
Method Article
Caracterización totalmente autónoma y colección de datos de cristales de macromoléculas biológicas
En este artículo
Resumen
Aquí, se describe cómo utilizar la selección automática y opciones de recopilación de datos disponibles en algunos beamlines del sincrotrón. Los científicos enviaran muestras de cryocooled el sincrotrón, las propiedades de difracción son evaluadas, los conjuntos de datos son recogidos y procesados y, cuando sea posible, una solución de la estructura se lleva a cabo, todo ello sin intervención humana.
Resumen
Haces de rayos x de alta brillantez juntados con automatización han llevado al uso de líneas de base de sincrotrón macromolecular Cristalografía de rayos x (MX) para los más exigentes proyectos en biología estructural. Sin embargo, mayoría de las instalaciones aún requiere la presencia de un científico en el sitio para realizar los experimentos. Una nueva generación de líneas automatizadas, dedicada a la caracterización automática de y recopilación de datos, cristales de macromoléculas biológicas se ha desarrollado recientemente. Estas líneas representan una nueva herramienta para los biólogos estructurales a la pantalla los resultados de ensayos de cristalización inicial o la colección de un gran número de conjuntos de datos de difracción, sin los usuarios tener que controlar la línea de sí mismos. A continuación os mostramos cómo montar un experimento para la detección automática y recogida de datos, cómo se realiza un experimento en la línea, como los conjuntos de datos resultantes se procesan y cómo, cuando sea posible, la estructura cristalina de la macromolécula biológica se soluciona.
Introducción
Determinar la estructura tridimensional de proteínas específicas es fundamental en biología. La información que se deriva de hacer así que arroja luz sobre la función biológica y sobre la forma y la especificidad de los sitios activos o vinculantes contenida en la molécula en estudio. En muchos casos, esto permite que los mecanismos de acción para determinar o, en su caso, posibles moléculas terapéuticas a desarrollar. MX es la técnica más utilizada para obtener información estructural, pero un cuello de botella es la determinación de las condiciones óptimas para obtener cristales bien diffracting. Por lo tanto, ensayos de cristalización se llevan a cabo en numerosas condiciones diferentes y son luego evaluados, para encontrar los mejores cristales para recopilación de datos de difracción. La automatización de la instalación de ensayos de cristalización1 claramente ha ayudado en este sentido. Sin embargo, los pasos subsecuentes (montaje de cristal, proyección de difracción y recopilación de datos de difracción) generalmente llevan a cabo manualmente, teniendo un montón de tiempo, esfuerzo y recursos. La automatización de difracción proyección y recopilación de datos, por lo tanto, significa una ganancia enorme en tiempo y eficacia.
Difracción proyección y recopilación de datos de MX más a menudo se lleva a cabo en beamlines del sincrotrón MX en la cual automatización ha facilitado en gran medida este proceso. Sin embargo, en la mayoría de los casos, es necesario para el científico para estar presente en la línea durante un experimento o para operar remotamente. Recientemente, una nueva generación de líneas totalmente automatizadas de MX ha sido desarrollados2. Aquí, los usuarios no necesitan estar presentes, ya sea físicamente o a distancia, durante una sesión experimental. Esto permite a los científicos a dedicar más tiempo a menos tareas de rutina, en lugar de pasar días enteros y a menudo noches, proyección de cristales y que recoge datos de difracción. Primera línea totalmente automatizada de todo el mundo es el masivo automatizado muestra selección instalación integrada (macizo de 1, ID30A-1)2,3 en la instalación Europea de radiación sincrotrón (ESRF). Tiene un entorno de muestra única en la que un dewar de alta capacidad que contiene la muestra funciona en tándem con un cambiador de muestras robótico que actúa también como goniómetro4,5 de la línea. MACIZO-1 es una línea de undulator equipado con un conteo de fotón único híbrido pixel detector6, que opera en una longitud de onda fija de 0,969 Å (12.84 keV) con un intenso haz de rayos x (2 x 1012 fotones/s). El tamaño de la viga en la posición de la muestra se puede ajustar entre un mínimo de 10 μm (viga redonda) a un máximo de 100 μm μm x 65 (horizontal por el tamaño vertical de la viga). En promedio, la línea puede procesar, en un completamente automático de moda 120 cristales (véase abajo), en 24 h. La operación de la línea se basa en una serie de flujos de trabajo7, cada una de ellas toma decisiones inteligentes basadas en los resultados de los anteriores pasos del flujo de trabajo, para la medición de los mejores datos posibles de la muestra bajo estudio. En particular, la evaluación de las características de la difracción de una muestra individual toma en flujo y volumen de cristal cuenta y asegura, donde el cristal es más grande que el haz de rayos x, que sólo la mejor región del cristal se utiliza para datos posteriores colección. Conjuntos de datos de difracción están, por lo tanto, optimizados para resolución máxima con radiación minimizar daños2,3. Exigentes protocolos de recogida de datos, tales como las estrategias de colección de datos (múltiples posiciones) pseudo-helicoidal ambos nativos y sola longitud de onda difracción anómala (SAD) recogida de datos, también están disponibles8.
Experimentos completamente automáticos en macizo-1 implican cryocooling y montaje de los cristales en una montura magnética muestra adecuada para el estándar de equipo de línea deseada pernos de columna9, entrando en los parámetros experimentales deseados en el ' difracción Plan' de la tabla en el sistema integrado de proteína Cristalografía líneas (ISPyB)10, un sistema de gestión de información en la web para los experimentos de MX y enviar las muestras a la línea. En el ESRF, todos los costos del transporte de las muestras de la línea son apoyados por la oficina del usuario ESRF (véase la página web del ESRF11 para más detalles). MACIZO-1, no hay restricciones se colocan en el tamaño del lazo o calidad cristalina. Al elegir un plan de difracción de un cristal determinado, el usuario puede utilizar la configuración predeterminada o elija entre los flujos de trabajo específicos, que pueden personalizarse para cada muestra. Varios flujos de trabajo programados están disponibles. En el flujo de trabajo de3 MXPressE, el lazo que contiene la muestra primero se alinea con la posición de la muestra utilizando centrado óptico. A continuación, basada en rayos X centrado asegura que la mejor región del cristal se centra en el haz de rayos x. Estrategias de recogida de datos entonces se calculan utilizando eEDNA, un marco para el desarrollo de aplicaciones basadas en plugin especialmente para análisis de datos en línea en el campo de experimentos de rayos x, teniendo en cuenta cristal volumen y el flujo en tiempo real en la línea. Tras la recogida de un conjunto de datos de difracción completo, esto es procesado mediante una serie de tuberías de proceso de datos automático12 y los resultados estarán disponibles para inspección y descarga en ISPyB. El flujo de trabajo de3 MXPressE SADpretende en cristales que contienen selenio-metionina de la proteína diana y explota el hecho de que la energía de funcionamiento del macizo-1 es justo sobre el borde Se K. Aquí, la estrategia de recolección de datos MXPressE eEDNA está optimizada para recopilación de datos triste (es decir, de alta redundancia y con la resolución en donde Rfusión entre pares de Bijvoet es inferior a 5%). Para las propiedades de difracción de una serie de cristales sin recopilación posterior de la pantalla, el flujo de trabajo de3 MXScorepuede utilizarse para producir una evaluación de la calidad de los cristales analizados. En el flujo de trabajo de3 MXPressI, de los datos de rotación de 180 ° se recogen usando oscilaciones de 0,2 ° y usando el ángulo phi inicial y la resolución determinada por una estrategia eEDNA. MXPressO 3 incluye una resolución de preobserved en el flujo de trabajo (por defecto: dmin = 2 Å). Para hacer una evaluación inicial de los cristales resultantes de una ensayo de cristalización, el flujo de trabajo de3 MXPressMse ofrece. Esto realiza un acoplamiento de la alto-dosis para explorar sobre la orientación más amplia de apoyo muestra con ninguna colección de datos o centrado. Recientemente, dos nuevos experimento flujos de trabajo, MXPressP y MXPressP_SAD, que realizan recopilaciones de datos pseudohelical, han sido puestas en ejecución8. Puede seguirse la ejecución de todos los pasos en los flujos de trabajo en línea y en tiempo real por el usuario, a través de ISPyB.
A continuación os mostramos cómo preparar un experimento totalmente automatizado de MX en macizo-1 y cómo recuperar y analizar los datos resultantes de la experiencia. Por ejemplo, utilizamos proteína de glicina mitocondrial humano escote sistema H (GCSH). Esta proteína que contiene ácido lipoico participa en el sistema de la hendidura de la glicocola responsable de la degradación de la glicina. Este sistema incluye la proteína P, una decarboxilasa de la glicocola dependiente del fosfato de piridoxal, la proteína T, una enzima que requiere tetrahidrofolato y la proteína L, una deshidrogenasa lipoamida. GCSH transfiere el grupo metilamina de glicina de la proteína de P a la proteína T. Defectos en la proteína H son la causa de hyperglycinemia de nonketotic (NKH) en los seres humanos13.
Protocolo
Nota: La producción, purificación y cristalización de GCSH se describen en archivo adicional 1.
1. breve descripción de la preparación sin conexión y montaje de cristal
- Posición de un lazo de nylon o de otro soporte de montaje de cristal ya fijado a un pin de la espina dorsal debajo de uno o más cristales y sacarlos de la solución de precipitación (20 μl de 0,5 M sodio formiato pH 4.0 + 25 μl de solución de proteína).
- Extraer el líquido a granel en el crystal(s) tocando el Monte con una mecha de papel al aspirar apagado cualquier exceso de líquido.
- Sumerja el crystal(s) en la solución crioprotectora que contiene la solución de precipitación más glicerol al 30%; Luego, retire el soporte de cristal y el crystal(s).
- Extraer el líquido a granel en el crystal(s) tocando el Monte con una mecha de papel al aspirar apagado cualquier exceso de líquido.
- Hundir el Monte en un frasco de lomo relleno con nitrógeno líquido y almacenarlo, junto con los otros cristales dispuestos del mismo modo, en un laboratorio de Biología Molecular europeo (EMBL) / ESRF cambiador puck9 a temperatura de nitrógeno líquido de la muestra.
Nota: Las crystal(s) es estables en esta condición hasta que el beamtime está disponible.
2. solicitando beamtime en macizo-1
- Solicitar lo antes posible en el homepage ESRF (en http://www.esrf.eu/UsersAndScience/UserGuide/Applying) beamtime.
Nota: Hay un número de modos posibles de acceso a las líneas del ESRF MX. Los laboratorios pueden solicitar colectivamente como parte de un bloque de asignación grupo (bolsa), que beamtime para 2 años. Si los grupos desean aplicar individualmente, pueden solicitar acceso, que les permite un acceso rápido a las líneas después de la revisión por pares del balanceo. La propuesta del grupo revisada y autorizada por el grupo de seguridad del ESRF que puede solicitar información adicional. Si la propuesta es aceptada, se comunicará un experimento número y una contraseña. Investigación propietaria puede realizarse mediante la compra de beamtime. - Completar la seguridad requiere de formación en línea (en http://www.esrf.eu/UsersAndScience/UserGuide/Preparing/SafetyTraining).
- Libro beamtime en el calendario del macizo-1.
Nota: Es posible reservar un máximo de 50 titulares de muestra a ser analizada por turno. - Llene la forma para declarar un experimento por correo (http://www.esrf.eu/UsersAndScience/UserGuide/Preparing/new-a-form), junto con la información de seguridad, para las muestras que se van a medir.
3. creación de un plan de difracción en ISPyB
Nota: El plan de difracción tiene toda la información necesaria para una muestra en ISPyB y puede contener información adicional para adaptar el experimento realizado para cada muestra.
- Abierto ISPyB (en https://exi.esrf.fr/).
- Seleccione MX experimentos.
- Inicie sesión con el número de experimento y la contraseña.
- Haga clic en envío | Agregar nuevo y proporcionar la información necesaria. Haga clic en Guardar.
- Haga clic en Agregar paquete y rellene la información solicitada. Haga clic en Guardar.
- A continuación, haga clic en Agregar contenedordar el código de barras del disco como el nombre y elegir el disco de la columna vertebral. Haga clic en Guardar.
- Haga clic en el símbolo de envase y Editary rellene la información necesaria, como el nombre de proteína, recomendado: flujo de trabajo, posición de cristal en la disco, etc., con respecto a las muestras.
- Elegir la proteína (por ejemplo, GCSH o lisozima) que ha sido aprobada por el grupo de seguridad ESRF.
- Introduzca un nombre de muestra único para identificar cada muestra individual. Es posible opcionalmente escanear el código de barras pin. El resto de la información a continuación es opcional.
- Introduzca la información opcional.
- Para cada muestra individual, introduzca el tipo de experimento (es decir, MXPressE_SAD, SCORE o MXPressO, etc., por defecto MXPressE) bajo Tipo de la exp. Esto define qué flujo de trabajo automática se utilizará para procesar cada cristal. Dado que los cristales GCSH son agujas, elija MXPressP.
- Entrar en un grupo del espacio (por ejemplo, P1, C2 y P212121), si se conoce. Si está presente, este se utilizará para los cálculos de estrategia de colección de datos y por las tuberías de procesamiento automático de datos disponibles.
- Introduzca la resolución deseada (por defecto: dmin = 2.0 Å). Esto define la distancia del detector de cristal de acoplamiento inicial análisis, caracterización y recolección de datos por defecto.
- Establecer la resolución del umbral deseado (por ejemplo, 1.5 Å o 2.3Å), para evitar la colección de conjuntos de datos completo de cristales que no difractan a este límite. Esto puede ahorrar tiempo de análisis y espacio de almacenamiento de datos.
- Establecer la integridad requerida (por defecto: 0.99). Establecer la necesaria multiplicidad (por defecto: 4). Si más de un cristal se encuentra en el soporte de la muestra, establecer el número máximo de cristales para ser analizados. El valor predeterminado es 1 o 5 para MXPressP.
- Seleccione el tamaño apropiado de la viga (por defecto: 50 μm). Si no se selecciona un valor específico, el centro de rayos X y cálculos de estrategia de colección de datos se realizará con un tamaño de haz de 50 μm.
Nota: Durante cualquier colección subsecuente de conjuntos completos de datos, el tamaño de la viga se adaptará automáticamente. - Poner en el grupo, si se conoce, en la columna de grupo espacio forzado. Ajustar la sensibilidad de la radiación de los cristales (0.5 – 2.0 de baja a alta sensibilidad, con un valor predeterminado de 1).
- Si lo desea, ajustar el ángulo de rotación total a recogerse para la colección de conjunto de datos completo (por defecto: el ángulo de giro total determinado por eEDNA).
- Guardar los valores. Haga clic en volver a los envíos. Presione para enviar envío a ESRF.
- Imprimir la etiqueta de envío y enviar las muestras. Los usuarios deben arreglar una camioneta con un servicio de mensajería, utilizando los datos de cuenta ESRF.
Nota: Es muy importante seleccionar la etiqueta de devolución incluyen permitir el regreso sin costura de muestras (véase https://www.esrf.eu/MXDewarReimbursement).
4. recolección, visualización y recuperación
Nota: En el día del experimento, las muestras se transfieren a la macizo-1 alta capacidad Dewar (HCD). Los científicos de la línea ejecutar la recolección de datos, que puede ser seguida por usuarios remotamente. Para cada tipo de muestra diferente los usuarios reciben un correo electrónico informándole que ha comenzado la recolección de datos. Como se señaló anteriormente, puede seguirse la ejecución de todos los pasos en los flujos de trabajo en línea y en tiempo real por el usuario a través de ISPyB, que los resultados pueden ser vistos y descargados.
- Para cada muestra analizada, examinar los resultados del experimento automático en ISPyB (https://exi.esrf.fr/).
- Sesión, utilizando el experimento número y contraseña y haga clic en la deseada sesión experimental en ID30A-1.
- Seleccionar la preferida (mayor puntuación) autoprocessing de la tubería (por ejemplo, granadas o XDS_APP) y descargar los datos escritos en el grupo correcto con la más alta integridad y mayor resolución haciendo clic en Resultados de recoger la última y, a continuación, en Descargar.
Nota: Todas las imágenes de malla, línea y caracterización están en un subdirectorio para cada muestra, llamado /MXPressE_01. El ESRF ejecuta automáticamente cinco paquetes de procesamiento independiente, es decir, EDNA_proc12, granadas12, XDS_APP14, autoPROC15y XIA216. Integración de datos se basa en XDS, con la excepción de XIA2, que se basa en esferas. Todos los paquetes se ejecutan también en los modos anómalos y nonanomalous, que permite la detección automática de una señal anómala, si está presente en los datos, para ser utilizado en SAD phasing protocolos. Cada paquete utiliza diferentes parámetros y árboles de decisión, lo que significa que algunos paquetes de operar mejores con algunas muestras. Sin embargo, esto puede hacer para un gran número de resultados cuando se contabiliza el número de paquetes y grupos del espacio posible. Los resultados son, por lo tanto, clasificados basado aproximadamente en resolución y otras métricas de calidad, tales como Rfusión en la shell de resolución más baja, CC(1/2) e integridad. Esto tiene como objetivo guiar al usuario a los mejores conjuntos de datos, pero todos los resultados y grupos del espacio posible deben inspeccionarse cuidadosamente.
- Descomprimimos la carpeta descargada, que incluirá todos los archivos de registro y XDS_ASCII combinar. HKL y archivos .mtz combinado y escala.
Nota: en caso de que la estructura (en formato PDB) de la proteína de interés o un homólogo del cierre fue subida a ISPyB al inicio del experimento, la tubería autoprocessing ESRF automáticamente realizará un reemplazo molecular (MR) ejecutar utilizando esta estructura como la modelo de búsqueda en la mejor puntuación de la solución. Los resultados de la tubería del Señor aparecen en ISPyB y pueden encontrarse en la carpeta de datos (por ejemplo, /data/visitor/mx2112/id30a1/20180711/PROCESSED_DATA/GCSH/GCSH-x5/autoprocessing_GCSH-x5_run1_1/grenades_fastproc/user_nohet.pdb_ mrpipe_dir /). Aquí, el modelo final se llamará coot1.pdb y el sidechains.mtz de datos de reflexión. Tenga en cuenta que la tubería podría disminuir la simetría de la célula (reducción de la célula primitiva) con el fin de aumentar la probabilidad de encontrar una solución. En este caso, la tubería del Señor escribió la solución en una celda monoclínico (C2) en lugar de una celda ortorrómbica (C2221). Detalles sobre cómo realizar un reemplazo molecular ejecutan manualmente (ejemplificado por la segunda mejor puntuación autoprocessing solución) están incluidos en los Archivos complementarios.
Resultados
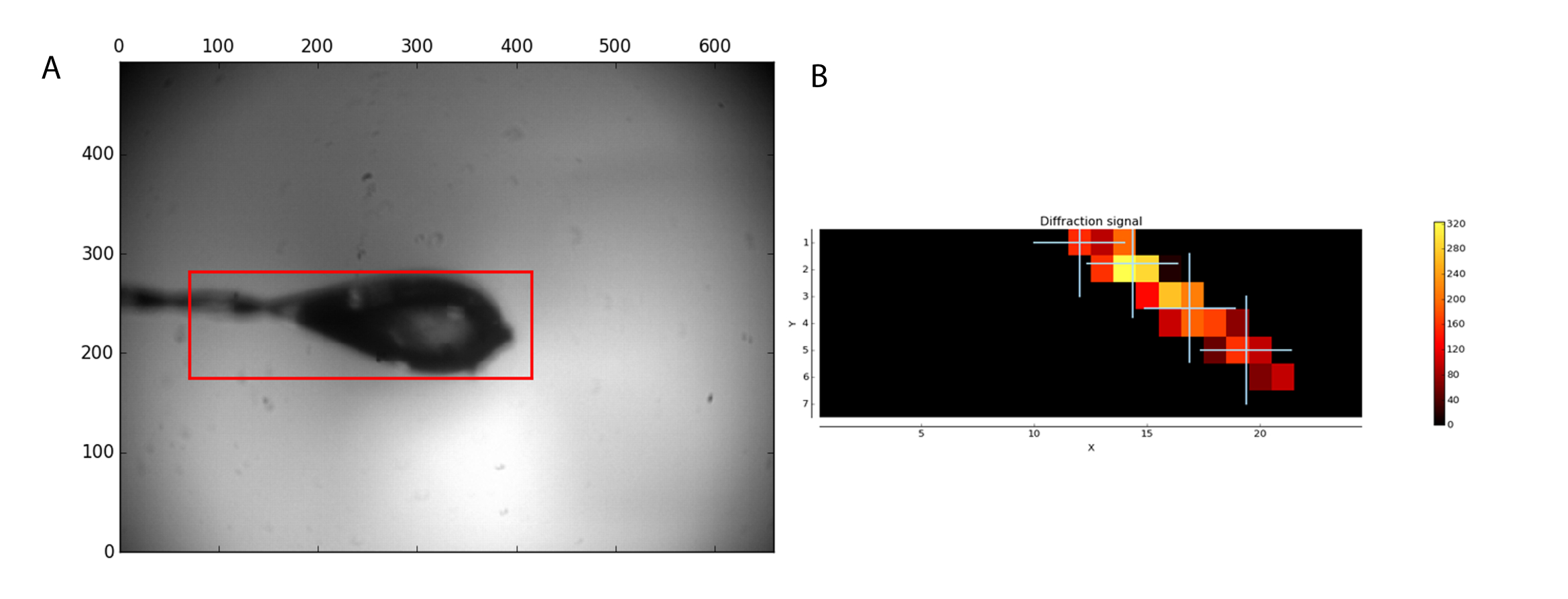
El flujo de trabajo de MXPressP solía en la línea IRT 1 macizo, completamente automática, Monte centro en el haz de rayos x, caracterizar y recopilar conjuntos de datos de difracción completo de una serie de cristales de GCSH humana. Las muestras fueron montadas y el bucle analizados para que un área de exploración (figura 1, izquierda). Tras el análisis de la difracción, se seleccionaron cuatro puntos dentro del cristal para la recogida de datos (figura 1, derecha). Posterior procesamiento por tuberías de análisis automatizado de los datos, incluyendo la tubería del Señor, dado conjuntos de datos de alta calidad (tabla 1) que se encontró una solución de Señor. Esta última permite evaluar rápidamente si el conjunto de datos obtenido y el modelo de búsqueda usados son adecuados para la eliminación por sustitución molecular. Además, la presencia de ligandos puede ser juzgada, lo que permite al usuario centrarse sólo en los conjuntos de datos más prometedores para su posterior análisis. Determinación de estructura manual por Señor rindió un mapa de densidad electrónica de alta calidad después de automatizado de un solo ciclo de refinamiento (Figura 2a). Para este conjunto de datos, la tubería automatizada corte los datos en un 1.32 Å de resolución; sin embargo, los usuarios todavía pueden decidir cortar los datos con una resolución menor para llegar a estadísticas de diferente calidad (CC1/2, < I/σ(I) >, Rmeas) en el shell de resolución más alta. La estructura cristalina de la estructura humana de GCSH es similar a la de la proteína bovina (3KlR)16.
Densidad de electrones continua es visible para la cadena entera del aminoácido, además de la etiqueta de histidina de la N-terminal. De las cuatro substituciones que distinguen GCSH humana y bovina, tres son fácilmente identificables en la densidad del electrón (Ile/Val66 Asp/Glu98 y Leu/Phe149; Figura 2b -d). Esto es menos claro para la substitución del Asp/Lys125 para que la densidad del electrón de la cadena lateral se resuelve sólo parcialmente debido a la flexibilidad (Figura 1e). El modelo obtenido actualmente tiene Rtrabajar y Rgratis valores de 20.4% y 23,8%, respectivamente y puede optimizarse aún más por los ciclos más de modelo y manuales de construcción y refinamiento.
| GRANADAS de la tubería | Tubería XDS_APP | |
| Procesamiento y recolección de datos | ||
| Fuente de rayos x / haz de línea | ESRF / MACIZO 1 | |
| Longitud de onda (Å) | 0,966 | |
| Resolución (Å) | 41,88-1.48 (1.53 – 1,48) | 41.86-1.32 (1,39 – 1.32) |
| Total/Unique reflexiones | 127670 / 28644 | 177332 / 40134 |
| (12178 / 2775) | (23772 / 5714) | |
| Grupo del espacio para la indización, escalamiento y fusión | C222 | C2221 |
| Dimensiones de la célula | ||
| a, b, c (Å) | 42.20, 83.75, 95.85 | 42.19, 83.72, 95,82 |
| Mosaicity | 0.05 | 0.05 |
| Rmeas (%) | 10.0 (110.7) | 11.1 (198.2) |
| < I/σ(I) > | 9.6 (1.3) | 7.6 (0.7) |
| CC1/2 (%) | 99.7 (53.9) | 99.7 (19,1) |
| Exhaustividad (%) | 99.6 (99.6) | 99.5 (98,6) |
| Multiplicidad | 4.5 (4.4) | 4.4 (4.2) |
| Reemplazo molecular y el refinamiento del modelo preliminar | ||
| Grupo del espacio para la eliminación | C2 | C2221 |
| Dimensiones de la célula | ||
| a, b, c (Å) | 83,74, 42.18, 95.82 | 42.19, 83.72, 95,82 |
| Α, Β, Γ (°) | 90, 90.03, 90 | 90, 90, 90 |
| Modelo de la búsqueda para Sr. (PDB) | 3KLR | 3KLR |
| Moléculas de la proteína / ASU | 2 | 1 |
| Residuos de la proteína | 250 | 125 |
| Rtrabajo/rlibre (%) después refinamiento 1 | 24.3 / 26.5 | 20.4 / 23.8 |
| Longitud de enlace RMSD (Å) después refinamiento 1 | 0.01 | 0.01 |
| Ángulo de enlace RMSD (°) después refinamiento 1 | 1.2 | 1,83 |
| Rotamer aislados (%) después refinamiento 1 | 1.07 | 4.29 |
| Ramachandran favorecida/permitido/no permitido (%) después refinamiento 1 | 95.93 / 4.07 / 0 | 95.12 / 4.88 / 0 |
Tabla 1: estadísticas de colección, refinamiento y validación de datos de difracción de rayos x. Valores para el shell de resolución más alta se dan en soportes.

Figura 1: muestra de análisis antes de la recolección de datos. (A) la región seleccionada para análisis aparece un cuadro rojo. (B) el análisis de imágenes de difracción se muestra como un mapa de calor. Cuatro posiciones en el cristal encuentra fueron seleccionados para la recolección de datos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: validación Visual de los mapas de densidad de electrones obtenidos después refinamiento. Mapas de densidad electrónica contorneadas en 2 x nivel del r.m.s. alrededor de (un) Trp143, (b) Val66 (Ile de GCSH humana) y (c) Glu98 (Asp en humano GCSH) y mapas de contorno en 1 x y aproximaciones sucesivas nivel alrededor de (d) Phe149 (Leu en humano GCSH) y (e) Lys125 (Asp en humano GCSH). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Líneas completamente automáticas proporcionan automatizado caracterización y recopilación de datos de gran número de cristales macromoleculares sin la presencia de un científico, ya sea en la línea o a distancia, ser necesario. Utilizando líneas completamente automatizadas tiene muchas ventajas comparados con operación manual. Por ejemplo, la muestra automatizada centrado, basado en el acoplamiento de rayos x y línea de análisis, es más preciso que el realizado con el ojo humano como no es afectado por efectos térmicos u ópticos. De hecho, estas exploraciones de malla y línea de proporcionan datos adicionales (es decir, dimensiones detalladas del cristal y la mejor región del cristal de diffracting) que son importantes para determinar el tamaño correcto Haz a utilizar para la recolección de datos, especialmente para los pequeños cristales 18— y a menudo resultan en una mejora de la calidad de los datos de difracción obtenidos. Además, aprovechándose de los parámetros definidos por el usuario en la configuración de los experimentos automáticos, los pasos en los flujos de trabajo específicos se pueden adaptar para satisfacer mejor el sistema bajo estudio, optimizando así aún más la tasa de éxito del experimento.
Tomando juntos, la fiabilidad de los flujos de trabajo disponibles, el acceso directo a la línea (usuarios auto programar, usando un calendario [ver arriba]) y el enfoque completamente automatizado del macizo-1 proporciona un riguroso, alto rendimiento y ahorro de tiempo alternativa a los clásicos experimentos MX y la posibilidad de implementar aplicaciones en flujos de trabajo automáticos y procedimientos más avanzados. En un futuro próximo, cartografía de cristal en 3D19 será implementado para mejorar la precisión de centrado de rayos x, mientras que los protocolos más complejos, tales como experimentos de deshidratación20de cristal, se pueden automatizar. Se espera que datos completamente autónoma se convertirá en un método estándar en MX, proporcionar datos de alta calidad para pantallas de fragmento de molécula pequeña, optimizar la proyección de un gran número de mal diffracting cristales y automáticamente la fase de información para resolver estructuras de cristal de novo. En combinación con la evolución de la cosecha automatizada de cristales21, la posibilidad de solución de estructura de cristal de proteína como un servicio automatizado bien podría convertirse en una realidad.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Los autores agradecen el ESRF para beamtime.
Materiales
| Name | Company | Catalog Number | Comments |
| Beamline MASSIF-1 | ESRF | ||
| BL21DE3 | New England Biolabs | C2527I | |
| chloramphenicol | Roth | 3886.1 | |
| Concentrators: Amicon Ultra-4 Ultracel -30K | Merck Millipore | UFC803024 | |
| Dialyzing membrane | Spectrumlabs | 132655 | |
| DMSO | Sigma-Aldrich | D8418 | |
| Dnase | Roche | 11284932001 | |
| DTT | Euromedex | EU0006-B | |
| EDTA- free protease inhibitors | Roche | 4,693,159,001 | |
| glycerol | VWR Chemicals Prolabo | 14388.29T | |
| His-trap HP | GE healthcare | 17-5247-01 | |
| imidazole | Sigma-Aldrich | 56750-500G | |
| IPTG | Euromedex | EU0008-B | |
| LB medium | Sigma-Aldrich | L3022 | |
| lipoic acid | Sigma-Aldrich | T5625 | |
| loop | Hampton Research | HR8-124 | |
| lysozyme | Roche | 10 837 059 001 | |
| MonoQ 5/50 GL | GE healthcare | 17-5166-01 | |
| NaCl | Fisher Chemical | S/3160/60 | |
| Sonicator vibra cell 75/15 | SONICS | ||
| SPINE pucks | MiTeGen | SKU: M-CSM003-0001A | |
| Tris base | Euromedex | 26-128-3094-B | |
| Sodium Formate | Sigma-Aldrich | 1064430500 | |
| GCSH purification buffer | 20 mM TRIS pH 8, 200 mM NaCl | ||
| GCSH cryo-protection buffer | 0.25 M Sodium Formate pH 4, 30% glycerol | ||
| Programs: | |||
| MxCube | Gabadinho, J. et al. MxCuBE : a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700-707, doi: 10.1107/S0909049510020005 (2010) | local development | |
| ISPyB | ESRF | Solange Delagenière, Patrice Brenchereau, Ludovic Launer, Alun W. Ashton, Ricardo Leal, Stéphanie Veyrier, José Gabadinho, Elspeth J. Gordon, Samuel D. Jones, Karl Erik Levik, Seán M. McSweeney, Stéphanie Monaco, Max Nanao, Darren Spruce, Olof Svensson, Martin A. Walsh, Gordon A. Leonard; ISPyB: an information management system for synchrotron macromolecular crystallography, Bioinformatics, Volume 27, Issue 22, 15 November 2011, Pages 3186-3192, https://doi.org/10.1093/bioinformatics/btr535 | local development |
| MXCube2 | ESRF | Gabadinho, J. et al. MxCuBE : a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700-707, doi: 10.1107/S0909049510020005 (2010). De Santis, D., Leonard, G. Notiziario Neutroni e Luce di Sincrotrone,Consiglio Nazionale delle Ricerche. (19), 24-226 (2014). | local development |
| BES workflow server | Brockhauser, S. et al. The use of workflows in the design and implementation of complex experiments in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 68 (8), 975-984, doi: 10.1107/S090744491201863X (2012). | ||
| DOZOR | ESRF | Bourenkov and Popov, unpublished | local development |
| BLISS beamline control | Guijarro, M. et al. BLISS - Experiments Control for ESRF EBS Beamlines. Proceedings of the 16th Int. Conf. on Accelerator and Large Experimental Control Systems, ICALEPCS2017, Barcelona, Spain. doi: 10.18429/jacow-icalepcs2017-webpl05 (2018). | local development | |
| AUTO processing of images | Monaco, S. et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. Journal of Applied Crystallography. 46 (3), 804-810, doi: 10.1107/S0021889813006195 (2013) | local development | |
| BEST and EDNA | Incardona, M.-F., Bourenkov, G.P., Levik, K., Pieritz, R.A., Popov, A.N., Svensson, O. EDNA : a framework for plugin-based applications applied to X-ray experiment online data analysis. Journal of Synchrotron Radiation. 16 (6), 872-879, doi: 10.1107/S0909049509036681 (2009). | local development | |
| CCP4 | Winn, M.D. et al. Overview of the CCP 4 suite and current developments. Acta Crystallographica Section D Biological Crystallography. 67 (4), 235-242, doi: 10.1107/S0907444910045749 (2011). | ||
| Phaser MR | McCoy, A.J., Grosse-Kunstleve, R.W., Adams, P.D., Winn, M.D., Storoni, L.C., Read, R.J. Phaser crystallographic software. Journal of Applied Crystallography. 40 (4), 658-674, doi: 10.1107/S0021889807021206 (2007). | ||
| Coot | Emsley, P., Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 60, 2126-32 (2004). | ||
| refmac5 | Murshudov, G.N., Vagin, A.A., Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 53, 240--255 (1997). | ||
| Matthews | Matthews, B.W. Solvent content of protein crystals. Journal of Molecular Biology. 33 (2), 491-497 (1968). |
Referencias
- Hui, R., Edwards, A. High-throughput protein crystallization. Journal of Structural Biology. 142 (1), 154-161 (2003).
- Bowler, M. W., et al. MASSIF-1: a beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of Synchrotron Radiation. 22 (6), 1540-1547 (2015).
- Svensson, O., Malbet-Monaco, S., Popov, A., Nurizzo, D., Bowler, M. W. Fully automatic characterization and data collection from crystals of biological macromolecules. Acta Crystallographica Section D Biological Crystallography. 71 (8), 1757-1767 (2015).
- Cipriani, F., et al. CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films. Acta Crystallographica Section D Biological Crystallography. 68, 1393-1399 (2012).
- Nurizzo, D., et al. RoboDiff: combining a sample changer and goniometer for highly automated macromolecular crystallography experiments. Acta Crystallographica Section D Structural Biology. 72 (8), 966-975 (2016).
- Bowler, M. W., Svensson, O., Nurizzo, D. Fully automatic macromolecular crystallography: the impact of MASSIF-1 on the optimum acquisition and quality of data. Crystallography Reviews. 22 (4), 233-249 (2016).
- Brockhauser, S., et al. The use of workflows in the design and implementation of complex experiments in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 68 (8), 975-984 (2012).
- Svensson, O., Gilski, M., Nurizzo, D., Bowler, M. W. Multi-position data collection and dynamic beam sizing: recent improvements to the automatic data-collection algorithms on MASSIF-1. Acta Crystallographica Section D Structural Biology. 74, 433-440 (2018).
- Cipriani, F., et al. Automation of sample mounting for macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 62 (10), 1251-1259 (2006).
- Delageniere, S., et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 27 (22), 3186-3192 (2011).
- Monaco, S., et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. Journal of Applied Crystallography. 46 (3), 804-810 (2013).
- Kikuchi, G., Motokawa, Y., Yoshida, T., Hiraga, K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences. 84 (7), 246-263 (2008).
- Sparta, K. M., Krug, M., Heinemann, U., Mueller, U., Weiss, M. S. XDSAPP2.0. Journal of Applied Crystallography. 49 (3), 1085-1092 (2016).
- Vonrhein, C., et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallographica Section D Biological Crystallography. 67 (4), 293-302 (2011).
- Winter, G. xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography. 43 (1), 186-190 (2010).
- Higashiura, A., et al. High-resolution X-ray crystal structure of bovine H-protein at 0.88 Å resolution. Acta Crystallographica Section D Biological Crystallography. 66 (6), 698-708 (2010).
- Sanishvili, R., et al. A 7 µm mini-beam improves diffraction data from small or imperfect crystals of macromolecules. Acta Crystallographica Section D Biological Crystallography. 64 (4), 425-435 (2008).
- Bowler, M. W., et al. Diffraction cartography: applying microbeams to macromolecular crystallography sample evaluation and data collection. Acta Crystallographica Section D Biological Crystallography. 66 (8), 855-864 (2010).
- Bowler, M. W., et al. Automation and Experience of Controlled Crystal Dehydration: Results from the European Synchrotron HC1 Collaboration. Crystal Growth & Design. 15 (3), 1043-1054 (2015).
- Zander, U., et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallographica Section D Structural Biology. 72 (4), 454-466 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados