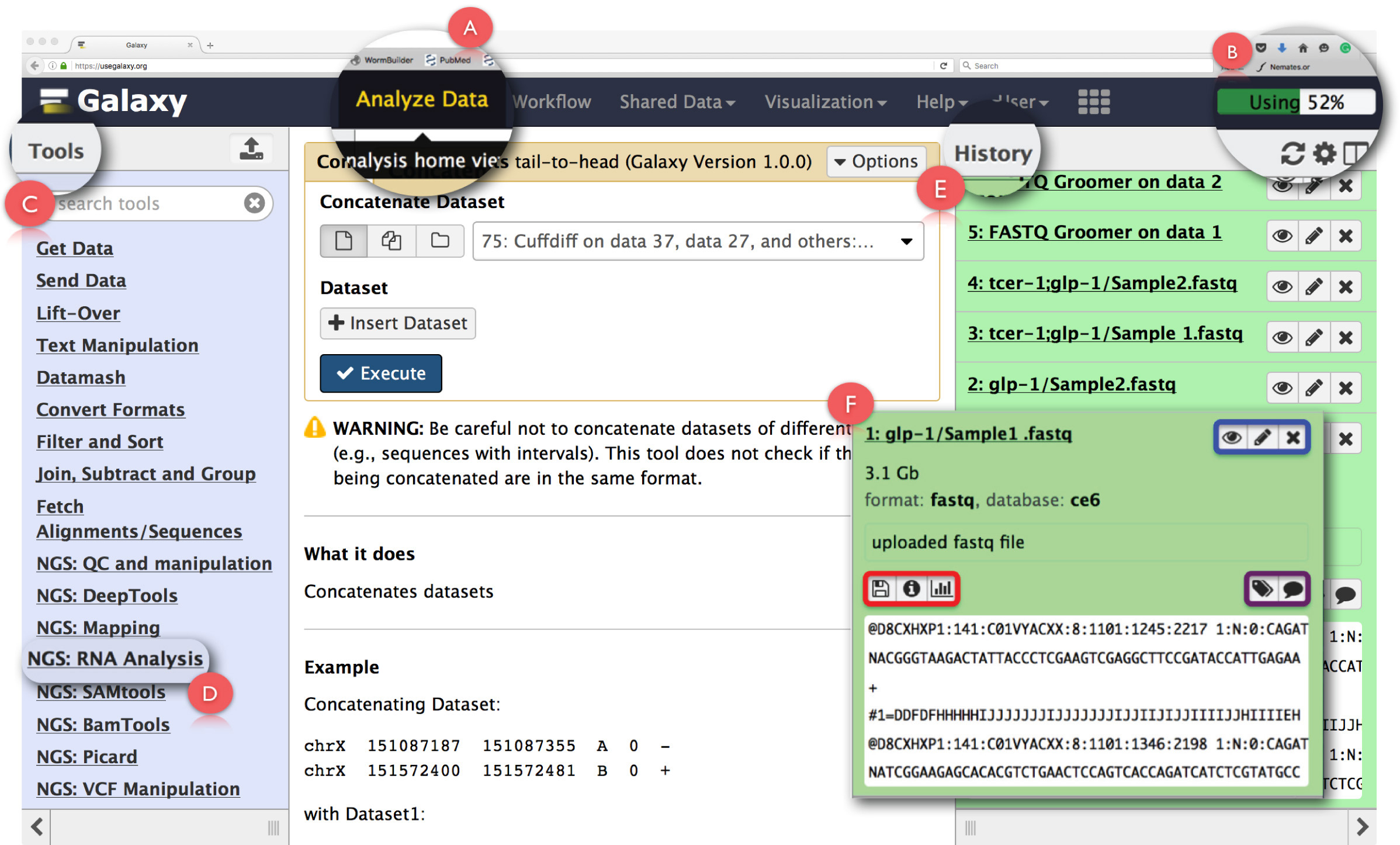
Figura 2: Estructura de la Galaxy panel de interfaz y funciones clave de ARN-Seq usuario. Las principales características de la página se expanden y se destacaron. (A) pone de relieve la función 'Analizar datos' en la cabecera de la página web utilizada para el acceso Análisis de Inicio Ver. (B) es la 'barra de progreso' que indica el espacio en el servidor Galaxy utilizado por la operación. (C) es el 'Herramientas Sección' que enumera todas las herramientas que se pueden ejecutar en la interfaz Galaxy. (D) muestra los 'NGS: Análisis de ARN' sección herramienta utilizada para el análisis de ARN-Seq. (E) representa el panel 'Historia' que muestra todos los archivos generados utilizando Galaxy. (F) muestra un ejemplo del cuadro de diálogo que se abre al hacer clic en cualquier archivo en la sección de Historia. Dentro de (F), la caja azul destaca iconos que se pueden utilizar para ver, editthe atributos o eliminar el conjunto de datos, el cuadro morado destaca iconos que se pueden utilizar para 'editar' las etiquetas conjunto de datos o anotación, y, el cuadro rojo indica iconos descargar los datos, ver detalles de la tarea realizada o volver a ejecutar la operación. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Introducción a Galaxy
NOTA: Galaxy se puede ejecutar en un servidor público gratis con una plataforma basada en la web que proporciona acceso a la nube y el almacenamiento gratuito limitado. También se puede descargar y ejecutar localmente en la máquina del usuario o grupos de computación organizadas por instituciones, sino procesamiento local, puede ser restringido por los límites de almacenamiento de datos y las limitaciones de potencia de procesamiento de máquinas de los usuarios. Detalles sobre la descarga e instalación se pueden alcanzar en https://wiki.galaxyproject.org/Admin/GetGalaxy . En este protocolo se describe el uso basado en la web de la tubería Galaxy.- Después de descargar y almacenar los datos de NGS en la máquina del usuario, el acceso al Galaxylaxy.org/" target = "_blank"> https://usegalaxy.org/.
- Registrar una cuenta de usuario haciendo clic en 'Usuario' en la cabecera de la página, inicio de sesión y comenzar a familiarizarse con el panel de interfaz de usuario.
NOTA: Se recomienda que los usuarios de primera vez utilizan el tutorial 'Haga clic aquí' proporcionado en la página de inicio para familiarizarte con el conjunto básico de Galaxy ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) . - Haga clic en 'Analizar datos' (Figura 2A) en el panel de cabecera para acceder al 'Análisis de Inicio Ver', que es también la pantalla de inicio sobre el Galaxy.
NOTA: La cabecera también contiene otros enlaces cuyos detalles se puede ver, con solo pasar el puntero del ratón sobre ellos. La esquina superior derecha de la cabecera tiene una barra de progreso que supervisa espacio utilizado para las tareas (Figura 2B). - dolamer en 'NGS: Análisis de ARN' tarea en el 'Herramientas Menú' en el panel izquierdo (Figura 2C) para acceder a todas las herramientas necesarias para el análisis de datos de RNA-seq.
NOTA: El 'Menú Herramientas' cataloga todas las operaciones que Galaxy ofrece. Este menú se divide basado en tareas y haciendo clic en cualquiera abrirá una lista de todas las herramientas necesarias para llevar a cabo esa tarea. - Crear nueva historia de análisis haciendo clic en el icono de engranaje en la parte superior del panel 'Historial' de la derecha (Figura 2E). Elija 'Crear nuevo' opción en el menú emergente. Dar a esta 'Historia' un nombre adecuado para identificar el análisis.
NOTA: El panel de 'Historial' muestra todos los archivos cargados para su análisis, así como todos los archivos de salida que se generan mediante la ejecución de tareas sobre el Galaxy. Al hacer clic en un nombre de archivo en este panel se abre un cuadro de diálogo con información detallada sobre la tarea realizaday un fragmento del conjunto de datos (Figura 2F). Iconos de este cuadro permiten al usuario 'vista', 'editar los atributos' o 'eliminar' el conjunto de datos (Figura 2F, resaltada en azul). Además, el usuario puede también 'editar' etiquetas conjunto de datos o anotación (Figura 2F, resaltado en púrpura), 'de descarga de los datos, 'ver detalles' de la tarea, 'repetición' la tarea o incluso 'visualizar' el conjunto de datos de esta cuadro de diálogo (Figura 2F, resaltada en rojo). - Haga clic en la función 'Subir archivo' en 'Obtener datos' en el 'menú Herramientas' para subir archivos FASTQ primas.
NOTA: Al hacer clic en esta o cualquier otra herramienta abre una breve descripción de la operación, y la propia prueba, en el panel central 'Análisis Interfaz'. Este panel ata juntos el'Herramientas' en el panel izquierdo y el 'Archivos de entrada' desde el panel de la derecha 'Historia' (Figura 2E). En este caso, los archivos de entrada de la 'historia' se seleccionan y otros parámetros definidos para ejecutar una tarea determinada. El conjunto de datos de salida resultante de cada prueba se guarda de nuevo en 'Historia'. Se incluye con la prueba en el panel "Análisis de interfaz" son explicaciones para todos los parámetros disponibles para el funcionamiento de una herramienta dada junto con una lista detallada de todos los archivos de salida de la herramienta genera. - Después se abre la tarea en el 'Análisis de interfaz', haga clic en 'Seleccionar archivo local' o 'Seleccionar archivo de FTP' (carga rápida), vaya a la carpeta que contiene los archivos de secuenciación y seleccione el conjunto de datos apropiada para ser cargado.
- Permitir Galaxy en 'Auto-detectar' la (ajuste predeterminado) subido el tipo de archivo. Seleccione 'C. ELEgans 'en el menú desplegable para el genoma.
- Haga clic en 'Inicio' para iniciar la carga de datos. Una vez subido el archivo, se guarda en el panel de 'Historial' y se puede acceder desde allí.
- Si los archivos de datos de secuenciación múltiples se producen para una sola muestra, combinarlos con la función 'Concatenate'. Para ello, abre la opción 'Gestión de texto' en el 'Menú de Herramientas'.
- Haga clic en la herramienta 'Concatenate', seleccione los archivos que necesitan ser combinado en el cuadro desplegable en el medio de 'interfaz de Análisis' y haga clic en 'Ejecutar'.
NOTA: Los archivos de salida producidos usando esta tarea se generan en el formato FASTQ. El programa de mapeo tiene un límite de 16.000.000 de secuencias por archivo FASTQ y cuando se alcanza este límite se genera un nuevo archivo de FASTQ para las secuencias restantes. el '; Se necesita herramienta Concatenate' en tales casos para combinar los conjuntos de datos. - Convertir los archivos de formato FASTQ cargados en el formato requerido para fastqsanger Galaxy análisis de RNA-Seq mediante el uso de la 'FASTQ peluquero' herramienta 'encontrado en los NGS: control de calidad y la manipulación de la sección (véase el archivo suplementario).
- Elija el conjunto de datos FASTQ apropiado en el 'Archivo para el novio' opción y ejecutar la herramienta utilizando parámetros por defecto.
NOTA: Los archivos de salida producidos usando esta tarea se generan en el formato fastqsanger.
- Pruebas de Calidad de Datos fastqsanger-Control
- Comprobar la calidad de la fastqsanger subido lee con la función 'FastQC' situado bajo 'NGS: control de calidad y la manipulación' en el menú 'Herramientas'.
- Elija el archivo de datos fastqsanger preparado en el menú desplegable de 'Short leer los datos de la biblioteca actual' y ejecutar la herramienta utilizando parámetros por defecto.
NOTA: Prestar especial atención a la calidad de las lecturas y la presencia de cualquiera de las secuencias adaptadoras. Los adaptadores se retiran generalmente como parte del procesamiento de datos RNA-Seq mensaje por proveedores de NGS pero en algunos casos, se pueden dejar atrás. Para la explicación de los estándares de calidad ir a http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - Consulte con el proveedor de NGS y si adaptadores están presentes, recortarlas con la función 'clip' de la 'NGS: control de calidad y la manipulación' menú de tareas.
NOTA: Los archivos de salida producidos usando esta tarea se generan en el formato txt prima, así como en html que se puede abrir en cualquier navegador web.
- Análisis de datos con Tuxedo suite
- Sombrero de copa
- Descargar la última versión de C. elegans fasta genoma de referencia y GTF (Transferencia de genes Format) archivos de archivo de carga', como se describió anteriormente en 2.2.6.
- Abrir los 'NGS: Análisis de ARN' sección y haga clic en la herramienta 'TopHat' para mapear la secuenciación lee en el genoma de referencia descargado.
- Seleccione la respuesta adecuada en el menú desplegable a la pregunta '¿Es este único extremo o de datos de extremo emparejado?'
- Elija el archivo FASTQ apropiado.
- Seleccione 'Use un genoma de la historia' en el siguiente menú desplegable y elegir genoma de referencia descargado en el paso 2.4.1.1.
- Seleccione 'Default' para el resto de parámetros y haga clic en 'Ejecutar'.
NOTA: Entre los archivos de salida producidas usando esta tarea, el archivo '' Golpea aceptadas se utiliza para las etapas siguientes.
- Gemelos y Cuffmerge
- Seleccione la 'Cuffherramienta en los enlaces NGS: Sección de Análisis de ARN 'para ensamblar las transcripciones, estimar su abundancia y analizar la expresión diferencial.
- En el primer menú desplegable, seleccione los 'éxitos aceptadas (formato BAM)' mapeado de archivos obtenidos del análisis TopHat.
- En el segundo menú desplegable, establecer anotación de referencia al archivo gtf descargado en el paso 2.4.1.1.
- Seleccione 'Sí' en la opción 'Realizar la corrección de sesgo' y ejecutar la tarea con la configuración predeterminada para todos los demás parámetros.
NOTA: Entre los archivos de salida producidas usando esta tarea, el archivo '' Aceptados Las transcripciones se utiliza para las etapas siguientes. - Herramienta abierta 'Cuffmerge' en los NGS: Análisis de ARN '' para fusionar las transcripciones reúnen en haces de producción propia en todas las muestras de RNA-Seq.
NOTA: La primera caja en la herramienta de auto-puebla y las listas de todo el archivos gtf producidos por gemelos. - Seleccione el archivo 'Montado Transcripciones' para todas las cepas / condiciones ensayadas, incluyendo repeticiones biológica de la misma cepa / condición (véase la discusión para réplicas biológicas).
- Seleccione 'Sí' para 'Uso de referencia de anotación' y selecciona el archivo descargado en el paso gtf 2.4.1.1.
- En el cuadro siguiente, de nuevo seleccione 'Sí' para la opción 'Usar Sequence Data' y elija el archivo completo del genoma fasta descargado en el paso 2.4.1.1.
- Manteniendo el resto de parámetros por defecto, haga clic en 'Ejecutar'.
NOTA: Cuffmerge genera un archivo de salida gtf sola.
- Cuffdiff
- Vaya a la herramienta 'Cuffdiff' en los NGS: Análisis de ARN 'sección. En el menú 'Transcripciones', seleccione el archivo de salida resultante de la fusión de Cuffmerge.
- Etiquetalas condiciones 1 y 2 con los nombres dos cepas / condición.
NOTA: Cuffdiff puede realizar comparaciones entre más de dos cepas o condiciones, así como experimentos de tiempo. Sólo tiene que utilizar la opción 'agregar nuevas condiciones' para añadir cada nuevo cepas / condición, según sea necesario. - Para cada cepa / condición, bajo 'replicados' Select individuo archivos de salida 'Resultados aceptadas' de TopHat que corresponden a las diferentes réplicas biológicas de que la cepa / condición. Mantenga pulsada la tecla 'cmd', si se utiliza un ordenador Macintosh, y la tecla 'Ctrl', si se utiliza un PC, para seleccionar varios archivos.
- Deje todas las demás opciones como parámetros por defecto. Haga clic en 'Ejecutar' para ejecutar la tarea.
NOTA: Cuffdiff genera numerosos archivos de salida en un formato de tabla como la lectura final del análisis de ARN-Seq. Estos incluyen los archivos con el seguimiento de las transcripciones, FPKM genes (combinadosvalores FPKM de transcripciones que comparten una identidad de genes), transcritos primarios y secuencias de codificación. Todos los archivos de datos generados se pueden ver en cualquier aplicación de hoja de cálculo y contienen atributos similares, tales como nombre del gen, locus, el cambio (en escala de log2), así como datos estadísticos en comparaciones entre las cepas / condiciones, incluyendo el valor p y q valores doblar. Los datos de estos archivos se pueden clasificar sobre la base de la significación estadística de las diferencias o doble cambio en la expresión génica (magnitud y dirección del cambio, como en UP o hacia abajo genes regulados) y manipulados según los requisitos de los usuarios. Si se necesita la conversión entre diferentes identificadores de genes (por ejemplo, Wormbase ID gen vs. número cósmido), herramientas disponibles en Biomart ( http://www.biomart.org/ ) puede ser utilizada.
- Sombrero de copa
3. ontología de genes (GO) Análisis plazo utilizando DAVID
- DAVID acceso desde el sitio web hTTP: //david.ncifcrf.gov/. Haga clic en 'Iniciar análisis' en la cabecera de la página web. En 'Paso 1', copiar y pegar la lista de genes obtenidos de Galaxy en la caja A. En 'Paso 2', seleccione 'Wormbase Gene ID' como identificador para los genes de entrada.
NOTA: DAVID reconoce categorías de anotación más disponibles públicamente, por lo que otros identificadores de genes (como Entrez ID gen o gen símbolo) también se puede utilizar. - En 'Paso 3', seleccione 'lista de genes' (genes a analizar) bajo 'Tipo de lista' y luego haga clic en 'Enviar Lista' icono.
NOTA: 'Análisis Asistente', se abrirá a una lista de todas las herramientas DAVID hipervínculos que se pueden ejecutar en la lista de genes cargado (Figura 3). Haga clic en estos enlaces para acceder a los módulos correspondientes pertinentes de acuerdo a los requerimientos del usuario. Para identificar las herramientas adecuadas para una tarea determinada, haga clic en "¿Qué herramientas utilizar DAVID? 'Enlace de la' ; Análisis página Asistente'. Haga clic en el enlace 'Iniciar análisis' en la cabecera para volver a la página principal del 'Análisis Asistente' en cualquier momento durante el análisis.

Figura 3: Disposición de la DAVID Análisis Asistente página web y ejemplos de salidas de operación. Web interfaz de usuario del 'Análisis Asistente' se enumeran las herramientas utilizadas para analizar la lista de genes de subida para el enriquecimiento en base a varios parámetros. Al hacer clic sobre estas herramientas de informes de los datos analizados en una nueva página web. Ejemplos de los informes tabulares generados a partir de 'Gene Clasificación Funcional', 'Tabla de anotación funcional' y 'Clustering anotación funcional' se muestran como inserciones (flechas).> Haga clic aquí para ver una versión más grande de esta figura.
- Herramienta de anotación funcional 1: La agrupación funcional de anotación
- Haga clic en el módulo 'Funcional anotación La agrupación' para ir a la página de resumen. Mantenga las categorías de anotación por defecto y haga clic en 'Funcional anotación La agrupación' para generar grupos de anotación términos similares clasificados según su puntuación de enriquecimiento.
- Haga clic en el nombre de cada término con hipervínculo para leer los detalles al respecto y 'RT' (términos relacionados) para listar otros términos similares relacionados con la categoría.
- Haga clic en la barra de color púrpura a la lista de los genes asociados con un término y el rojo 'G' para listar todos los genes asociados con todos los términos dentro de un grupo.
- Haga clic en el icono verde para ver una vista bidimensional de todos los genes y los términos de un clúster.
NOTA: Las tres últimas columnas muestran los resultados analíticos y estadísticos para cadatérmino. Los resultados de este y todos los demás análisis se pueden descargar en un formato .txt haciendo clic en el enlace 'Descargar Archivo'.
- Herramienta de anotación funcional 2: Tabla de anotación funcional
- Volver a la página de resumen y haga clic en 'Funcional anotación Gráfico' para identificar términos biológicos significativamente sobrerrepresentados (por ejemplo, la actividad del factor de transcripción o de actividad quinasa) asociados a la lista de genes.
- Haga clic en el nombre plazo para obtener información más detallada y 'RT' (términos relacionados) para listar otros términos relacionados.
- Haga clic en la barra de color púrpura para listar todos los genes asociados del correspondiente categoría individual.
NOTA: Las dos últimas columnas lista de resultados de las pruebas de estadística para cada categoría.
- Herramienta de anotación funcional 3: Tabla funcional de anotación
- Volver a la página de resumen y haga clic en 'Functional Tabla de anotación "para ver una lista de todas las anotaciones asociadas a los genes en una lista sin ningún tipo de cálculos estadísticos.
Nota: Esta herramienta puede ser útil para el análisis del gen por gen de una lista o para observar los genes específicos, altamente interesantes.
- Volver a la página de resumen y haga clic en 'Functional Tabla de anotación "para ver una lista de todas las anotaciones asociadas a los genes en una lista sin ningún tipo de cálculos estadísticos.
- Gen Herramienta Clasificación Funcional
- Volver a 'Análisis Asistente' y haga clic en el módulo 'Gene Clasificación Funcional' para segregar la lista de genes de entrada en grupos relacionados funcionalmente de genes clasificados según su 'enriquecimiento Resultado', una medida del enriquecimiento global del grupo de genes en la lista.
- Haga clic en el nombre plazo para obtener información más detallada y 'RG' para revelar los genes relacionados funcionalmente del grupo de genes
- Haga clic en el rojo 'T' (informes plazo) a la lista de biología asociado y el icono verde para ver una vista bidimensional de todos los genes y los términos.
- -Gen nombreVisor de lotes
- Volver a 'Análisis Wizard' y haga clic en 'Gene-Nombre de lote Visor' traducir 'Wormbase genes identificadores' en sus correspondientes nombres de genes. (WBGene00022855 = tCER-1).
- Haga clic en el nombre de genes para obtener más información específica de genes.
- Haga clic en el 'RG' (genes relacionados) que aparece junto a cada gen para revelar los genes prevé que sea funcionalmente relacionado con el gen de interés.
4. Carga de datos RAW en el NCBI secuencia de lectura del archivo (SRA)
- Acceder a la página web de la SRA en Iniciar sesión en enlace NCBI' o registrar una nueva cuenta.
- Haga clic en 'Bioproject'.
- Haga clic en la 'presentación' bajo el 'Uso Bioproject' título de la izquierda.
- Seleccionar la opción 'Nueva Presentación'. Detalles de la actualización del remitente. Continúe a través de las siete fichas restantes, Rellenando los detalles del experimento y los datos siendo cargado. Haga clic en 'Enviar' cuando esté terminado.
NOTA: En la quinta pestaña 'muestra biológica', deje la ranura para 'muestra biológica' vacío. - Actualizar la página resultante haciendo clic en el enlace 'Mis envíos'. Los datos presentados se mostrarán con un número de presentación asignado, breve descripción y estado de carga.
- Haga clic en 'muestra biológica' en la parte superior de esta página, en el cuadro 'iniciar una nueva presentación' y crear una 'nueva presentación'. Presentar presentaciones separadas para cada muestra.
- Al igual que en el caso de 'Bioproject' en 4.4, actualizar los detalles del remitente y continuar con el resto de las fichas de llenado en los detalles de cada ficha. Una vez completada la revisión y haga clic en 'Enviar'.
- Navegue a http: //www.ncbi.nlm.nih.gov / SRA para crear la 'secuencia de lectura del archivo (SRA)' presentación final.
- Haga clic en 'Inicio de sesión a SRA' bajo 'Introducción'.
- En la siguiente página, haga clic en el enlace 'NCBI PDA'. Un vínculo 'Actualizar Preferencias' abrirá. Complete el formulario y haga clic en 'Guardar preferencias'.
- En la página resultante, haga clic en el enlace 'Crear un nuevo trabajo'. Introduzca un nombre adecuado bajo 'Alias' y haga clic en 'Guardar'. Se creará una tabla con la identificación de envío y otros detalles.
- Haga clic en 'New Experiment' y registrar al menos una biblioteca secuenciación único para cada 'muestra biológica'.
- Designar y vincular el creado con anterioridad 'Bioproject' y de identificación de la petición 'muestra biológica'. Se creará un 'nuevo experimento'.
- Haga clic en 'New Run' en la parte inferior de la páginaDespués del experimento SRA se ha hecho e identificar los archivos de datos que necesitan ser vinculados a la misma.
- Calcular la suma MD5 de cada archivo de datos. Para hacer esto en un terminal de Macintosh, vaya a Aplicaciones / Utilidades / Terminal. En terminal, tipo en 'md5' (sin las comillas) seguido por un espacio. Arrastrar y soltar los archivos que necesitan ser cargado en el terminal del buscador y haga clic en 'Enter'.
- Terminal devolverá una suma MD5 alfanumérico. Introducir esto como parte del proceso de envío de la carga de archivos. Utilice el nombre de usuario y la contraseña proporcionada por el sistema para subir archivos a través de FTP.
Resultados
En C. elegans, la eliminación de las células madre de línea germinal (GSCS) se extiende la vida útil, mejora la resistencia al estrés, y eleva la grasa corporal 24, 28. Pérdida de GSCs, ya sea provocado por ablación por láser o por mutaciones, tales como GLP-1, provoca prolongación de la vida a través de la activación de una red de factores de transcripción 29. Uno de tales fa...
Discusión
Importancia de la plataforma de secuenciación Galaxy biología de hoy
El Proyecto Galaxy se ha convertido en fundamental para ayudar a los biólogos y sin la formación de bioinformática para procesar y analizar los datos de secuenciación de alto rendimiento de una manera rápida y eficiente. Una vez considerada una tarea hercúlea, esta plataforma accesible al público que ha hecho correr algoritmos bioinformáticos complejos para analizar los datos NGS un proceso sencillo, fiable y fácil....
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores desean expresar su agradecimiento a los laboratorios, grupos e individuos que han desarrollado Galaxy y David, y por lo tanto hechas NGS ampliamente accesible para la comunidad científica. La ayuda y el asesoramiento brindado por sus colegas de la Universidad de Pittsburgh durante nuestra formación bioinformática es reconocido. Este trabajo fue apoyado por una Fundación Médica Ellison Nueva Académico en el envejecimiento premio (AG-NS-0879-12) y una subvención de los Institutos Nacionales de Salud (R01AG051659) a AG.
Materiales
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
Referencias
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22 (2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085 (2011).
- Consortium, C. e. S. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383 (2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639 (2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788 (2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158 (2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados