
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
のトランスクリプトーム解析
要約
ギャラクシーとDAVIDバイオインフォマティクストレーニングなしの研究者が分析し、RNA-配列データを解釈できるようにする一般的なツールとして浮上しています。私たちは、 線虫の研究者は、アクセスをRNA-配列実験を行い、銀河を使用してデータセットを処理し、DAVIDを用いた遺伝子リストから意味のある生体情報を取得するためのプロトコルを記述します。
要約
次世代シーケンシング(NGS)技術は、生物学的な調査の性質に革命をもたらしてきました。これらのうち、RNAシークエンシング(RNA-配列)は、遺伝子発現分析およびトランスクリプトームマッピングのための強力なツールとして浮上しています。しかし、RNA-配列データセットを処理することは、高度な計算の専門知識を必要とし、生物学の研究者のための固有の課題を提起します。このボトルネックは、バイオインフォマティクスのスキルを持たないユーザは、RNA-配列データを分析することを可能にするオープンアクセスギャラクシープロジェクト、および注釈用のデータベース、可視化、および統合されたディスカバリー(DAVID)、助け遺伝子オントロジー(GO)用語分析スイートによって軽減されました大規模なデータセットからの生物学的意味を導き出します。しかし、初めてのユーザーとバイオインフォマティクスアマチュア、これらのプラットフォームでの自己学習や習熟のための時間がかかり、困難なことができます。私たちは、 線虫の研究者は、ワームのRNAを単離するのに役立ちます簡単なワークフローを記述し、RNA-配列実験を行いますそして、ギャラクシーとDAVIDのプラットフォームを使用してデータを分析します。このプロトコルは、濃縮のためにスクリーニングすることができる遺伝子のリストを生成するために、各ステップでのパラメータをユーザに案内する、生NGSデータ、品質管理チェック、位置合わせ、及び示差遺伝子発現分析にアクセスするための様々なギャラクシーモジュールを使用するための段階的な指示を提供しますDAVIDを用いた遺伝子クラスまたは生物学的プロセス。全体的に、我々はこの記事が初めてRNA-配列の実験と同様に少数のサンプルを実行している頻繁にユーザーを行っC.エレガンスの研究者に情報を提供することを期待しています。
概要
ヒトゲノムの最初のシーケンシング、フレッド・サンガーのジデオキシヌクレオチド配列決定法を用いて行っ、10年かかった、と推定米国$ 3 10億、2を要しました 。しかし、創業以来十年余りで、次世代シーケンシング(NGS)技術は、それが可能2週間以内とUS $ 1,000ヒトゲノム全体を配列決定することになりました。ゲノム配列決定プロジェクトが急速に当たり前になりつつあり、コストの大幅な削減に伴い、信じられないほど効率的にシーケンシング・データ収集の増え続ける速度を許可する新しいNGS機器は、想像を絶する方法で、現代生物学に革命を起こしています。また、これらの開発は、このようなRNA配列決定(RNA-SEQ)、ゲノムワイドなエピジェネティック修飾の研究、DNA-タンパク質相互作用を介して遺伝子発現解析などの多くの他の分野における進歩を亜鉛めっきしており、ヒト宿主における微生物多様性をスクリーニングします。 RNA-SeのNGSベース特に、qはそれが可能精度と感度との包括的トランスクリプトームを識別し、マッピングするために作られており、および発現プロファイリングのための選択の方法として、マイクロアレイ技術を交換しました。マイクロアレイ技術が広く用いられているが、そのようなクロスハイブリダイゼーションを確実に測定することができる発現変化の制限された範囲として、既存の既知のゲノム情報とアレイ、および他の欠点への依存によって制限されます。 RNA-配列は、他方で、、その明確なDNAマッピング性質に低いバックグラウンドノイズを生成しながら、既知および未知の両方の転写物を検出するために使用され得ます。一緒に、このような酵母、ハエ、虫、魚やマウスなどのモデル生物が提供する数多くの遺伝的ツールとRNA-配列は、多くの重要な最近の生物医学の発見のための基盤を務めています。しかし、ストレージの制限、処理、およびすべてのほとんどは、メートルなど、より広い科学コミュニティにアクセスできなくNGSを作る重要な課題のまま、配列決定データの大量のeaningfulバイオインフォマティクス解析。
シーケンシング技術と指数データ蓄積の急速な進歩は、研究者は、アクセス分析し、この情報を理解することができます計算プラットフォームのための大きい必要性を作成しました。初期のシステムは、コンピュータプログラミングの知識に大きく依存していたのに対し、非プログラマがアクセスし、洗練された分析を許可しなかったデータを視覚化することができ、このようなNCBIなどのゲノムブラウザ。 Webベースのオープンアクセスプラットフォーム、ギャラクシー( https://galaxyproject.org/ )は、この間隙を充填しNGSデータを処理して、簡単に複雑なのスペクトルを実行するために、研究者を可能に貴重なパイプラインであることが証明されましたバイオインフォマティクス分析します。ギャラクシーはアントンNekrutenko(ペンシルベニア州立大学)、ジェームズ・テイラーの研究室で、最初に設立され、維持されている(ジョンズホプキンス大学)F "> 3。ギャラクシーは。それRNA-配列研究に関わるすべてのステップを含む無数のバイオインフォマティクスのニーズ、のための『ワンストップショップ』を作る計算タスクの広い範囲を提供していますそのサーバー上のどちらかのデータ処理を実行するためにユーザーをItallowsローカルに自分のマシン上で。データとワークフローを再現し、共有することができます。オンラインチュートリアル、ヘルプセクション、およびウィキページ ( https://wiki.galaxyproject.org/Supportギャラクシープロジェクト専用)は、一貫したサポートを提供しています。しかし、初めてのユーザーのために、特になしバイオインフォマティクストレーニングを持つものは、パイプラインが困難な表示されることと自己学習や習熟の過程は時間がかかることがあります。また、生物学的システムを検討し、実験や方法の詳細は、インパクトを使用しますいくつかのステップでは、分析の決定、およびこれらの命令なしにナビゲートすることは困難。
全体的にRN -配列ギャラクシーワークフローは、データのアップロード及びRNA-配列データ解析10の異なる段階のために必要な様々なツールの集合であり、Tuxedoのスイート4、5、6、7、8、9を用いて分析を行っ品質チェックから成り11、12、13、14。典型的なRNA-配列実験は、実験の部(試料調製、mRNA単離およびcDNAライブラリーの調製)、NGSおよびバイオインフォマティクスデータ解析から成ります。これらのセクション、ギャラクシー・パイプラインに含まれるステップの概要を、 図1に示されています。
3fig1.jpg」/>
図1:RNA-配列のワークフローの概要。 2つのウォーム株(AとB、オレンジと緑の線、矢印、それぞれ)の遺伝子発現プロファイルを比較するために、RNA配列の実験に関わる実験及び計算ステップの実例。ギャラクシー利用の様々なモジュールは、赤色で示さ我々のプロトコルに対応するステップとボックスで示されています。各種操作の出力は、青色で示されたファイルフォーマットに灰色で書かれています。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
タキシードSuiteの最初のツールは、「 トップハット 」と呼ばれるアライメントプログラムです。これは、NGS入力は小さな断片に読み取り、参照ゲノムにマップし破壊します。この2段階プロセスはそれがそのアライメントさもなければジすることができるイントロン領域にまたがる読み出し保証しますsruptedまたはを占め、マッピングされている逃しました。これは、カバレッジを増加させ、新規なスプライス部位の同定を容易にします。 トップハット出力は2つのファイル(ゲノム位置を含むスプライス部位に関する情報を)BEDファイルと(各読み取りのマッピングの詳細を)BAMファイルとして報告されます。次に、BAMファイルは「 カフス」と呼ばタキシードスイートで、その後のツールを使用して、各サンプル内の個々の転写物の豊かさを推定するために、参照ゲノムに対して整列されます。 カフス機能の完全長転写物の断片または全ての遺伝子のための入力データで可能なすべてのスプライスバリアントにまたがる「transfrags」を報告してアライメントをスキャンすることもできます。これに基づいて、それは、配列決定される各サンプルの(すべての遺伝子について遺伝子当たり生成されたすべての転写物のアセンブリ)「トランスクリプトーム」を生成します。これらのカフスアセンブリは、その後崩壊し、再と一緒にマージされていますフェレンスゲノムは次のツール、「Cuffmerge」を使用して、下流の差分分析のための単一の注釈ファイルを生成します。最終Cuffmerge出力ファイルに各試料のトップハット出力を比較することにより、 サンプル間の最後に、「Cuffdiff」ツール測定示差遺伝子発現( 図1)。 カフリンクスは FPKM / RPKM使用する転写物の存在量を報告した値を(フラグメントを/マッピングされた百万分の転写物のパーキロベースは読み取り読み込み)。遺伝子の長さ(カウントがレベルを比較するために、遺伝子の長さについて正規化されなければならないので、遺伝子は、異なる長さを有する(参照ゲノムに整列サンプルからの読み取りの平均数)これらの値は、深さの生NGSデータの正規化を反映して遺伝子間)。 FPKMとRPKMはFPKMをするために使用され、一方、すべての読み取りが、単一のフラグメントに対応するシングルエンドのRNA配列のために使用されてRPKMと本質的に同じですそれは、2つの同一の断片に対応することができる読み出すという事実を占めるように、ペアエンドRNA-配列。最終的に、これらの分析の結果は、差動的条件および/または試験された株の間に発現された遺伝子のリストです。
成功したギャラクシーの実行が完了すると「遺伝子リスト」が生成されると、次の論理的なステップは、データセットから意味のある知識を推定するために分析するより多くのバイオインフォマティクスが必要です。多くのソフトウェアパッケージは、DAVID(注釈、可視化と統合発見のためのデータベース)15として一般的に利用可能なウェブベースの計算パッケージを含む、このニーズに応えるために登場しました。 DAVIDは、その統合された生物学的な知識ベースにアップロードされた遺伝子のリストを比較し、遺伝子リストに関連する生物学的注釈を明らかにすることで、高スループットの研究から大きな遺伝子リストに生物学的な意味を割り当てることが容易。これは、IDEにすなわち 、テストは、濃縮分析が続いています任意の生物学的プロセスまたは遺伝子クラスが統計的に有意な様式で遺伝子リスト(S)に過剰出現した場合ntify。それは広いため、統合された知識ベースと「遺伝子リスト10」を 、16由来のゲノムの中に豊かな生物学的なテーマを検出するために、研究者を可能にする強力な分析アルゴリズムの組み合わせの一般的な選択肢となっています。さらなる利点は、任意のシーケンシングプラットフォームと非常にユーザーフレンドリーなインターフェースで作成された遺伝子リストを処理する能力が含まれます。
線虫線虫(Caenorhabditis elegans)もこのような小さなサイズ、透明体、シンプルなボディープラン、遺伝的および分子解剖の文化や偉大な従順の容易さなどの多くの利点のために知られている遺伝モデルシステムです。ワームは、既知のヒト相同体17で40%保存された遺伝子まで含む、小さな単純でよく注釈付きのゲノムを有します。実際、C.エレガンス最初のゲノム完全18を配列決定した後生動物、およびRNA-配列は、生物のトランスクリプトーム19、20をマッピングするために使用された第一の種の一つでした。初期のワームの研究では、技術21、22の発展に貢献したハイスループットRNAキャプチャ、ライブラリ準備およびシーケンシングだけでなく、バイオインフォマティクスパイプラインのためのさまざまな方法で実験を関与します。近年では、ワームでRNA-配列ベースの実験が当たり前になってきました。しかし、伝統的なワームの生物学者のためのRNA-配列データのコンピュータ分析によってもたらされる課題は、技術のより大きな、より良い利用のための主な障害のまま。
本稿では、 線虫から生成されたハイスループットRNA-配列データを分析するギャラクシー・プラットフォームを使用するためのプロトコルを記載しています。多くの初めて小SCAのためのルユーザー、RNA-配列実験を実施するための最もコスト効率と簡単な方法は、実験室でRNAを分離し、シーケンシングcDNAライブラリーとNGS自体の製造のために、市販の(または社内)NGS施設を利用することです。したがって、まず、RNA配列のためのC.エレガンス RNA試料の単離、定量および品質評価に必要な手順を詳述しています。次に、位置合わせ、アセンブリ、および遺伝子発現の示差定量続くポストシークエンス品質管理チェックのテストから始まる、NGSデータの分析のためにギャラクシー・インターフェースを使用するための手順を提供します。加えて、我々はDAVIDを用いて、生物学的濃縮の研究のための銀河から生じた遺伝子リストを精査する方向が含まれています。ワークフローの最後のステップとして、私たちは、このようなNCBIのシーケンス読むアーカイブ(SRA)(などの公開サーバへの上にRNA-配列データをアップロードするための手順を説明します。http://ワットww.ncbi.nlm.nih.gov/sra)科学界にそれが自由にアクセスできるようにします。全体的に、我々はこの記事では、ワーム初めてRNA-配列実験を行っ生物学者だけでなく、少数のサンプルを実行している頻繁にユーザーに包括的かつ十分な情報を提供することを期待しています。
プロトコル
1. RNA単離
- 予防策
- 存在する任意のRNアーゼを除去するために市販のRNaseスプレーを使用して全体の作業表面、器具及びピペットを拭います。
- 定期的にプロトコルの異なる段階中に新鮮なものでそれらを変更し、すべての回で手袋を着用してください。
- 唯一のフィルターチップを使用し、RNAの分解を避けるために、可能な限り氷の上にすべてのサンプルを保ちます。
注:NGSプラットフォームから最適なデータを得るためには、高品質のRNAを開始することが重要です。 RNA単離および調製方法は、サンプルの起源に依存して配列決定し、研究者の好みの方法を変えます。いくつかの市販のキットは、この目的のために使用することができ、またはRNAはまた、RNA抽出の標準的なフェノール - クロロホルム法を用いて単離することができます。いずれかの方法では、上記の予防措置は、汚染とOBTを最小限にするためにプロセス全体に従うべきですAIN原始RNAサンプル。
- 収穫ワーム
- 、株あたり1,000〜1,500、年齢をマッチさせたC.エレガンスの成虫を得るために、次亜塩素酸塩漂白処理23によりワームの人口を同期します。
- 30秒間テーブルトップ遠心機で325×gでM9緩衝液とスピンを使用してプレートワームを洗い流します。ワームのペレットを残しM9バッファーを吸引除去します。細菌のキャリーオーバーを排除するために、少なくとも三回、この手順を繰り返します。
- ウォームペレットを、溶解緩衝液〜500μL(市販のキットを使用している場合)またはトリゾール追加(フェノールおよびグアニジンイソチオシアネートの単相性溶液を、フェノール場合:1.3.3に記載クロロホルム抽出が行われる)ワーム組織を破壊します、RNアーゼを不活性化し、核酸を安定化させます。
注:プロトコルは、-80℃で貯蔵続いて液体窒素中でサンプルを凍結フラッシュすることによりここに一時停止することができます。
- RNAの単離
- 20秒のサイクルで45%の振幅で超音波処理ワームサンプル。 'ON' と40秒。 'OFF'(ひずみあたり8-12サイクル)。すべての回で、氷上でサンプルを保管してください。
注:超音波処理器プローブがバッファに浸漬されており、全体で一定のレベルに保たれていることを確認してください。サンプルの泡立ちを避け、徹底的に-間のサンプルプローブを清掃してください。超音波処理サイクルを用いる超音波処理の種類に応じて変えることができます。超音波処理条件は最初の実験を開始する前に、試験試料に最適化されていることをお勧めします。 - 市販のキットを使用する場合、所定のプロトコルに従ってRNA単離を進めます。フェノール - クロロホルム法を使用して、RNA単離のために、以下の手順を実行します。
- 遠心分離を10分間16,000×gで試料を超音波処理しました。 4℃で
- 1.5 mLのRNaseフリーのマイクロチューブに上清を移し、(RNA / DNA単離試薬の体積番目 1/5)のクロロホルム100μLを加えます。
あぶない:クロロホルムは有毒です。この物質を取り扱う際の暴露を最小限に抑え、吸入を避けるために、化学フードで働いています。 - 60秒 - 30徹底的にサンプルをボルテックス。サンプルは、3分間室温で放置します。
- 15分間11,750×gで遠心します。 4°Cで。 DNA含有白いインタフェースを吸引しないように注意しながら、新しいRNaseフリーのマイクロチューブにのみ上部の水層を転送します。繰り返しは、1.3.6を通じて1.3.4を繰り返します。
- 250μLの2-プロパノール(水性相又は1/2 RNA / DNA単離試薬体積の70%)を添加し、混合するためのチューブを反転させます。チューブは10分間室温で座るか、-80°Cで一晩のままにしてみましょう。
- 10分間11,750×gで遠心分離サンプル。 4°Cで。ペレットが邪魔されないように、チューブの底に数μLを残して、非常に慎重に上清を除去。
- (RNaseフリー水を使用して作製)75%エタノール500μLでペレットを洗浄し、5分間、16,000×gでスピンダウン。 AT 4°C。
- ペレットを乱すことなく、できるだけ多くの上清を取り除きます。空気は数分間フード内でペレットを乾燥させます。
- RNaseフリー水30μLを加え、10分間加熱することにより、RNAペレットを溶解するのに役立ちます。 60°Cで。
- バイオアナライザーを使用してRNAの質と量を確認してください。
注:バイオアナライザはRNAの質の尺度としてR NA I ntegrity N個のアンバー(RIN)を生成します。少なくとも8のRINは、RNA配列の試料の推奨閾値(高い方が良い)です。 RNAの量と質はまた、分光光度法で確認することができますだけでなく、RNAの完全性の視覚的評価が続くべきです。これを行うには、28Sおよび18SリボソームRNAバンドの適切な分離を得るために十分な長さを1.2%アガロースゲル上でサンプルを実行します。二つの別個のバンド(18S rRNAのための1.75キロバイトおよびC.エレガンスの場合に28S rRNAの3.5 KB)の存在は、RNAの品質の許容される尺度です。 - 市へ〜100 ng /μLでRNAを使用しますシーケンシングライブラリーの調製のためのベンダー/ NGS施設へのp。
注:RNAサンプルは、シークエンシングサービスプロバイダにドライアイスで出荷する必要があります。ほとんどのプロバイダは、ライブラリの準備の前に独立したRNAの品質管理テストを行います。
- 20秒のサイクルで45%の振幅で超音波処理ワームサンプル。 'ON' と40秒。 'OFF'(ひずみあたり8-12サイクル)。すべての回で、氷上でサンプルを保管してください。
2. RNA-配列データ分析
- 生シーケンシングデータのダウンロード
- 「 ファイル転送プロトコル」(FTP)を使用してNGSプロバイダからfastq.gz形式でエンコードされた圧縮された生FASTQ配列決定データをダウンロードします。

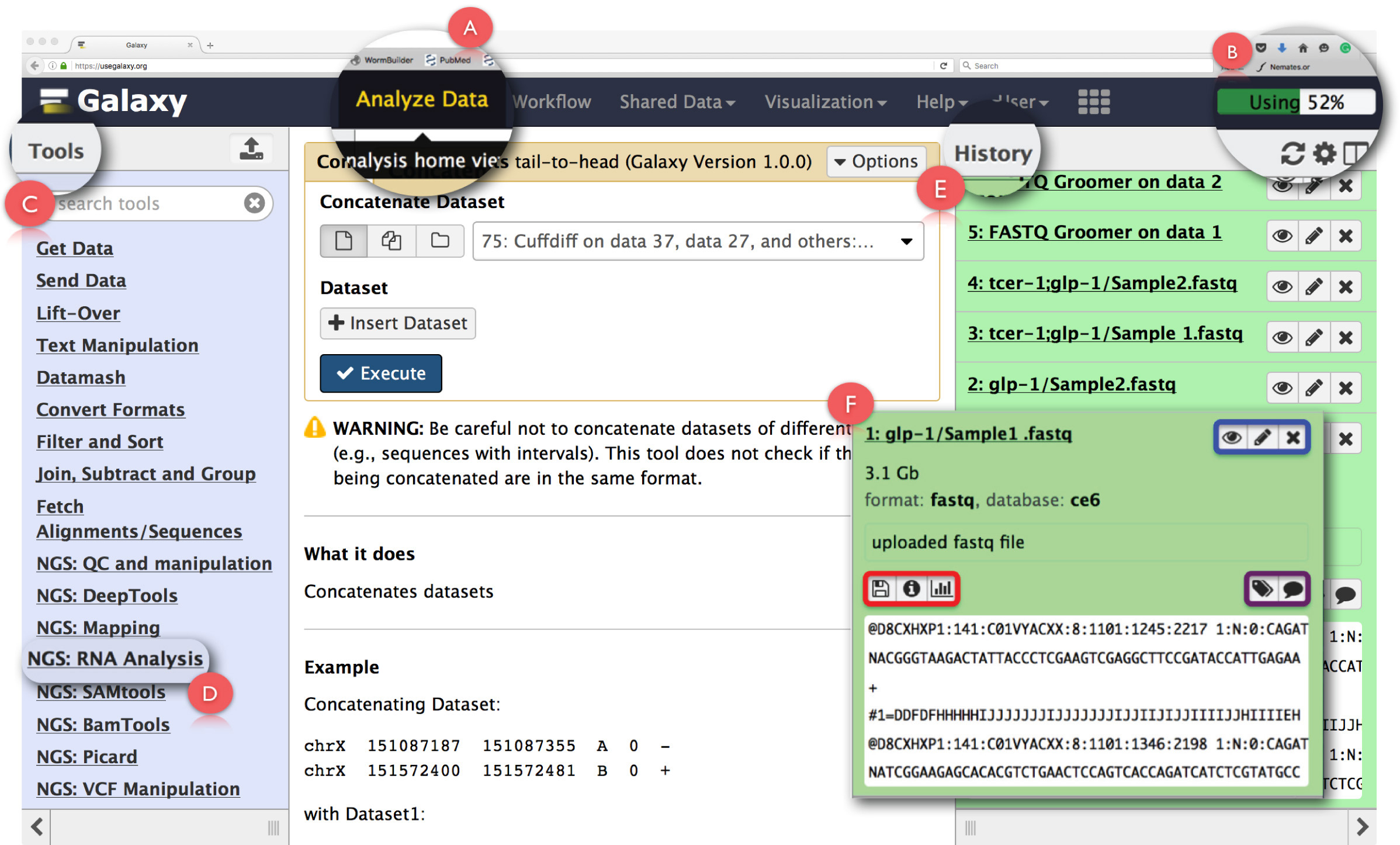
図2:銀河のユーザーインターフェイスパネルとキーRNA-配列機能のレイアウト。ページの主な機能は拡張され、強調表示されます。 (A)は、アクセスに使用されるウェブページのヘッダ内のデータを分析 」機能を強調します分析ホームビュー。 (B)は、操作によって利用ギャラクシーサーバー上のスペースを示す「 プログレスバー 」です。 (C)は、銀河インターフェイス上で実行することができ、すべてのツールが一覧表示されます「 ツールセクション 」です。 RNA-配列分析のために使用されるツールのセクション:(D)は、「RNA 分析NGS」を示しています。 (E)は、銀河を使用して生成されたすべてのファイルを一覧表示します「 歴史」パネルを示しています。 (F)は、 履歴]セクションで任意のファイルをクリックしたときに開くダイアログボックスの一例を示しています。 (F)の中で、青いボックスを表示するために使用することができ、アイコン、editthe属性を強調したり、紫色の箱は、データセットタグや注釈「編集」に使用することができ、アイコンが強調表示され、データセットを削除 、赤いボックスは、アイコンを示しデータをダウンロードするには、タスクの詳細の表示を行っまたは操作を再実行してください 。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
- ギャラクシー入門
注:ギャラクシーは、クラウドへのアクセスおよび無料の限られたストレージを提供するウェブベースのプラットフォームを使用して無料公開サーバ上で実行することができます。またダウンロードすると、ユーザーのマシンや機関が、ローカル処理によってホストされている計算クラスタ上でローカルに実行することができ、データ・ストレージの制限やユーザーのマシンの処理能力の限界によって制約される可能性があります。ダウンロードとインストールの詳細については、にアクセスすることができhttps://wiki.galaxyproject.org/Admin/GetGalaxy 。このプロトコルでは、我々はギャラクシーパイプラインのWebベースの使用方法について説明します。- ユーザーのマシン上のNGSデータをダウンロードして保存した後、アクセスギャラクシーでlaxy.org/」ターゲット= "_blank"> https://usegalaxy.org/。
- ページ、ログインのヘッダーに「ユーザー」をクリックしてユーザーアカウントを登録し、ユーザーインターフェースパネルに慣れることから始めます。
注:これは、初めてのユーザーがホームページで提供「ここからスタート」チュートリアルはギャラクシーの基本的なセットアップに慣れ取得する利用することをお勧めします( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) 。 - また、銀河の起動画面で「分析ホームビュー」にアクセスするために、ヘッダーパネルで「データの分析」( 図2A)をクリックします。
注:ヘッダにもその詳細は、それらの上にマウスポインタを置くと見ることができる他のリンクを収容します。ヘッダの右上隅は、タスク( 図2B)のために利用されるスペースを監視するプログレスバーを有しています。 - CRNA-seqのデータ解析に必要なすべてのツールにアクセスするには、左側のパネル( 図2C)の「ツールメニュー」でタスク:「RNA分析NGS」になめます。
注:「ツールメニューのカタログギャラクシーが提供するすべての操作。このメニューには、いずれかのタスクとクリックに基づいて分割され、そのタスクを達成するために必要なすべてのツールのリストを開きます。 - 右( 図2E)の「 歴史 」パネルの上部にある歯車のアイコンをクリックすることで、新たな分析の履歴を作成します。選択してポップアップメニューからオプション「 新規作成 」を。この「 歴史に 」分析を識別するための適切な名前を付けます。
注:「歴史」パネルには、銀河でタスクを実行することによって生成されたすべての出力ファイルだけでなく、分析のためにアップロードされたすべてのファイルを表示します。このパネルでファイル名をクリックすると、実行されるタスクに関する詳細な情報をダイアログボックスを開きますデータセット( 図2F)のスニペット。このボックス内のアイコンは、データセット( 図2Fは 、青色で強調表示された)「 属性を編集 」または「 削除 」、「 ビュー 」をユーザに可能にします。さらに、ユーザはこのからのデータセットをさえも、「 編集 」データセットタグや注釈( 図2Fは 、紫色で強調表示)、タスクの「 ダウンロード 」データ、「 詳細の表示 」、「 再実行 」タスクまたは「を可視化する 」ことができますダイアログボックス( 図2Fは 、赤色で強調しました)。 - 生FASTQファイルをアップロードするには「ToolsMenuから 」内のデータを取得]の下の「 ファイルのアップロード」機能をクリックしてください。
注:このをクリックするか、他のツールは、中央の「分析インターフェース」パネルで、操作の簡単な説明、およびテスト自体を開きます。このパネルは一緒にひも左のパネルと右の「 歴史 」パネルから「入力ファイル」( 図2E)から「ツール」。ここでは、「 歴史 」からの入力ファイルを選択し、他のパラメータは、与えられたタスクを実行するように定義されています。すべてのテストの結果の出力データセットは、「 歴史 」に書き戻さ保存されます。ツールが生成するすべての出力ファイルの詳細なリストと一緒に与えられたツールを実行するために利用可能なすべてのパラメータの説明は「 分析インターフェイス 」パネルでテストして含まれます。 - タスクは「分析インターフェース」で開いた後、または、(より速いアップロード)「FTPファイルを選択して」シーケンスファイルを含むフォルダに移動し、アップロードする適切なデータセットを選択して「 ローカルファイルを選択 」をクリックします。
- アップロードされたファイルの種類(デフォルト設定)「 自動検出 」にギャラクシーを許可します。 「C.エルを選択ゲノムのためのプルダウンメニューでegans "。
- データのアップロードを開始するために、「 スタート 」をクリックします。ファイルがアップロードされると、それは「 歴史 」パネルに保存され、そこからアクセスすることができます。
- 複数のシーケンスデータファイルが単一のサンプルのために製造されている場合は、「 連結し 」ツールを使用してそれらを結合。これを行うには、「ツールメニュー」の「 テキスト操作」オプションを開きます。
- 「 分析・インターフェース」の途中でドロップダウンボックスから結合して「 実行」をクリックする必要のあるファイルを選択し、「連結し」ツールをクリックします。
注:このタスクを使用して作成された出力ファイルは、FASTQ形式で生成されます。マッピングプログラムは、FASTQファイルごと1600万シーケンスの限界があり、その制限に達したときに新しいFASTQファイルは残りの配列のために生成されます。 ";を連結」ツールは、データセットを結合するような場合に必要とされています。 - 「:QCと操作NGS」セクション(補足ファイルを参照してください)アップロードFASTQ形式のファイルは、下にある「FASTQトリマー」ツールを使って、銀河RNA-配列分析に必要なfastqsanger形式に変換します。
- オプション「 新郎へのファイル」の下で、適切なFASTQデータセットを選択し、デフォルトパラメータを使用してツールを実行します。
注:このタスクを使用して作成された出力ファイルはfastqsanger形式で生成されます。
- データ品質管理テストfastqsanger
- 下にある「FastQC」ツール使って読み込むアップロードfastqsangerの品質を確認してください:「ツール」メニューの「NGSをQCと操作 」。
- ショア 」のドロップダウンメニューから手入れfastqsangerデータファイルを選択します。t「は現在のライブラリからデータを読み込み、デフォルトパラメータを使用してツールを実行します。
注:読み込み、任意のアダプター配列の存在の品質に特別な注意を払ってください。アダプタは、通常NGSプロバイダによってポストRNA-配列データの処理の一部として除去されるが、いくつかの例では、残されてもよいです。品質基準の説明についてはへ行くhttp://www.bioinformatics.babraham.ac.uk/projects/fastqc/ 。 - NGSプロバイダに確認してくださいとアダプタが存在する場合、「NGS:QCと操作」から「 クリップ」ツールを使用してトリムタスクメニュー。
注:このタスクを使用して作成された出力ファイルは、生のTXT形式でだけでなく、任意のWebブラウザで開くことができるHTMLで生成されます。
- タキシードSuiteとのデータ分析
- トップハット
- の最新バージョンをダウンロード C。エレガンス参照ゲノムのFASTAおよび2.2.6で説明したようにGTF(遺伝子導入形式) のアップロードファイルからファイル」。
- 「:RNA分析NGS」セクションをし、シークエンシングは、ダウンロードした参照ゲノムに読み込み、マップするために「 トップハット」ツールをクリックして開きます。
- 質問へのドロップダウンメニューから適切な答えを選択して「このシングルエンドまたはペアエンドデータです?」
- 適切なFASTQファイルを選択します。
- [次へ]を選択し、ドロップダウンメニューで「履歴からゲノムを使用してください」とステップ2.4.1.1でダウンロードした参照ゲノムを選択してください。
- 他のパラメータについては「デフォルト」を選択し、「 実行」をクリックします。
注:このタスクを使用して作成された出力ファイルの中で、「 受理ヒット 」ファイルは、後続のステップのために使用されています。
- カフスとCuffmerge
- 「カフを選択NGSリンクでツール『:転写物を組み立てるためのRNA分析』セクションでは、異なる発現のための彼らの豊かさとテストを見積もります。
- 最初のドロップダウンメニューでは、 トップハット分析から得られたマッピングされた「 受理ヒット(BAM形式)」ファイルを選択してください。
- 2番目のドロップダウンメニューでは、ステップ2.4.1.1でダウンロードGTFファイルへの参照アノテーションを設定します。
- 「 バイアス補正を実行」オプションのために「はい」を選択し、他のすべてのパラメータのデフォルト設定を使用してタスクを実行します。
注:このタスクを使用して作成された出力ファイルの中で、「 受け入れられた転写物」ファイルは、後続のステップのために使用されています。 - 開く「Cuffmerge」ツール」NGS:RNA分析「 組み立て転写物 」生成のためのすべてのRNA配列のサンプルをマージします。
注:ツール自己を移入し、リストの最初のボックスのすべて カフリンクスによって生成GTFファイル。 - (生物学的複製のための議論を参照してください)同じ系統/条件の生物学的複製を含む、試験した全ての株/条件、のための「組み立て転写物」ファイルを選択します。
- 「 使用リファレンス注釈」の「はい」を選択し、ステップ2.4.1.1でダウンロードしたGTFファイルを選択します。
- 以下のボックスに、再び「 使用シーケンスデータ 」オプションのために「はい」を選択し、ステップ2.4.1.1でダウンロードした全ゲノムFASTAファイルを選択します。
- 「実行」をクリックし、デフォルトとして他のパラメータを維持します。
注:Cuffmergeは、単一のGTFの出力ファイルを生成します。
- Cuffdiff
- セクション:「RNA分析NGS」の「Cuffdiff」ツールに移動します。 「 トランスクリプト」メニューでは、Cuffmergeからマージされた出力ファイルを選択します。
- ラベル二つの株/条件名と条件1と2。
注:Cuffdiffつ以上の株または条件間の比較を行うだけでなく、時間経過実験することができます。単純に、必要に応じて、それぞれの新しい株/条件を追加するために「 追加新条件」オプションを使用します。 - それぞれの株/条件については、下には、その歪み/条件の異なる生物学的複製に対応するトップハットから個人の受理ヒット"出力ファイルを選択して「 複製します」。 Macintoshコンピュータを使用している場合、「CMD」キーを押しながら、「CTRL」キー、複数のファイルを選択するには、PCを使用している場合。
- デフォルトパラメータとして他のすべてのオプションのままにしておきます。タスクを実行するために、「実行」をクリックします。
注:Cuffdiffは RNA-配列解析の最終読み出しとして、表形式で、多くの出力ファイルを生成します。これらを組み合わせた転写物、遺伝子に関するFPKMの追跡を持つファイル(含めますFPKM遺伝子アイデンティティを共有する転写物の値)、主要転写物およびコード配列。生成されたすべてのデータファイルは、任意のスプレッドシートアプリケーションで閲覧し、そのような遺伝子名、遺伝子座、(LOG2規模の)倍数変化ならびにp値とq値を含む株/条件、の間の比較の統計データと同様の属性を含むことができます。これらのファイル内のデータは、統計的有意差に基づいてソートまたは遺伝子発現(上方または調節遺伝子を下方制御のように変化の大きさと方向)の変化を折ると、ユーザーの要件に応じて操作することができます。異なる遺伝子の識別子との間の変換が必要な場合( 例えば 、Wormbaseコスミド数に対する遺伝子ID)は、Biomart(上の利用可能なツールhttp://www.biomart.org/を )利用することができます。
- トップハット
3.遺伝子オントロジー(GO)用語分析DAVIDを使用して
- ウェブサイトの時間からのアクセスDAVIDttps://david.ncifcrf.gov/。 Webページのヘッダーに「分析の開始」をクリックします。 「ステップ1」で、「ステップ2」においてボックスAにギャラクシーから得られた遺伝子のリストをコピーして貼り付け、入力された遺伝子の識別子として「Wormbase遺伝子ID」を選択します。
注:DAVIDが最も公的に利用可能な注釈の種類を認識するので、(例えばのEntrez遺伝子ID又は遺伝子記号など)他の遺伝子識別子を使用することもできます。 - 「ステップ3」では、(遺伝子を分析するために)「リスト型」の下の「遺伝子リスト」を選択し、「リストの送信」をアイコンをクリックしてください。
注:「 分析ウィザード」、アップロードされた遺伝子リスト( 図3)上で実行することができ、すべてのハイパーリンクDAVIDツールを一覧表示する開きます。ユーザーごとの要件として、関連する対応するモジュールにアクセスするためにこれらのリンクをクリックしてください。 どのDAVIDツールを使用する 」をクリックし、与えられたタスクのための適切なツールを特定するには? 「オンリンク」 ; 分析ウィザード」のページ。分析中の任意の時点で「 分析ウィザードのホーム・ページに戻るには、ヘッダー内の「分析の開始」リンクをクリックしてください。

図3:DAVID 分析ウィザードウェブページと動作出力の例のレイアウト。 「 分析ウィザード 」Webユーザー・インターフェースは、さまざまなパラメータに基づいて、濃縮のためにアップロード遺伝子リストを分析するために使用するツールを示します。これらのツールをクリックすると、新しいWebページで分析されたデータを報告します。 「 遺伝子機能分類 」、「 機能アノテーションチャート 」と「 機能的注釈クラスタリング」から生成された表形式レポートの例は、インセット(矢印)として示されています。>この図の拡大版をご覧になるにはこちらをクリックしてください。
- 機能的な注釈ツール1:機能注釈クラスタリング
- 概要ページに移動するには「 機能注釈クラスタリング 」モジュールをクリックします。デフォルトの注釈のカテゴリを維持し、その濃縮スコアによってランク付けと同様のアノテーション用語のクラスタを生成するには「 機能注釈クラスタリング 」をクリックします。
- カテゴリに関連する他の同様の用語をリストアップし、それについての詳細と「RT」(関連用語)を読み取るために、各用語のハイパーリンク名をクリックします。
- クラスタ内のすべての用語に関連するすべての遺伝子を一覧表示する用語に関連する遺伝子と赤い「G」を一覧表示するには、紫色のバーをクリックしてください。
- クラスタ内のすべての遺伝子との用語の2次元ビューを表示するには、緑色のアイコンをクリックしてください。
注:最後の3つの列は、それぞれの分析と統計結果を一覧表示します期間。この他のすべての分析の結果は、「ダウンロードファイル」リンクをクリックすることにより、.txt形式でダウンロードすることができます。
- 機能的な注釈ツール2:機能アノテーションチャート
- 概要ページに戻り、遺伝子リストに関連付けられて大幅に過剰表示生物学用語( 例えば転写因子活性またはキナーゼ活性)を識別するために、「 機能的な注釈チャート」をクリックします。
- その他の関連する用語をリストするために、より詳細な情報と「RT」(関連用語)を取得するために用語の名前をクリックしてください。
- 個々のカテゴリを、対応するすべての関連する遺伝子を一覧表示するには、紫色のバーをクリックしてください。
注:最後の2列には、各カテゴリの統計的検定の結果を一覧表示します。
- 機能注釈ツール3:機能アノテーション表
- 概要ページに戻り、Functio」をクリック任意の統計計算せずに、リスト上の遺伝子に関連付けられているすべての注釈の一覧を参照するには最終注釈テーブル'。
注:このツールは、リストの遺伝子ごとの遺伝子分析のために役立つことができたり、特定の、非常に興味深い遺伝子を見て。
- 概要ページに戻り、Functio」をクリック任意の統計計算せずに、リスト上の遺伝子に関連付けられているすべての注釈の一覧を参照するには最終注釈テーブル'。
- 遺伝子機能分類ツール
- 「 分析ウィザード」に戻り、自分の「エンリッチメントスコア」、リスト中の遺伝子群の全体的な濃縮の指標に従ってランク付けされた遺伝子の機能的に関連のグループに入力された遺伝子のリストを分離するために「 遺伝子機能分類 」モジュールをクリックしてください。
- 遺伝子群の機能的に関連する遺伝子を明らかにし、より詳細な情報と「RG」を取得するために用語の名前をクリックしてください
- すべての遺伝子との用語の2次元ビューを見るために関連する生物学と緑のアイコンを一覧表示するには、赤「T」(用語レポート)をクリックします。
- 遺伝子名バッチビューア
- 「 分析ウィザード」に戻り、それに対応する遺伝子名に「Wormbase遺伝子のID」を翻訳するために「 遺伝子名のバッチビューア」をクリックします。 (WBGene00022855 = TCER-1)。
- より多くの遺伝子固有の情報を取得するために遺伝子名をクリックします。
- 遺伝子を明らかにするために、各遺伝子の横にある[RG '(関連遺伝子)のリンクをクリックして、目的の遺伝子に機能的に関連すると予測しました。
NCBIシーケンス読むアーカイブへのアップロード4. RAWデータ(SRA)
- NCBI」リンクにサインインするにSRAのWebページにアクセスするか、新しいアカウントを登録します。
- 「Bioproject」をクリックします。
- 左側の「 使用Bioproject]という見出しの下にある「 提出」をクリックします。
- オプション「 新しい提出」を選択します。提出者の詳細情報を更新します。残りの7つのタブを続行実験の詳細に記入し、データがアップロードされています。完成したときに「 送信」をクリックします。
注:5番目の「 生体試料」タブで、「 生体試料」が空のためのスロットを残します。 - 「私の提出」リンクをクリックすることで、結果のページを更新します。提出されたデータは、割り当てられたサブミッション番号、簡単な説明とアップロードの状態で表示されます。
- ボックスの新しい投稿を開始」と「新しい提出」を作成して、このページの上部にある[生体試料」をクリックします。各サンプルの別々の提出を提出してください。
- 4.4の「Bioproject」と同様に、提出者の詳細を更新し、各タブの詳細に充填タブの残りを続行。一度審査を完了し、「 送信」をクリックします。
- 移動します。http://www.ncbi.nlm.nih.goV / SRAは、最終的な「配列読むアーカイブ(SRA)」の提出を作成します。
- 「はじめに」の下にある「SRAへのログイン」をクリックします。
- 次のページで「NCBI PDA」リンクをクリックしてください。 「アップデートの環境設定」リンクが開きます。フォームに必要事項を記入し、「保存の設定」をクリックしてください。
- 表示されたページで、「新規作成服従」リンクをクリックしてください。 「 エイリアスの下、適切な名前を入力し、[保存]をクリックします。提出IDおよびその他の詳細とテーブルが作成されます。
- 「新実験」をクリックし、各「生体試料」のため少なくとも一つのユニークなシーケンシングライブラリを登録。
- 以前に作成した「BioProject」と「生体試料」提出のIDを指定してリンクします。 「新しい実験は」を作成できます。
- ページの下部にある「新しいファイル名を指定して実行」をクリックします後SRA実験が行われ、それにリンクする必要があり、データファイルを識別されています。
- 各データファイルのMD5サムを計算します。マッキントッシュ端末でこれを行うには、 アプリケーション/ユーティリティ/ターミナルに移動します。端末では、(引用符)「MD5」でタイプスペースが続きます。ドラッグアンドファインダーからの端末にアップロードする必要のあるファイルをドロップすると「入力」をクリックします。
- ターミナルは、英数字MD5の合計を返します。ファイルアップロードのための申請プロセスの一部としてこれを入力します。 FTPを使用してファイルをアップロードするために、システムによって提供されたユーザ名とパスワードを使用してください。
結果
C.エレガンスでは、生殖細胞系列幹細胞(GSCs)の除去は、寿命を延長する、ストレスの回復力を高め、体脂肪24、28が上昇します 。レーザアブレーションによって、又はそのようなGLP-1などの変異によってもたらさGSCsの損失、のいずれかが、転写のネットワーク29を因...
ディスカッション
現代生物学の銀河シーケンシング・プラットフォームの意義
ギャラクシープロジェクトは、高速かつ効率的な方法で、高スループット配列決定データを処理し、分析するためのバイオインフォマティクストレーニングなし生物学者を助けることに尽力となっています。一度、この一般的に利用可能なプラットフォームは、単純で信頼性が高く、簡単なプロセスNGSデータを分...
開示事項
著者は、開示することは何もありません。
謝辞
著者は、ギャラクシーとDAVIDを開発し、ひいては科学コミュニティのためのNGSが広くアクセス行っている研究室、グループや個人に感謝の意を表したいと思います。当社のバイオインフォマティクストレーニング中にピッツバーグ大学の同僚によって提供されるヘルプやアドバイスが認められています。この作品には賞(AG-NS-0879から12)と国立衛生研究所AGに(R01AG051659)からの助成金を高齢でエリソン医学財団新奨学生によってサポートされていました。
資料
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
参考文献
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22 (2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085 (2011).
- Consortium, C. e. S. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383 (2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639 (2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788 (2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158 (2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved