
Method Article
Fully Autonomous Characterization and Data Collection from Crystals of Biological Macromolecules
In This Article
Summary
Here, we describe how to use the automated screening and data collection options available at some synchrotron beamlines. Scientists send cryocooled samples to the synchrotron, and the diffraction properties are screened, the data sets are collected and processed and, where possible, a structure solution is carried out—all without human intervention.
Abstract
High-brilliance X-ray beams coupled with automation have led to the use of synchrotron-based macromolecular X-ray crystallography (MX) beamlines for even the most challenging projects in structural biology. However, most facilities still require the presence of a scientist on site to perform the experiments. A new generation of automated beamlines dedicated to the fully automatic characterization of, and data collection from, crystals of biological macromolecules has recently been developed. These beamlines represent a new tool for structural biologists to screen the results of initial crystallization trials and/or the collection of large numbers of diffraction data sets, without users having to control the beamline themselves. Here we show how to set up an experiment for automatic screening and data collection, how an experiment is performed at the beamline, how the resulting data sets are processed, and how, when possible, the crystal structure of the biological macromolecule is solved.
Introduction
Determining the three-dimensional structure of specific proteins is crucial in biology. The information that is derived from doing so sheds light on the biological function and on the shape and specificity of active and/or binding sites contained in the molecule under study. In many cases, this allows mechanisms of action to be determined or, where appropriate, potential therapeutic molecules to be developed. MX is the technique most commonly used to obtain structural information, but a bottleneck is the determination of the optimal conditions to obtain well-diffracting crystals. Therefore, crystallization trials are carried out in numerous different conditions and are then screened, to find the best crystals to be used for diffraction data collection. The automation of the setup of crystallization trials1 has clearly helped in this regard. However, the subsequent steps (i.e., crystal mounting, diffraction screening, and diffraction data collection) are usually carried out manually, taking up a lot of time, effort, and resources. The automation of diffraction screening and data collection would, therefore, mean an enormous gain in time and efficiency.
Diffraction screening and data collection in MX is most often carried out at synchrotron MX beamlines at which automation has largely facilitated this process. However, in most cases, it is necessary for the scientist to be present at the beamline during an experiment or to operate it remotely. Recently, a new generation of completely automated MX beamlines has been developed2. Here, users do not need to be present, either physically or remotely, during an experimental session. This allows scientists to spend more time on less routine tasks, rather than spending entire days, and often nights, screening crystals and collecting diffraction data. The world's first fully automated beamline is the Massively Automated Sample Selection Integrated Facility (MASSIF-1, ID30A-1)2,3 at the European Synchrotron Radiation Facility (ESRF). It has a unique sample environment in which a high-capacity sample-containing dewar operates in tandem with a robotic sample changer that also acts as the beamline's goniometer4,5. MASSIF-1 is an undulator beamline equipped with a single-photon-counting hybrid pixel detector6, that operates at a fixed wavelength of 0.969 Å (12.84 keV) with an intense X-ray beam (2 x 1012 photons/s). The beam size at the sample position can be adjusted between a minimum of 10 µm (round beam) to a maximum of 100 µm x 65 µm (horizontal by vertical beam size). On average, the beamline can process, in a completely automatic fashion (see below), 120 crystals in 24 h. The operation of the beamline is based on a series of workflows7, each of which takes intelligent decisions based on the outcome of previous steps in the workflow, to ensure the measurement of the best possible data from the sample under study. In particular, the evaluation of the diffraction characteristics of an individual sample takes into account crystal volume and flux and ensures, where the crystal is larger than the X-ray beam, that only the best region of the crystal is used for subsequent data collection. Diffraction data sets are, thus, optimized for maximum resolution with minimized radiation damage2,3. Demanding data collection protocols, such as pseudo-helical (multi-position) data collection strategies for both native and single-wavelength anomalous diffraction (SAD) data collection, are also available8.
Completely automatic experiments at MASSIF-1 involve cryocooling and mounting the crystals on a magnetic sample mount suitable for the desired beamline equipment standard pins SPINE9, entering the desired experimental parameters in the 'diffraction plan' table in the Integrated System for Protein Crystallography beamlines (ISPyB)10, a web-based information management system for MX experiments, and sending the samples to the beamline. At the ESRF, all costs of the transport of the samples to/from the beamline are supported by the ESRF User Office (see the website of the ESRF11 for details). At MASSIF-1, no restrictions are placed on the loop size or crystal quality. When choosing a diffraction plan for a given crystal, the user can either use default settings or choose from specific workflows, which can be customized for each sample. Several preprogrammed workflows are available. In the MXPressE3 workflow, the sample-containing loop is first aligned to the sample position using optical centering. Then, X-ray-based centering ensures that the best region of the crystal is centered to the X-ray beam. Data collection strategies are then calculated using eEDNA, a framework for developing plugin-based applications especially for online data analysis in the X-ray experiments field, taking into account crystal volume and the real-time flux at the beamline. Following the collection of a full diffraction data set, this is then processed using a series of automatic data processing pipelines12 and the results are made available for inspection and download in ISPyB. The MXPressE SAD3 workflow is aimed at selenomethionine-containing crystals of the target protein and exploits the fact that the operating energy of MASSIF-1 is just above the Se K edge. Here, the MXPressE eEDNA data collection strategy is optimized for SAD data collection (i.e., high redundancy, and with the resolution set to where the Rmerge between Bijvoet pairs is below 5%). To screen the diffraction properties of a series of crystals without subsequent data collection, the MXScore3 workflow can be used to produce a full quality assessment of the crystals analyzed. In the MXPressI3 workflow, 180° of rotation data are collected using 0.2° oscillations and using the starting phi angle and the resolution determined by an eEDNA strategy. MXPressO3 includes a preobserved resolution into the workflow (default: dmin = 2 Å). To make an initial assessment of the crystals resulting from a crystallization trial, the MXPressM3 workflow is offered. This performs a high-dose mesh scan over the widest orientation of sample support with no data collection or centering. Recently, two new experiment workflows, MXPressP and MXPressP_SAD, which perform pseudohelical data collections, have been implemented8. The execution of all steps in all workflows can be followed online and in real-time by the user, via ISPyB.
Here we show how to prepare a fully automated MX experiment at MASSIF-1 and how to retrieve and analyze the data resulting from the experiment. As an example, we use human mitochondrial glycine cleavage system protein H (GCSH). This lipoic acid-containing protein is part of the glycine cleavage system responsible for the degradation of glycine. This system further includes the P protein, a pyridoxal phosphate-dependent glycine decarboxylase, the T protein, a tetrahydrofolate-requiring enzyme, and the L protein, a lipoamide dehydrogenase. GCSH transfers the methylamine group of glycine from the P protein to the T protein. Defects in the H protein are the cause of nonketotic hyperglycinemia (NKH) in humans13.
Protocol
NOTE: The production, purification, and crystallization of GCSH are described in Supplementary File 1.
1. Brief description of the offline preparation and crystal mounting
- Position a nylon loop or another crystal mounting support already fixed to a SPINE pin under one or more crystals and lift them out of the precipitation solution (20 µL of 0.5 M sodium formate pH 4.0 + 25 µL of protein solution).
- Remove the bulk liquid around the crystal(s) by touching the mount with a paper wick to suck off any excess liquid.
- Soak the crystal(s) in the cryoprotective solution containing the precipitation solution plus 30% glycerol; then, remove both the crystal support and the crystal(s).
- Remove the bulk liquid around the crystal(s) by touching the mount with a paper wick to suck off any excess liquid.
- Plunge the mount into a SPINE vial filled with liquid nitrogen and store it, along with any other crystals similarly prepared, in a European Molecular Biology Laboratory (EMBL)/ESRF sample changer puck9 at liquid nitrogen temperature.
NOTE: The crystal(s) are stable in this condition until the beamtime is available.
2. Requesting beamtime on MASSIF-1
- Request beamtime as early as possible on the ESRF homepage (at http://www.esrf.eu/UsersAndScience/UserGuide/Applying).
NOTE: There are a number of possible modes of access to the ESRF MX beamlines. Laboratories can apply collectively as part of a Block Allocation Group (BAG), to have beamtime allocated for 2 years. If groups wish to apply individually, they can apply for rolling access, which allows them rapid access to the beamlines after peer review. The group’s proposal will be reviewed and cleared by the ESRF Safety Group who may request additional details. If the proposal is accepted, an experiment number and password will be communicated. Proprietary research can be performed by purchasing beamtime. - Complete the required safety training online (at http://www.esrf.eu/UsersAndScience/UserGuide/Preparing/SafetyTraining).
- Book beamtime on the MASSIF-1 Calendar.
NOTE: It is possible to book up to a maximum of 50 sample holders to be analyzed per shift. - Fill in the A-form to declare a mail-in experiment (http://www.esrf.eu/UsersAndScience/UserGuide/Preparing/new-a-form), along with the required safety information, for the samples that are to be measured.
3. Creation of a diffraction plan in ISPyB
NOTE: The diffraction plan holds all the information needed for a sample in ISPyB and can contain additional information to tailor the experiment performed for each sample.
- Open ISPyB (at https://exi.esrf.fr/).
- Choose MX experiments.
- Log in with the experiment number and password.
- Click on Shipment | Add new and provide the necessary information. Click Save.
- Click Add parcel and fill in the information requested. Click Save.
- Then, click Add container, give the puck barcode as the name, and choose SPINE puck. Click Save.
- Click on the container symbol and Edit, and fill in the necessary information, like protein name, preferred workflow, crystal position in the puck, etc., concerning the samples.
- Choose the protein (for example, GCSH or lysozyme) that has been approved by the ESRF safety group.
- Enter a unique sample name to identify each individual sample. It is possible to optionally scan the pin barcode. The rest of the information below is optional.
- Enter the optional information.
- For each individual sample, enter the experiment type (i.e., MXPressE_SAD, SCORE, or MXPressO, etc., default MXPressE) under Exp. Type. This defines which automatic workflow will be used to process each crystal. Given that the GCSH crystals are needles, choose MXPressP.
- Enter a space group (for example, P1, C2, or P212121), if known. If present, this will be used for data collection strategy calculations and by the automatic data processing pipelines available.
- Enter the desired resolution (default: dmin = 2.0 Å). This defines the crystal-to-detector distance for the initial mesh scans, characterization, and default data collection.
- Set the desired threshold resolution (for example, 1.5 Å or 2.3Å), to prevent the collection of full datasets from crystals which do not diffract to this limit. This can save data storage space and analysis time.
- Set the required completeness (default: 0.99). Set the required multiplicity (default: 4). If more than one crystal is contained on the sample support, set the maximum number of crystals to be analyzed. The default value is 1 or 5 for MXPressP.
- Select the appropriate beam size (default: 50 µm). If a specific value is not selected, the X-ray-centering and data collection strategy calculations will be performed with a beam size of 50 µm.
NOTE: During any subsequent collection of complete data sets, the beam size will be adapted automatically. - Put in the space group, if known, in the forced space group column. Set the radiation-sensitivity of the crystals (0.5–2.0 for low to high sensitivity, with a default value of 1).
- If desired, set the total rotation angle to be collected for the full dataset collection (default: the total rotation angle determined by eEDNA).
- Save the values. Click on Return to Shipments. Press Send Shipment to ESRF.
- Print the shipping label and send the samples. Users should arrange a pickup with a courier, using the ESRF account details.
NOTE: It is very important to select Include return label to allow the seamless return of samples (see https://www.esrf.eu/MXDewarReimbursement).
4. Data collection, viewing, and retrieval
NOTE: On the day of the experiment, samples are transferred to the MASSIF-1 High Capacity Dewar (HCD). Beamline scientists then launch the data collection, which can be followed by users remotely. For each different sample type users receive an e-mail informing them that the data collection has started. As previously noted, the execution of all steps in all workflows can be followed online and in real-time by the user via ISPyB, from which the results can be viewed and downloaded.
- For each sample analyzed, examine the results of the automatic experiment in ISPyB (https://exi.esrf.fr/).
- Log in, using the experiment number and password, and click on the desired experimental session at ID30A-1.
- Select the preferred (top-scoring) autoprocessing pipeline (for example, Grenades or XDS_APP) and download the data written out in the correct space group with the highest completeness and highest resolution by clicking on Last Collect Results and, then, Download.
NOTE: All mesh, line, and characterization images are in a subdirectory for each sample, called /MXPressE_01. The ESRF automatically runs five separate processing packages, namely EDNA_proc12, Grenades12, XDS_APP14, autoPROC15, and XIA216. Data integration is based on XDS, with the exception of XIA2, which is based on DIALS. All packages are also run in anomalous and nonanomalous modes, allowing the automatic detection of an anomalous signal, if present in the data, to be used in SAD phasing protocols. Each package uses different parameters and decision trees, meaning that some packages run better with certain samples. However, this can make for a large number of results when the number of packages and possible space groups is accounted for. The results are, therefore, ranked based roughly on resolution and other quality metrics, such as Rmerge in the lowest resolution shell, CC(1/2), and completeness. This is aimed at guiding the user to the best data sets, but all possible space groups and results should be inspected carefully.
- Unzip the downloaded folder, which will include all log files and unmerged XDS_ASCII.HKL and merged and scaled .mtz files.
NOTE: In case the structure (in PDB format) of the protein of interest or a close homolog was uploaded to ISPyB at the start of the experiment, the autoprocessing pipeline at ESRF will automatically perform a molecular replacement (MR) run using this structure as the search model on the best scoring solution. The results of the MR pipeline are displayed in ISPyB and can be found in the processed data folder (for example, /data/visitor/mx2112/id30a1/20180711/PROCESSED_DATA/GCSH/GCSH-x5/autoprocessing_GCSH-x5_run1_1/grenades_fastproc/user_nohet.pdb_mrpipe_dir/). Here, the final model will be named coot1.pdb and the reflection data sidechains.mtz. Note that the pipeline might reduce the symmetry of the cell (primitive cell reduction) in order to increase the likelihood of finding a solution. In the case here, the MR pipeline wrote out the solution in a monoclinic cell (C2) rather than in an orthorhombic cell (C2221). Details on how to perform a molecular replacement run manually (exemplified for the second best-scoring autoprocessing solution) are included in the Supplementary Files.
Results
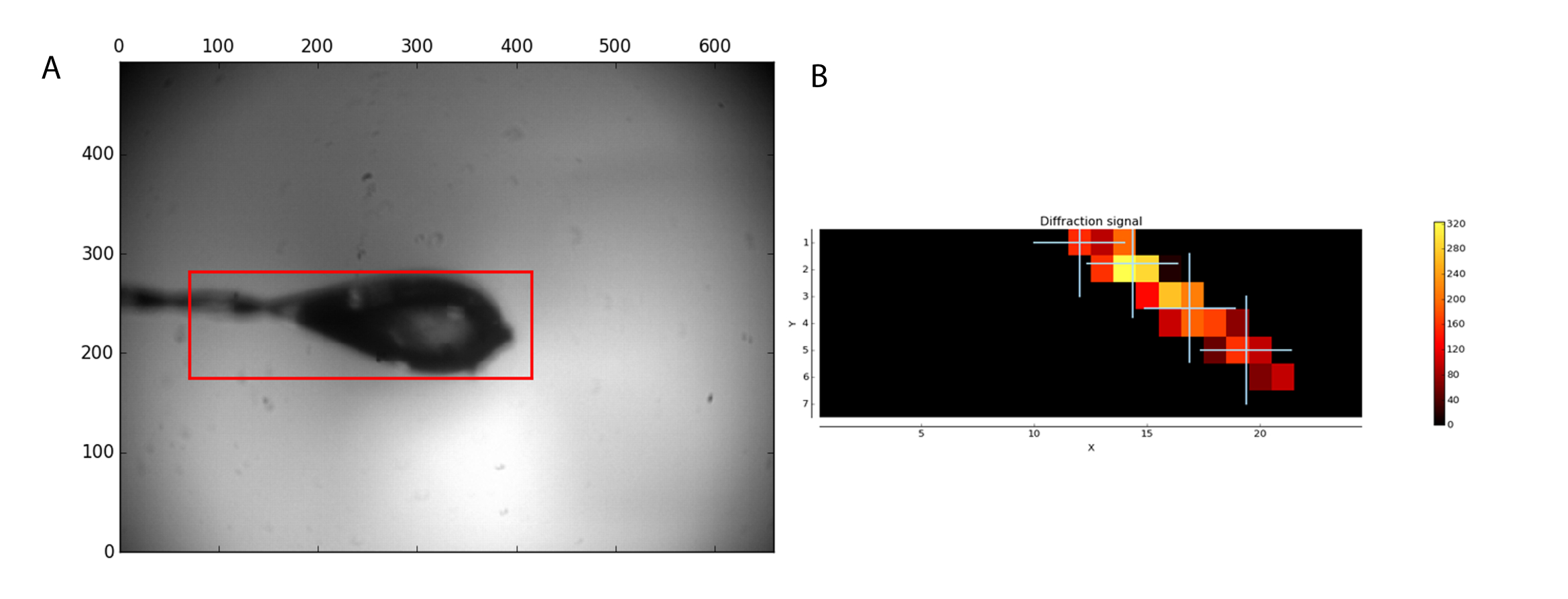
The MXPressP workflow was used at the ESRF beamline MASSIF-1 to, fully automatically, mount, center in the X-ray beam, characterize, and collect full diffraction data sets from a series of crystals of human GCSH. The samples were mounted and the loop analyzed for an area to scan (Figure 1, left). After the diffraction analysis, four points were selected within the crystal for data collection (Figure 1, right). Subsequent processing by automated data analysis pipelines, including the MR pipeline, yielded high-quality datasets (Table 1) for which an MR solution was found. The latter allows users to rapidly evaluate whether the obtained dataset and the used search model are suitable for phasing by molecular replacement. In addition, the presence of ligands can be judged, thus permitting the user to focus only on the most promising datasets for further analysis. Manual structure determination by MR yielded a high-quality electron density map after a single automated refinement cycle (Figure 2a). For this dataset, the automated pipeline cut the data at a 1.32 Å resolution; however, users can still decide to cut the data at a lower resolution to arrive at different quality statistics (CC1/2, <I/σ(I)>, Rmeas) in the highest resolution shell. The crystal structure of human GCSH structure is similar to that of the bovine protein (3KlR)16.
Continuous electron density is visible for the entire amino acid chain, apart from the N-terminal histidine tag. Of the four substitutions that distinguish human and bovine GCSH, three are readily identifiable in the electron density (Ile/Val66, Asp/Glu98, and Leu/Phe149; Figure 2b-d). This is less clear for the Asp/Lys125 substitution for which the electron density of the side chain is only partially resolved due to flexibility (Figure 1e). The currently obtained model has Rwork and Rfree values of 20.4% and 23.8%, respectively, and can be further optimized by further cycles of automated and manual model building and refinement.
| GRENADES pipeline | XDS_APP pipeline | |
| Data collection and processing | ||
| X-ray source / Beam line | ESRF / MASSIF-1 | |
| Wavelength (Å) | 0.966 | |
| Resolution (Å) | 41.88 – 1.48 (1.53 – 1.48) | 41.86 - 1.32 (1.39 – 1.32) |
| Total/Unique reflections | 127670 / 28644 | 177332 / 40134 |
| (12178 / 2775) | (23772 / 5714) | |
| Space group for indexing, scaling and merging | C222 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 42.20, 83.75, 95.85 | 42.19, 83.72, 95,82 |
| Mosaicity | 0.05 | 0.05 |
| Rmeas (%) | 10.0 (110.7) | 11.1 (198.2) |
| <I/σ(I)> | 9.6 (1.3) | 7.6 (0.7) |
| CC1/2 (%) | 99.7 (53.9) | 99.7 (19.1) |
| Completeness (%) | 99.6 (99.6) | 99.5 (98.6) |
| Multiplicity | 4.5 (4.4) | 4.4 (4.2) |
| Molecular replacement and preliminary model refinement | ||
| Space group for phasing | C2 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 83.74, 42.18, 95.82 | 42.19, 83.72, 95,82 |
| α, β, γ (°) | 90, 90.03, 90 | 90, 90, 90 |
| Search model for MR (PDB) | 3KLR | 3KLR |
| Protein molecules / ASU | 2 | 1 |
| Protein residues | 250 | 125 |
| Rwork/Rfree (%) after 1st refinement | 24.3 / 26.5 | 20.4 / 23.8 |
| RMSD bond length (Å) after 1st refinement | 0.01 | 0.01 |
| RMSD bond angle (°) after 1st refinement | 1.2 | 1.83 |
| Rotamer outlier (%) after 1st refinement | 1.07 | 4.29 |
| Ramachandran favoured/allowed/disallowed (%) after 1st refinement | 95.93 / 4.07 / 0 | 95.12 / 4.88 / 0 |
Table 1: X-ray diffraction data collection, refinement, and validation statistics. Values for the highest resolution shell are given in brackets.

Figure 1: Sample analysis before data collection. (A) The region selected for scanning is shown by a red box. (B) The analysis of diffraction images is shown as a heat map. Four positions within the located crystal were selected for data collection. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Visual validation of electron density maps obtained after refinement. Electron density maps contoured at 2x r.m.s. level around (a) Trp143, (b) Val66 (Ile in human GCSH), and (c) Glu98 (Asp in human GCSH) and maps contoured at 1x r.m.s level around (d) Phe149 (Leu in human GCSH) and (e) Lys125 (Asp in human GCSH). Please click here to view a larger version of this figure.
{kind=link}
Discussion
Fully automatic beamlines provide automated characterization and data collection from large numbers of macromolecular crystals without the presence of a scientist, either at the beamline or remotely, being required. Using completely automated beamlines has many advantages compared to manual operation. For example, the automated sample centering, based on X-ray mesh and line scans, is more precise than that performed with the human eye as it is not affected by thermal or optical effects. Indeed, these mesh and line scans provide additional data (i.e., detailed dimensions of the crystal and the best diffracting region of the crystal) which are important in determining the correct beam size to use for data collection—especially for small crystals18—and often result in an improved quality of the obtained diffraction data. Moreover, by taking advantage of the user-defined parameters in the setup of automatic experiments, the steps in specific workflows can be tailored to best suit the system under study, thus further optimizing the experiment success rate.
Taking together, the reliability of the workflows available, the straightforward access to the beamline (users self-schedule, using a calendar [see above]), and the fully automated approach of MASSIF-1 provides a rigorous, high-throughput, and time-saving alternative to classical hands-on MX experiments and the potential to implement more advanced procedures and applications into automatic workflows. In the near future, crystal cartography in 3D19 will be implemented to improve the accuracy of X-ray centering, while more complex protocols, such as crystal dehydration experiments20, will be automated. It is hoped that fully autonomous data collection will become a standard method in MX, providing high-quality data for small-molecule fragment screens, optimizing the screening of large numbers of poorly diffracting crystals and automatically providing phase information to solve crystal structures de novo. In combination with developments in the automated harvesting of crystals21, the possibility of protein crystal structure solution as an automated service could well become a reality.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors thank the ESRF for beamtime.
Materials
| Name | Company | Catalog Number | Comments |
| Beamline MASSIF-1 | ESRF | ||
| BL21DE3 | New England Biolabs | C2527I | |
| chloramphenicol | Roth | 3886.1 | |
| Concentrators: Amicon Ultra-4 Ultracel -30K | Merck Millipore | UFC803024 | |
| Dialyzing membrane | Spectrumlabs | 132655 | |
| DMSO | Sigma-Aldrich | D8418 | |
| Dnase | Roche | 11284932001 | |
| DTT | Euromedex | EU0006-B | |
| EDTA- free protease inhibitors | Roche | 4,693,159,001 | |
| glycerol | VWR Chemicals Prolabo | 14388.29T | |
| His-trap HP | GE healthcare | 17-5247-01 | |
| imidazole | Sigma-Aldrich | 56750-500G | |
| IPTG | Euromedex | EU0008-B | |
| LB medium | Sigma-Aldrich | L3022 | |
| lipoic acid | Sigma-Aldrich | T5625 | |
| loop | Hampton Research | HR8-124 | |
| lysozyme | Roche | 10 837 059 001 | |
| MonoQ 5/50 GL | GE healthcare | 17-5166-01 | |
| NaCl | Fisher Chemical | S/3160/60 | |
| Sonicator vibra cell 75/15 | SONICS | ||
| SPINE pucks | MiTeGen | SKU: M-CSM003-0001A | |
| Tris base | Euromedex | 26-128-3094-B | |
| Sodium Formate | Sigma-Aldrich | 1064430500 | |
| GCSH purification buffer | 20 mM TRIS pH 8, 200 mM NaCl | ||
| GCSH cryo-protection buffer | 0.25 M Sodium Formate pH 4, 30% glycerol | ||
| Programs: | |||
| MxCube | Gabadinho, J. et al. MxCuBE : a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700-707, doi: 10.1107/S0909049510020005 (2010) | local development | |
| ISPyB | ESRF | Solange Delagenière, Patrice Brenchereau, Ludovic Launer, Alun W. Ashton, Ricardo Leal, Stéphanie Veyrier, José Gabadinho, Elspeth J. Gordon, Samuel D. Jones, Karl Erik Levik, Seán M. McSweeney, Stéphanie Monaco, Max Nanao, Darren Spruce, Olof Svensson, Martin A. Walsh, Gordon A. Leonard; ISPyB: an information management system for synchrotron macromolecular crystallography, Bioinformatics, Volume 27, Issue 22, 15 November 2011, Pages 3186-3192, https://doi.org/10.1093/bioinformatics/btr535 | local development |
| MXCube2 | ESRF | Gabadinho, J. et al. MxCuBE : a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700-707, doi: 10.1107/S0909049510020005 (2010). De Santis, D., Leonard, G. Notiziario Neutroni e Luce di Sincrotrone,Consiglio Nazionale delle Ricerche. (19), 24-226 (2014). | local development |
| BES workflow server | Brockhauser, S. et al. The use of workflows in the design and implementation of complex experiments in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 68 (8), 975-984, doi: 10.1107/S090744491201863X (2012). | ||
| DOZOR | ESRF | Bourenkov and Popov, unpublished | local development |
| BLISS beamline control | Guijarro, M. et al. BLISS - Experiments Control for ESRF EBS Beamlines. Proceedings of the 16th Int. Conf. on Accelerator and Large Experimental Control Systems, ICALEPCS2017, Barcelona, Spain. doi: 10.18429/jacow-icalepcs2017-webpl05 (2018). | local development | |
| AUTO processing of images | Monaco, S. et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. Journal of Applied Crystallography. 46 (3), 804-810, doi: 10.1107/S0021889813006195 (2013) | local development | |
| BEST and EDNA | Incardona, M.-F., Bourenkov, G.P., Levik, K., Pieritz, R.A., Popov, A.N., Svensson, O. EDNA : a framework for plugin-based applications applied to X-ray experiment online data analysis. Journal of Synchrotron Radiation. 16 (6), 872-879, doi: 10.1107/S0909049509036681 (2009). | local development | |
| CCP4 | Winn, M.D. et al. Overview of the CCP 4 suite and current developments. Acta Crystallographica Section D Biological Crystallography. 67 (4), 235-242, doi: 10.1107/S0907444910045749 (2011). | ||
| Phaser MR | McCoy, A.J., Grosse-Kunstleve, R.W., Adams, P.D., Winn, M.D., Storoni, L.C., Read, R.J. Phaser crystallographic software. Journal of Applied Crystallography. 40 (4), 658-674, doi: 10.1107/S0021889807021206 (2007). | ||
| Coot | Emsley, P., Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 60, 2126-32 (2004). | ||
| refmac5 | Murshudov, G.N., Vagin, A.A., Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 53, 240--255 (1997). | ||
| Matthews | Matthews, B.W. Solvent content of protein crystals. Journal of Molecular Biology. 33 (2), 491-497 (1968). |
References
- Hui, R., Edwards, A. High-throughput protein crystallization. Journal of Structural Biology. 142 (1), 154-161 (2003).
- Bowler, M. W., et al. MASSIF-1: a beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of Synchrotron Radiation. 22 (6), 1540-1547 (2015).
- Svensson, O., Malbet-Monaco, S., Popov, A., Nurizzo, D., Bowler, M. W. Fully automatic characterization and data collection from crystals of biological macromolecules. Acta Crystallographica Section D Biological Crystallography. 71 (8), 1757-1767 (2015).
- Cipriani, F., et al. CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films. Acta Crystallographica Section D Biological Crystallography. 68, 1393-1399 (2012).
- Nurizzo, D., et al. RoboDiff: combining a sample changer and goniometer for highly automated macromolecular crystallography experiments. Acta Crystallographica Section D Structural Biology. 72 (8), 966-975 (2016).
- Bowler, M. W., Svensson, O., Nurizzo, D. Fully automatic macromolecular crystallography: the impact of MASSIF-1 on the optimum acquisition and quality of data. Crystallography Reviews. 22 (4), 233-249 (2016).
- Brockhauser, S., et al. The use of workflows in the design and implementation of complex experiments in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 68 (8), 975-984 (2012).
- Svensson, O., Gilski, M., Nurizzo, D., Bowler, M. W. Multi-position data collection and dynamic beam sizing: recent improvements to the automatic data-collection algorithms on MASSIF-1. Acta Crystallographica Section D Structural Biology. 74, 433-440 (2018).
- Cipriani, F., et al. Automation of sample mounting for macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 62 (10), 1251-1259 (2006).
- Delageniere, S., et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 27 (22), 3186-3192 (2011).
- Monaco, S., et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. Journal of Applied Crystallography. 46 (3), 804-810 (2013).
- Kikuchi, G., Motokawa, Y., Yoshida, T., Hiraga, K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences. 84 (7), 246-263 (2008).
- Sparta, K. M., Krug, M., Heinemann, U., Mueller, U., Weiss, M. S. XDSAPP2.0. Journal of Applied Crystallography. 49 (3), 1085-1092 (2016).
- Vonrhein, C., et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallographica Section D Biological Crystallography. 67 (4), 293-302 (2011).
- Winter, G. xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography. 43 (1), 186-190 (2010).
- Higashiura, A., et al. High-resolution X-ray crystal structure of bovine H-protein at 0.88 Å resolution. Acta Crystallographica Section D Biological Crystallography. 66 (6), 698-708 (2010).
- Sanishvili, R., et al. A 7 µm mini-beam improves diffraction data from small or imperfect crystals of macromolecules. Acta Crystallographica Section D Biological Crystallography. 64 (4), 425-435 (2008).
- Bowler, M. W., et al. Diffraction cartography: applying microbeams to macromolecular crystallography sample evaluation and data collection. Acta Crystallographica Section D Biological Crystallography. 66 (8), 855-864 (2010).
- Bowler, M. W., et al. Automation and Experience of Controlled Crystal Dehydration: Results from the European Synchrotron HC1 Collaboration. Crystal Growth & Design. 15 (3), 1043-1054 (2015).
- Zander, U., et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallographica Section D Structural Biology. 72 (4), 454-466 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved