
Method Article
Uso de microscopía de expansión para agrandar físicamente embriones de Drosophila de montaje completo para obtener imágenes de superresolución
En este artículo
Resumen
Aquí, se presenta un protocolo para la implementación de microscopía de expansión en embriones tempranos de Drosophila para lograr imágenes de superresolución utilizando un microscopio confocal convencional de barrido láser.
Resumen
El caballo de batalla de la biología del desarrollo es el microscopio confocal, que permite a los investigadores determinar la localización tridimensional de moléculas marcadas dentro de muestras biológicas complejas. Mientras que los microscopios confocales tradicionales permiten resolver dos fuentes puntuales fluorescentes adyacentes situadas a unos pocos cientos de nanómetros de distancia, la observación de los detalles más finos de la biología subcelular requiere la capacidad de resolver señales del orden de decenas de nanómetros. Se han desarrollado numerosos métodos basados en hardware para la microscopía de superresolución que permiten a los investigadores eludir estos límites de resolución, aunque estos métodos requieren microscopios especializados que no están disponibles para todos los investigadores. Un método alternativo para aumentar el poder de resolución es agrandar isotrópicamente la propia muestra a través de un proceso conocido como microscopía de expansión (ExM), que fue descrito por primera vez por el grupo de Boyden en 2015. ExM no es un tipo de microscopía per se , sino más bien un método para hinchar una muestra preservando la organización espacial relativa de sus moléculas constituyentes. A continuación, la muestra expandida puede observarse con una resolución efectivamente aumentada utilizando un microscopio confocal tradicional. Aquí, describimos un protocolo para implementar ExM en embriones de Drosophila de montaje completo, que se utiliza para examinar la localización de Par-3, miosina II y mitocondrias dentro de las células epiteliales de superficie. Este protocolo produce un aumento de aproximadamente cuatro veces en el tamaño de la muestra, lo que permite la detección de detalles subcelulares que no son visibles con la microscopía confocal convencional. Como prueba de principio, se utiliza un anticuerpo anti-GFP para distinguir distintos grupos de miosina-GFP entre las cortezas celulares adyacentes, y la estreptavidina marcada con fluorescencia se utiliza para detectar moléculas biotiniladas endógenas para revelar los detalles finos de la arquitectura de la red mitocondrial. Este protocolo utiliza anticuerpos y reactivos comunes para el marcaje de fluorescencia, y debe ser compatible con muchos protocolos de inmunofluorescencia existentes.
Introducción
En biología celular y del desarrollo, ver para creer, y la capacidad de determinar con precisión los patrones de localización de las proteínas es fundamental para muchos tipos de experimentos. La microscopía confocal de barrido láser es la herramienta estándar para obtener imágenes de proteínas marcadas con fluorescencia en tres dimensiones dentro de muestras intactas. Los microscopios confocales convencionales son incapaces de distinguir (resolver) señales fluorescentes adyacentes que están separadas por menos de la mitad de la longitud de onda de la luz que emiten1. En otras palabras, dos fuentes puntuales deben estar separadas por al menos 200-300 nm en la dirección lateral (500-700 nm en la dirección axial) para resolverlas como dos señales distintas. Esta barrera técnica se conoce como límite de difracción, y es un obstáculo fundamental para los estudios de estructuras subcelulares complejas (por ejemplo, las redes citoesqueléticas o mitocondriales de actomiosina) con características espaciales por debajo del límite de difracción. Por lo tanto, las técnicas para aumentar el poder de resolución de los microscopios confocales convencionales son de interés general para la comunidad biológica.
Para eludir el límite de difracción, se han desarrollado una serie de diferentes tecnologías de microscopía de superresolución que permiten una resolución del orden de decenas de nanómetros o menos 1,2,3, revelando un mundo de complejidad biológica que antes solo era accesible a través de la microscopía electrónica. A pesar de las ventajas obvias de estos métodos basados en hardware, los microscopios de superresolución a menudo tienen requisitos específicos de etiquetado de muestras y largos tiempos de adquisición, lo que limita su flexibilidad, o simplemente pueden ser demasiado costosos para que algunos laboratorios accedan a ellos. Una alternativa a la superresolución basada en microscopios es la microscopía de expansión (ExM), que no es un tipo de microscopía per se, sino más bien un método para hinchar una muestra preservando la organización espacial relativa de sus moléculas constituyentes4. Las muestras expandidas isotrópicamente se pueden observar a una resolución efectivamente aumentada utilizando un microscopio confocal de fluorescencia tradicional. ExM fue descrito por primera vez por el grupo de Boyden en 20155, y desde entonces la técnica básica se ha adaptado para su uso en una variedad de experimentos 6,7,8. ExM también se ha adaptado para su uso en embriones de montaje completo, especialmente en Drosophila 9,10,11, C. elegans12 y pez cebra 13, lo que lo convierte en una poderosa herramienta para los biólogos del desarrollo.
ExM se basa en dos químicas de hidrogel diferentes: 1) hidrogeles de polielectrolito hinchables, que aumentan considerablemente de tamaño cuando se sumergen en agua14, y 2) hidrogeles de poliacrilamida, que tienen un espaciado de polímero extremadamente pequeño para permitir la expansión isotrópica de la muestra15. Si bien hay muchos protocolos ExM publicados, generalmente comparten los siguientes pasos: fijación de la muestra, etiquetado, activación, gelificación, digestión y expansión4. Por supuesto, las condiciones de fijación y las estrategias de marcaje con fluorescencia variarán en función de las necesidades del experimento y del sistema, y en algunos protocolos, el marcaje se produce después de la expansión. Las moléculas objetivo de la muestra deben cebarse (activarse) para unirse al hidrogel, lo que puede lograrse utilizando diferentes productos químicos4. Durante las etapas de gelificación, la muestra se satura con monómeros del futuro hidrogel (acrilato de sodio, acrilamida y el reticulante bisacrilamida), y el hidrogel se forma por polimerización de radicales libres catalizada por un iniciador, como el persulfato de amonio (APS), y un acelerador, como la tetrametilendiamina (TEMED)4. Después de la gelificación, la muestra se digiere enzimáticamente para homogeneizar la resistencia de la muestra al hinchamiento y asegurar la expansión isotrópica del hidrogel4. Finalmente, el hidrogel digerido se coloca en agua, lo que hace que se expanda hasta aproximadamente cuatro veces su tamaño lineal original4.

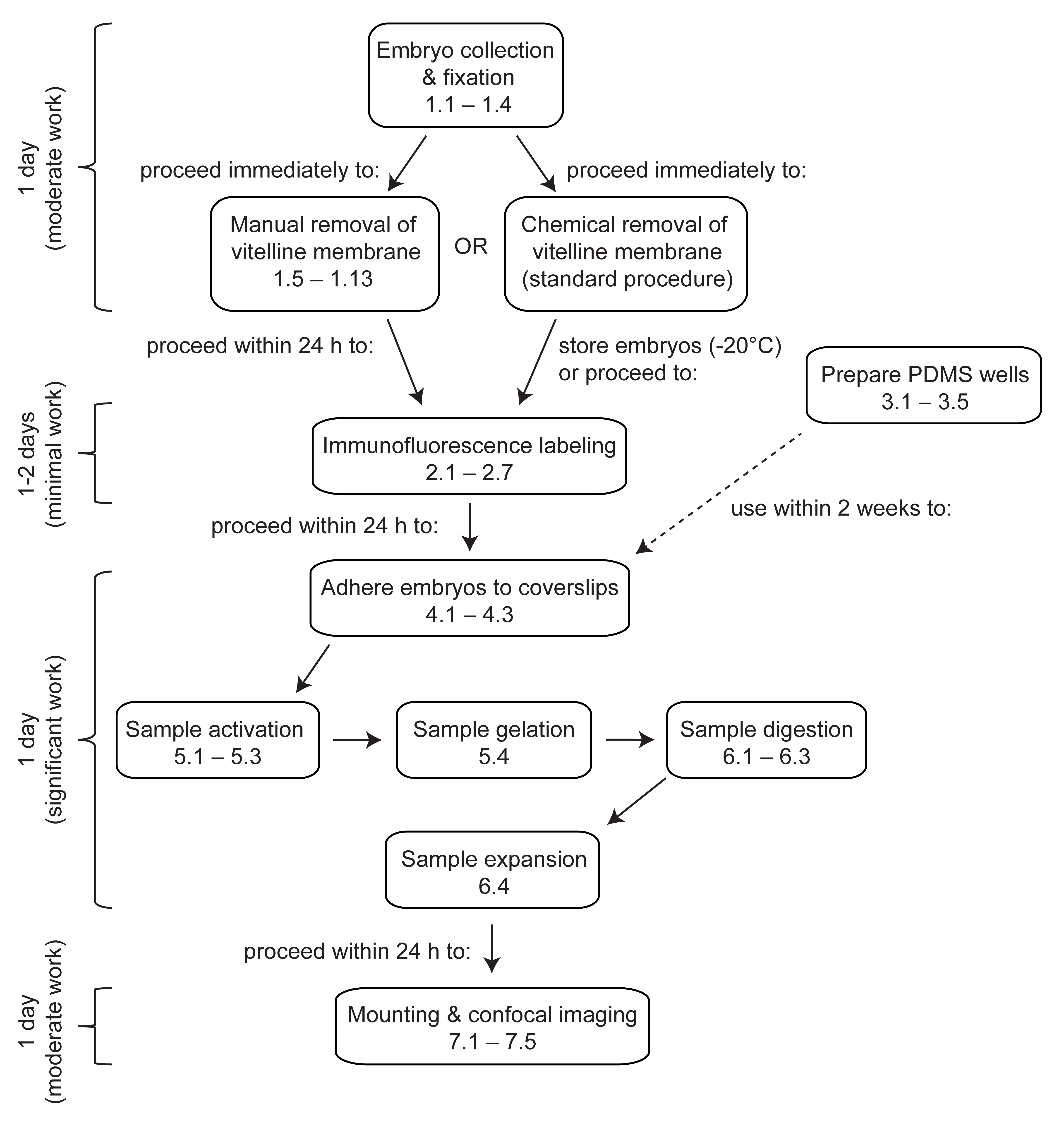
Figura 1: Visión general de la microscopía de expansión en embriones de Drosophila . ExM es un protocolo de varios pasos que tarda al menos 4 días en completarse. La recolección, fijación y devitellinización de embriones tarda 1 día o más, dependiendo de si se agrupan embriones de múltiples colecciones. El marcaje de inmunofluorescencia tarda 1 o 2 días, dependiendo de si los embriones se incuban durante la noche con los anticuerpos primarios. La activación, gelificación, digestión y expansión embrionaria se puede realizar en un solo día. Los geles se pueden montar y obtener imágenes inmediatamente después de la expansión, aunque por razones prácticas a menudo es deseable comenzar a obtener imágenes al día siguiente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Este protocolo describe cómo realizar ExM en embriones de Drosophila de montaje completo en etapa temprana a media16 para visualizar los patrones de localización de proteínas subcelulares a superresolución (Figura 1). Este método utiliza la química del éster N-hidroxisuccinimidil del ácido metilacrílico (MA-NHS) para activar y anclar las moléculas de proteína al hidrogel17, y es una modificación de un protocolo ExM publicado anteriormente para su uso en embriones y tejidos de Drosophila en etapa tardía11. Este protocolo utiliza pocillos de polidimetilsiloxano (PDMS) para moldear los hidrogeles y facilitar el intercambio de soluciones durante la activación y gelificación. Un método alternativo que no requiere la creación de pocillos de PDMS consiste en bajar los embriones adheridos a los cubreobjetos en gotas de solución de monómero que se encuentran sobre un trozo de película de sellado de laboratorio22. Además, este protocolo describe un método para eliminar manualmente la membrana vitelina impermeable que rodea a los embriones de Drosophila , que es un requisito previo para la tinción de inmunofluorescencia. Es importante destacar que este método de pelado manual de embriones se puede utilizar para seleccionar solo embriones de Drosophila en etapas adecuadas antes del etiquetado de la muestra, lo que aumenta en gran medida la probabilidad de terminar con muestras expandidas de la etapa y orientación correctas y, por lo tanto, hace que la recopilación de datos posteriores sea mucho más eficiente.
Protocolo
Este protocolo sigue las directrices de la Universidad de Arkansas (UARK) para la investigación en animales invertebrados, como Drosophila melanogaster, y fue aprobado por el Comité Institucional de Bioseguridad de la UARK (protocolo #20001).
1. Fijación y devitellinización de embriones de Drosophila
NOTA: El paso 1 describe un procedimiento (peeling manual) para la extracción manual de la membrana vitelina, una membrana impermeable transparente que rodea al embrión. Es importante destacar que el peeling manual permite la selección de embriones en estadios adecuados al comienzo del protocolo ExM, lo que aumenta en gran medida la probabilidad de obtener embriones en una orientación utilizable al final del protocolo ExM. Sin embargo, este protocolo ExM es completamente compatible con la recolección masiva de embriones y los procedimientos estándar para la eliminación de la membrana vitelina basada en metanol, en cuyo caso se puede pasar directamente al paso 2 (marcado de inmunofluorescencia).
- Prepare o compre varias agujas de vidrio finas. Las dimensiones reales de la punta de la aguja no son críticas, pero aseguran que las agujas sean lo suficientemente rígidas y afiladas como para perforar las membranas vitelinas de los embriones fijos. Fabricar agujas a partir de tubos capilares de vidrio (1 mm de diámetro exterior, 0,75 mm de diámetro interior) utilizando un extractor de micropipetas, como se prepararían las agujas para las microinyecciones de embriones18; Alternativamente, compre agujas pre-extraídas.
- Recoja embriones utilizando técnicas estándar de Drosophila 19 colocando >100 Drosophila adulta en un vaso de plástico ventilado sellado con una placa de jugo de frutas/agar20. Utilice ventanas de recolección cronometradas para enriquecer a los embriones de la etapa16 adecuada. Por ejemplo, para enriquecer para las etapas de gastrulación (etapa 6) y extensión convergente (etapa 7), cambie la placa de jugo de frutas, recoja los embriones durante 2 h a 25 °C, luego retire la placa y envejezca durante 2 h más a 25 °C para obtener embriones que tengan ~ 2-4 h de edad.
- Para eliminar el corion en forma de cáscara de huevo de los embriones, cubra la superficie de los platos de jugo de frutas con lejía al 50% (Tabla 1), libere los embriones de la superficie del agar agitándolos con un pincel pequeño y espere 3 minutos para que el corion se disuelva.
- Transferir los embriones decorionados con un pincel a un vial de centelleo de 30 mL que contenga 4 mL de heptano (fase superior orgánica) y 4 mL de tampón de fijación (fase inferior acuosa; Tabla 1). Diluir el formaldehído de un caldo recién preparado o recién abierto, y mezclar con 10x PBS y agua desionizada inmediatamente antes de añadir los embriones.
NOTA: Prepare el formaldehído a partir del poder del paraformaldehído o del formaldehído de grado EM al 16% en ampollas de vidrio. Se pueden utilizar existencias de formaldehído concentrado (por ejemplo, formaldehído al 37%), aunque los resultados pueden ser menos consistentes. - Los embriones se acumularán en la interfaz entre las fases orgánica y acuosa. Agregue tantos embriones como formen una sola capa en la interfaz. Si se agregan demasiados embriones a un vial, no se fijarán tan bien.
- Con cinta adhesiva fuerte, inmovilice los viales de centelleo de lado en una coctelera de mesa y agítelos durante 20 minutos a 220 rpm. Para una fijación óptima, mantenga una emulsión vigorosa entre las fases orgánica y acuosa durante toda la fijación.
- Durante el tiempo de fijación, prepare uno de los siguientes para cada una de las muestras.
- Tome una base de placa de Petri de plástico de 6 cm llena hasta la mitad con agar al 3% y marque un rectángulo de ~ 5 cm x 3 cm en el agar con una cuchilla de afeitar o bisturí. También se pueden utilizar platos de zumo de frutas/agar para este fin.
- Con una pequeña espátula de laboratorio, retire la losa de agar. Invierte la base de la placa de Petri y colócala en el banco. Coloque la losa de agar encima del plato invertido (Figura 2A).
- Tome la tapa de la placa de Petri y asegúrese de que esté seca. Con guantes, coloque un trozo de cinta adhesiva de doble cara dentro de la tapa (el trozo de cinta debe ser un poco más grande que la losa de agar; Figura 2B).
- Retire los viales de la coctelera, colóquelos en posición vertical sobre el banco y deje que las fases orgánica y acuosa se separen. Los embriones correctamente fijados permanecerán en la interfaz entre las dos fases.
- Transfiera los embriones fijados a la losa de agar utilizando una pipeta Pasteur de vidrio equipada con una pera de látex. Para evitar que los embriones se adhieran al interior de la pipeta, trate de mantener los embriones dentro del cuello estrecho de la pipeta y transfiera los embriones en varios lotes pequeños en lugar de todos a la vez. Una vez que todos los embriones estén en la losa de agar, elimine la mayor parte del heptano residual alrededor de los embriones con una pipeta P200. Realice este paso lo más rápido posible (<3 min) para evitar que los embriones fijados se sequen, lo que puede afectar negativamente a la morfología.
- Desde una altura de ~2 cm, deje caer la tapa con la cinta adhesiva de doble cara sobre la losa de agar para adherir los embriones a la cinta (Figura 2C). Retire suavemente la tapa de la losa de agar, colóquela boca abajo en el banco y luego agregue suficiente PBS-Tween (Tabla 1) para cubrir los embriones en la tapa.
- Utilizando un microscopio de disección estereoscópica con un aumento de aproximadamente 100x con iluminación indirecta, identifique los embriones en estadios adecuados utilizando marcadores morfológicos. Para los embriones en estadio 6, utilice marcadores como un surco cefálico visible y un mesodermo invaginado; Para los embriones en etapa 7, use marcadores como una banda germinal extendida; Para los embriones en etapa 11, use marcadores como una banda germinal completamente extendida y una segmentación visible a lo largo del eje16 de la cabeza a la cola.
- Para recolectar los embriones deseados, primero pinche la membrana vitelina (una membrana ovalada transparente alrededor del embrión) cerca del extremo anterior o posterior del embrión con una aguja de vidrio fina; La membrana se desinflará un poco a medida que se libere la presión. Luego, con unas pinzas finas o una sonda de metal, empuje suavemente el embrión en el otro extremo a través del orificio; La membrana vitelina permanecerá adherida a la cinta adhesiva de doble cara. Deje los embriones no deseados adheridos a la cinta.
- Recoger periódicamente los embriones desvitellinizados flotantes con una pipeta Pasteur de vidrio y trasladarlos a un tubo de microfuga de 1,5 ml.
- En este punto, realice uno de los siguientes pasos.
- Continúe directamente con los pasos de etiquetado de inmunofluorescencia. Los embriones desvitellinizados pueden pasar directamente a la solución de bloqueo (paso 2.2). No permita que los embriones permanezcan en PBS-Tween o en solución de bloqueo (Tabla 1) durante más de 16 h antes de continuar con el siguiente paso.
- Traslade los embriones al metanol para su almacenamiento. Retire la mayor cantidad posible de PBS-Tween y luego agregue 1 ml de metanol. Una vez que los embriones se hayan asentado, retire la mayor cantidad de metanol posible y agregue 1 ml de metanol fresco. Almacenar los embriones a -20 °C indefinidamente. El almacenamiento de metanol también permite la puesta en común de embriones de múltiples colecciones.
2. Marcaje de inmunofluorescencia
NOTA: Aparte de los pasos de incubación de anticuerpos, las cantidades exactas de líquido y los tiempos no son críticos en esta sección. Para realizar un enjuague o lavado, deje que los embriones se asienten en el fondo del tubo, elimine la mayor cantidad de líquido posible sin succionar los embriones y luego agregue ~ 1 ml de líquido nuevo; utilice una pipeta Pasteur de vidrio equipada con una bombilla de látex para una claridad y un control óptimos. Para el paso de enjuague, los embriones no se mecen, solo se dejan reposar; Para la etapa de lavado, los embriones se mecen en un nutator durante el tiempo indicado y luego se dejan reposar.
- Si los embriones no se almacenaron en metanol, continúe con el paso 2.2. Si los embriones se almacenaron en metanol, enjuague dos veces con PBS-Tween y luego lave durante 20 minutos dos veces con PBS-Tween.
- Lavar los embriones durante 30-60 min en 1 mL de solución bloqueante.
- Incubar los embriones con anticuerpos primarios diluidos en solución de anticuerpos (Tabla 1) durante 2 h a temperatura ambiente o preferiblemente durante la noche a 4 °C. Realizar este paso en el menor volumen posible (50-300 μL) para conservar los anticuerpos primarios; No es estrictamente necesario balancearse en un nutator.
- Aumentar la cantidad de anticuerpo primario utilizado en un experimento típico de inmunofluorescencia en al menos un 50% para ExM. Utilizar las siguientes concentraciones primarias de anticuerpos: 1:200 para el policlonal21 de cobaya anti-Par-3 y 1:100 para el policlonal de conejo anti-GFP.
- Retire la solución de anticuerpos primarios (guárdela a 4 °C si lo desea), enjuague dos veces con PBS-Tween y, a continuación, lávese durante 15 minutos cuatro veces con PBS-Tween.
- Incubar los embriones con anticuerpos secundarios fluorescentes en un volumen final de 300 μL (diluido en solución de anticuerpos) durante 1 h a temperatura ambiente en un nutator. Durante este paso, se puede agregar estreptavidina marcada con fluorescencia. A partir de este paso, proteger los embriones de una exposición excesiva y prolongada a la luz cuando sea posible, por ejemplo, cubriendo los tubos con una tapa de caja opaca o guardando las muestras en un cajón.
- Utilice las siguientes concentraciones: 1:500 para la IgG policlonal de cabra anti-conejo fusionada con Alexa Fluor 488; 1:500 para la cabra IgG anti-cobaya policlonal fusionada con Alexa Fluor 568; y 1:1000 para estreptavidina-Alexa Fluor 488.
- Retire y deseche la solución de anticuerpos secundarios. Enjuague los embriones dos veces con PBS-Tween y lávelos durante 15 minutos cuatro veces con PBS-Tween.
- En este punto, los embriones pueden almacenarse a 4 °C en la oscuridad, pero procesar las muestras lo más rápido posible (<24 h).
3. Preparación de pozos PDMS
NOTA: Los pocillos PDMS se pueden hacer con hasta 2 semanas de anticipación.
- Ajuste una incubadora o placa calefactora a 55 °C y una centrífuga que pueda hacer girar tubos cónicos a 15 °C.
- Para preparar la solución de PDMS (Tabla 1), coloque un tubo cónico de 50 ml en un recipiente secundario en una báscula y agregue 10 g de base de elastómero de silicona al tubo con una jeringa. A continuación, añade 1 g de agente de curado de elastómero de silicona e invierte el tubo varias veces para mezclar.
- Cree un tubo de equilibrio agregando una cantidad adecuada de agua a un segundo tubo cónico de 50 ml. Centrifugar la solución de PDMS a 500 x g durante 3 minutos a 15 °C y, a continuación, verterla en una placa de Petri de 10 cm a una profundidad de ~1 mm. Si es necesario, elimine las burbujas soplando suavemente sobre la solución con una manguera de aire. Deje que la solución de PDMS se solidifique durante la noche a 55 °C.
- Una vez que la losa de PDMS esté solidificada, con un bisturí, marque áreas cuadradas que sean ligeramente más pequeñas que un cubreobjetos de 22 mm x 22 mm. Dentro de cada cuadrado, marque y elimine un pozo cuadrado de ~ 8 mm de ancho.
- Transfiera cada pocillo cuadrado de PDMS a un cubreobjetos de 22 mm x 22 mm y adhiéralo firmemente (Figura 2D). La preparación de seis o más cubreobjetos debería producir un buen número de embriones expandidos para obtener imágenes.
4. Adhesión de los embriones a los cubreobjetos
- Aplique suficiente poli-L-lisina al 0,1% para cubrir la superficie del cubreobjetos dentro de cada pocillo (~50 μL) y colóquelos en una incubadora a 55 °C para que se sequen al aire. Repita este paso para aumentar la adhesividad.
- Enjuague brevemente los embriones una vez en 1x PBS para eliminar el detergente Tween, y luego transfiera >10 embriones a cada uno de los pocillos recubiertos de poli-L-lisina.
- Deje que los embriones se asienten en el fondo de los pocillos. Eliminar el exceso de líquido de los embriones adheridos con una pipeta Pasteur. Proceda inmediatamente al siguiente paso.
5. Activación y gelificación
NOTA: La activación se refiere a la adición de MA-NHS a los embriones, que modificará las proteínas y anticuerpos de la muestra para que puedan unirse al hidrogel. La gelificación se refiere a la generación de un hidrogel dentro y alrededor de los embriones en cada pocillo. Durante la gelificación, los embriones se impregnan con una solución de monómero y luego se tratan con una solución de gelificación para formar el hidrogel.
- Activar los embriones durante 1 h a temperatura ambiente llenando los pocillos con solución de activación (1 mM MA-NHS recién diluido en 1x PBS; Tabla 1). Cambie esta solución aproximadamente cada 10 minutos en el transcurso de 1 h.
- Enjuague los embriones con 1x PBS tres veces. Incubar los embriones en solución de monómeros (Tabla 1) durante 45 min a 4 °C.
- Mientras los embriones están asentados en la solución de monómero, prepare la solución de gelificación (Tabla 1). La preparación de ~ 2 ml de solución de gelificación es suficiente para cubrir los pocillos de PDMS de una placa de Petri completa de 10 cm. Asegúrese de agregar el APS al final, ya que iniciará la polimerización y comenzará la gelificación.
- Diluya el oxidante catalítico recién salido del polvo (por ejemplo, 1% de TEMPO p/v en agua). Combine 1.960 μL de solución de monómero con 30 μL de TEMED al 10% y 10 μL de TEMPO al 1%.
- Para evitar polimerizar todo el lote de solución de gelificación a la vez, trabaje en lotes pequeños. Divida la solución de gelificación (sin APS) en alícuotas de 125 μL entre los ocho tubos de una tira de PCR.
- Retire la solución de monómero de los tres pocillos de PDMS usando una aspiradora mientras tiene cuidado de no interrumpir los embriones. Añadir 5 μL de APS a uno de los tubos de PCR que contienen la solución de gelificación para iniciar la polimerización. Distribuya rápidamente la solución de gelificación polimerizante entre los pocillos (~40 μL por pocillo). Repita esto hasta que todos los pocillos y embriones estén cubiertos.
- Dejar que las muestras se gelifiquen durante 1,5-2,5 h a 37 °C. Agitar los hidrogeles cada cierto tiempo para controlar la polimerización. Los hidrogeles solidificados no se moverán. Los hidrogeles más gruesos tardarán más en completar la polimerización y solidificarse.
6. Digestión y expansión
NOTA: Los geles más gruesos y grandes tardarán más en expandirse, y el centro de los geles puede tardar varias horas en expandirse por completo; Esto se puede acelerar recortando los bordes del gel. A medida que los geles se expanden, su índice de refracción se volverá casi idéntico al del agua y se volverán muy difíciles de ver.
- Una vez completada la gelificación, retire los pocillos de PDMS del cubreobjetos mientras intenta no alterar los hidrogeles. Corta el exceso de material de hidrogel, si lo deseas.
- Transfiera los hidrogeles (aún adheridos a los cubreobjetos) individualmente a los pocillos de una placa de seis pocillos (Figura 2E). Tenga en cuenta que los hidrogeles pueden expandirse ligeramente durante la digestión.
- Cubrir completamente los geles con tampón de digestión (Tabla 1) durante 1 h a 37 °C. En general, 30 ml de tampón de digestión son suficientes para cubrir los geles en una placa de 6 pocillos.
- Después de la digestión, transfiera cada hidrogel individualmente a una placa de Petri de 6 cm deslizándolos fuera del cubreobjetos. Puede ser necesario usar un segundo cubreobjetos para desalojar el gel. Llene cada placa de Petri con agua desionizada para expandir el gel. Cambie el agua de tres a cuatro veces en el transcurso de 1-2 h hasta que los geles estén completamente expandidos (espere un aumento aproximado de cuatro veces en el ancho).
7. Montaje e imagen
NOTA: Los hidrogeles expandidos están compuestos casi en su totalidad de agua, lo que los hace casi transparentes y extremadamente frágiles. Los geles se pueden manipular utilizando cubreobjetos largos para moverlos y recogerlos. Monte y obtenga imágenes de solo uno o dos geles a la vez, ya que los geles liberarán agua gradualmente y comenzarán a deslizarse alrededor del cubreobjetos.
- Con una pipeta Pasteur, elimine la mayor cantidad posible de exceso de agua de la placa de Petri para minimizar el movimiento de los geles cuando se manipula.
- Coloque cada gel expandido, con los embriones en la superficie inferior, sobre un cubreobjetos grande (por ejemplo, de 24 mm x 40 mm) para obtener imágenes.
- Monte cada cubreobjetos con gel sobre el objetivo de un microscopio confocal de barrido láser invertido. Localice muestras correctamente organizadas y orientadas utilizando los modos de epifluorescencia o microscopía de campo claro en el microscopio con objetivos de aire de bajo aumento (5x o 10x) o aumento medio (20x).
- Para obtener imágenes en alta resolución, cambie a un objetivo de inmersión en aceite o agua de gran aumento (60x, 63x o 100x). La superficie de los embriones debe estar dentro del rango de enfoque del objetivo (<300 μm desde el cubreobjetos para un objetivo de 63x) para poder ser fotografiados.
- Recopile datos de muestras utilizando el modo confocal de escaneo láser en el microscopio. Asegúrese de recopilar imágenes no saturadas con un buen rango dinámico y utilice un número adecuado de píxeles por imagen para capturar la máxima información posible sobre la muestra23.

Figura 2: Desvitelinización manual y trabajo con hidrogeles . (A) Cortar una losa de agar de un plato de agar/jugo de frutas. (B) Colocar cinta adhesiva de doble cara dentro de la tapa de una placa de Petri de 6 cm. (C) Adherir embriones a la tapa sellada. (D) Una losa de PDMS con un pozo cuadrado adherido a un cubreobjetos de 22 mm x 22 mm. (E) Cubra con un pocillo PDMS dentro de una placa de 6 pocillos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Resultados
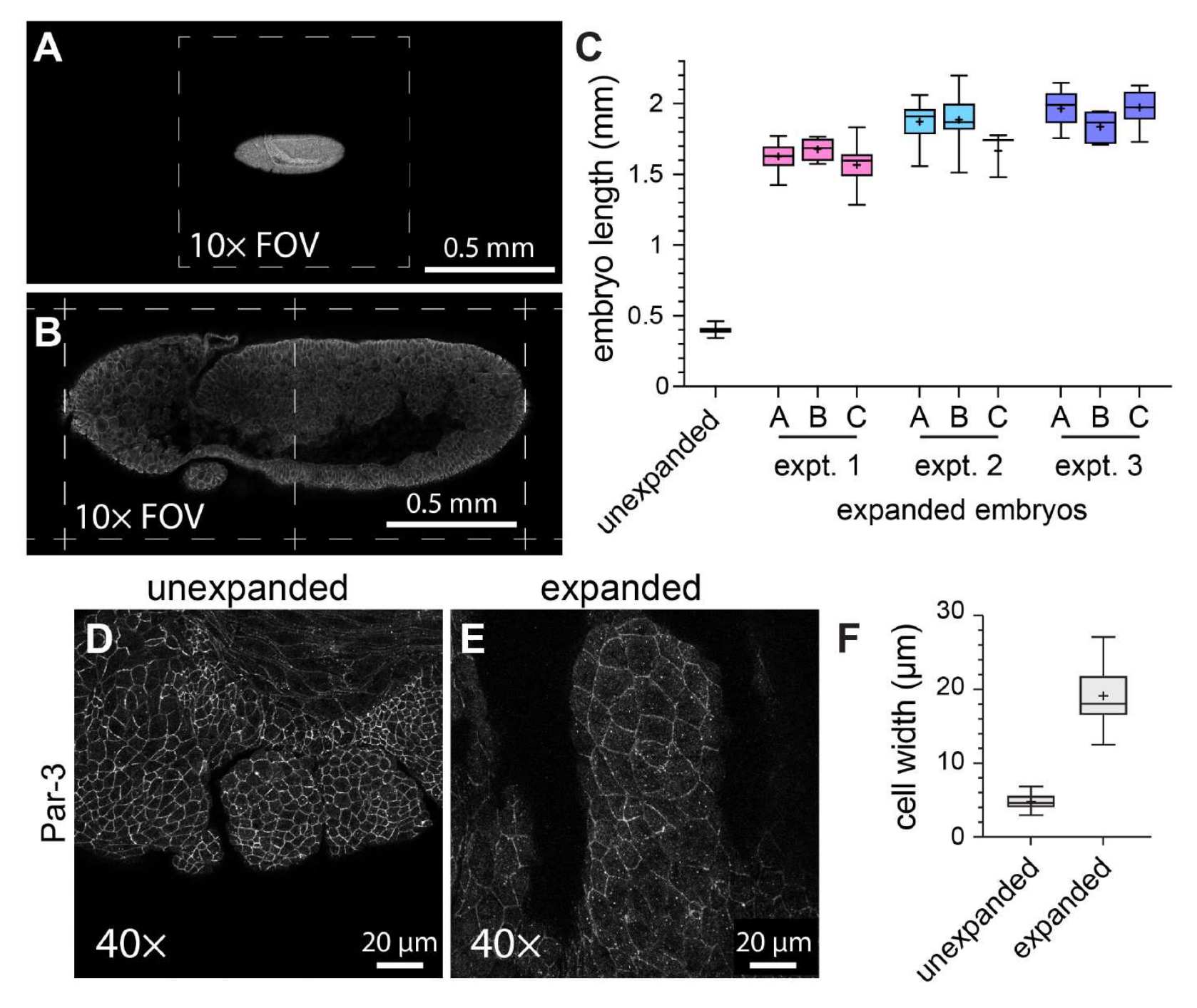
Para caracterizar la eficacia general de ExM en embriones de Drosophila de montaje entero, se midió la longitud del embrión a lo largo del eje cabeza-cola en embriones de control no expandidos frente a embriones expandidos (Figura 3A-C). Los embriones de control no expandidos se sometieron a las mismas condiciones de fijación y pasos de marcaje de inmunofluorescencia que los embriones expandidos, excepto que se montaron utilizando un medio de montaje solidificado antes de la obtención de imágenes. Los embriones individuales no expandidos abarcaron aproximadamente la mitad de un campo de visión cuando se utilizó un objetivo de 10x (Figura 3A). Por el contrario, los embriones expandidos abarcaron aproximadamente dos campos de visión completos cuando se utilizó el mismo objetivo de 10x (Figura 3B). Para evaluar cómo variaba el grado de expansión tanto dentro de los experimentos como entre ellos, se realizó el mismo protocolo ExM en tres ocasiones distintas, y se midió la longitud del embrión en tres geles diferentes dentro de cada experimento individual. La longitud media de la cabeza a la cola de los embriones control no expandidos fue de 398,8 μm (desviación estándar [DE] = 22,93 μm; n = 74; Figura 3C). Para el experimento 1, el experimento 2 y el experimento 3, la longitud media de los embriones fue de 1.596 μm (DE = 159,9 μm; n = 57), 1.868 μm (DE = 150,5 μm; n = 51) y 1.954 μm (DE = 120,3 μm; n = 44), respectivamente, lo que representa factores de expansión de 4,0, 4,7 veces y 4,9 veces, respectivamente (Figura 3C). La variación intraexperimental entre los geles fue mucho menos notable que la variación interexperimental, que fue de aproximadamente el 20% (Figura 3C). Para evaluar los efectos de ExM en la morfología celular y embrionaria, se utilizó un anticuerpo contra el componente de unión adherente Par-3 (Bazooka)21 para marcar las membranas celulares apicales, y obtuvimos imágenes de los segmentos bucales en desarrollo de embriones de Drosophila en etapa 11, una etapa con una estructura segmentada compleja (Figura 3D-F). En la muestra control, las células del segmento maxilar tenían un ancho promedio de 4,76 μm (DE = 1,053 μm, n = 25; Figura 3D,F). En las muestras expandidas de las que se obtuvieron imágenes utilizando el mismo objetivo de 40x y el mismo factor de zoom (1x), las células del segmento maxilar tenían una anchura media de 19,10 μm (DE = 3,966 μm, n = 18; Figura 3E,F), lo que representa una expansión de 4,0 veces. Por lo tanto, de acuerdo con informes anteriores11, pudimos expandir los embriones de Drosophila de montaje completo aproximadamente cuatro veces en dimensiones lineales utilizando ExM sin desgarro de la muestra ni distorsiones obvias en la morfología celular o tisular.

Figura 3: Expansión cuádruple de embriones de Drosophila. (A) Embriones de Drosophila no expandidos y (B) expandidos fotografiados con un objetivo de 10x (0,3 NA) con un zoom de 1x. Los campos de visión individuales (FOV) se indican con líneas discontinuas. Los embriones expresaron una versión marcada con GFP de la cadena ligera de miosina y se tiñeron con un anticuerpo anti-GFP. (C) Cuantificación de la longitud del embrión (a lo largo del eje cabeza-cola) en tres hidrogeles por experimento y a partir de tres experimentos ExM separados en comparación con controles no expandidos. (D,E) Segmentos maxilares de embriones de Drosophila en estadio 11 (D) no expandidos y (E) expandidos fotografiados con un objetivo de 40x (1,3 NA) con un zoom de 1x. Los contornos celulares (uniones adherentes) se detectaron con un anticuerpo anti Par-3/Bazooka (blanco). (F) Cuantificación de la anchura de la célula (eje largo) a partir de grupos equivalentes de células de (D) y (E). Los diagramas de caja de (C) y (F) muestran los rangos de percentiles 25, 50 y 75; los bigotes indican los valores mínimo y máximo; Los símbolos "+" indican la media. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Para demostrar que ExM se puede utilizar para resolver detalles subcelulares por debajo del límite de difracción típico, se obtuvieron imágenes del citoesqueleto de actomiosina en un control no expandido frente a embriones expandidos que se sometieron a una extensión convergente (etapa 7). Los eventos de remodelación tisular de la gastrulación y la extensión convergente están controlados en gran medida por cambios en la localización de la proteína motora miosina II24. Sin embargo, en el epitelio cilíndrico densamente empaquetado del ectodermo primitivo de Drosophila , es difícil observar muchos detalles finos del patrón de localización de la miosina II, incluso cuando se obtienen imágenes con un aumento de 158x (objetivo de 63x con un zoom óptico de 2,5x), un poder de resolución máximo típico para un microscopio confocal de barrido láser. Por ejemplo, debido a que la miosina II es una proteína cortical (ubicada directamente debajo de la membrana plasmática), los grupos de miosina II25 ubicados a ambos lados de los contactos célula-célula no se resolvieron en los embriones en etapa 7, y aparecieron como una sola línea donde se encontraron las células vecinas (Figura 4A). Por el contrario, en embriones expandidos en estadio 7, se pudieron observar líneas paralelas de miosina II en las uniones célula-célula, que representan grupos de proteínas corticales en células adyacentes (Figura 4B). La distancia entre las líneas paralelas de miosina II en muestras expandidas fue de 892,7 nm (DE = 0,171 nm, n = 12); cuando se divide por cuatro, esto produce una distancia predicha de ~ 220 nm entre las líneas de miosina en células adyacentes en embriones no expandidos, que de hecho está justo por debajo del límite de difracción para una señal detectada con Alexa 488 (emisión máxima de ~ 520 nm / 2 = 260 nm).
Además, también probamos si ExM podría usarse para resolver la arquitectura de la red mitocondrial en células densamente empaquetadas de embriones de Drosophila gastrulados (etapa 6). La función mitocondrial está estrechamente relacionada con la estructura de la red (es decir, orgánulos fusionados frente a orgánulos fragmentados), pero los detalles de la organización de la red mitocondrial son difíciles de visualizar utilizando la microscopía confocal convencional en tipos celulares que no son planos y/o delgados. Las mitocondrias son naturalmente ricas en moléculas biotiniladas y, por lo tanto, las mitocondrias se pueden marcar en el embrión temprano de Drosophila utilizando estreptavidina26 marcada con fluorescencia. En los embriones no expandidos en estadio 6 marcados con estreptavidina-Alexa 488, la señal aparecía como puntos citoplasmáticos que a menudo se superponían y eran difíciles de resolver (Figura 4C). Por el contrario, en los embriones expandidos en estadio 6, muchos más detalles finos de la red mitocondrial eran visibles y los puntos eran más fácilmente resolubles (Figura 4D)26,27. Estos resultados indican que ExM se puede utilizar para estudiar la organización de la red mitocondrial en tipos celulares que tradicionalmente no son adecuados para el análisis mitocondrial.

Figura 4: Detalles del citoesqueleto de actomiosina y las mitocondrias revelados por microscopía de expansión. (A,B) Localización de la miosina II en células de neuroectodermo (banda germinal) fotografiadas con un objetivo de 63x (1,4 NA) con un zoom de 2,5x en embriones no expandidos y (B) expandidos en estadio 7 (A) y (B) expandidos. Se detectó miosina II en los embriones que expresaban una versión transgénica marcada con GFP de la cadena ligera reguladora de la miosina II (sqh-GFP), que se detectó con un anticuerpo anti-GFP (rojo). En el embrión expandido se pueden resolver distintos grupos de miosina cortical localizados en células adyacentes (flechas blancas). (C,D) Redes mitocondriales en células de neuroectodermo fotografiadas con un objetivo del 63× (1,4 NA) con un zoom de 2,5x en embriones no expandidos (C) y expandidos (D) en estadio 6. Las mitocondrias se detectaron con estreptavidina-Alexa 488 (verde) y los contornos celulares se detectaron con un anticuerpo anti-Par-3/Bazooka (magenta). Los experimentos se realizaron con un microscopio confocal de barrido láser. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla 1: Recetas de solución. Composición de las soluciones utilizadas en este protocolo por orden de aparición. Todas las existencias son líquidas a menos que se indique lo contrario. Los productos químicos se resuspendieron o diluyeron en agua filtrada esterilizada en autoclave, a menos que se indique lo contrario. Haga clic aquí para descargar esta tabla.
Discusión
Desvitelinización manual
La mayoría de los protocolos de fijación de embriones de Drosophila implican la eliminación de la membrana vitelina mediante la agitación de los embriones fijados en una emulsión de metanol y heptano, lo que hace que las membranas se rompan a través de la ruptura osmótica26. Si bien la desvitelinización a base de metanol (estallido de metanol) es efectiva y apropiada para muchas aplicaciones, la desvitelinización manual (pelado a mano) ofrece algunas ventajas significativas. En primer lugar, el pelado manual permite elegir embriones en etapas precisas para desvellinizar y recolectar, lo que aumenta en gran medida la probabilidad de obtener embriones expandidos en una orientación utilizable al final del experimento. Este enriquecimiento es fundamental cuando se estudian aspectos específicos de los procesos de desarrollo rápido (por ejemplo, la invaginación del mesodermo o la extensión convergente), para los cuales los embriones en estadios apropiados pueden representar solo un pequeño porcentaje de todos los embriones, incluso dentro de una ventana de recolección de tiempo apretado. Por supuesto, para muchas aplicaciones, el estallido más tradicional de metanol a granel de embriones desde una ventana de recolección cronometrada será suficiente, y es posible que el peeling a mano no valga la pena el esfuerzo adicional. En segundo lugar, la unión de ciertos anticuerpos primarios y colorantes se ve afectada negativamente por la exposición previa de la muestra al metanol. Por esta razón, el peeling manual puede producir aumentos significativos en la calidad de la señal de inmunofluorescencia en comparación con las muestras reventadas con metanol, lo que lo convierte en una técnica general útil para los biólogos del desarrollo de Drosophila .
Microscopía confocal de alta resolución en embriones expandidos de Drosophila de montaje entero
Si bien la realización de microscopía confocal de alta resolución en muestras expandidas es conceptualmente la misma que en muestras no expandidas, ExM presenta algunos obstáculos técnicos. En particular, la orientación del embrión, que es aleatoria, se vuelve aún más importante a medida que aumenta el tamaño de la muestra, porque los objetivos de gran aumento y alta NA solo pueden enfocar la luz de las regiones de la muestra que están muy cerca del cubreobjetos27. Por lo tanto, por lo general, solo es posible enfocarse en las células en o cerca de la superficie del embrión que terminaron adyacentes al cubreobjetos cuando se formó el gel. La mejor manera de asegurarse de que haya especímenes de la orientación correcta al final es comenzar el protocolo ExM con una colección de embriones fijos en etapas compactas (p. ej., mediante el uso de pelado manual) y sembrar muchos embriones en cada pocillo (>10). Para visualizar las células en las profundidades del interior del embrión, puede ser necesario utilizar configuraciones de imagen más especializadas, como la microscopía de lámina de luz28. Además, encontramos que la calidad de la imagen se puede mejorar abriendo el orificio confocal a un tamaño mayor que una unidad aireada. Por supuesto, un aumento en el tamaño de un agujero de alfiler se producirá a costa de una disminución de la resolución máxima, pero en la práctica, incluso pequeños aumentos en el tamaño del agujero de alfiler pueden aumentar significativamente la intensidad de la señal (datos no mostrados). Los estudios futuros deben abordar sistemáticamente el tamaño del agujero de alfiler y la resolución efectiva en muestras ExM.
Variaciones de la ExM básica
El protocolo descrito aquí es un ejemplo relativamente simple de ExM que debería funcionar para muchas aplicaciones y ser fácil de implementar en la mayoría de los laboratorios de biología del desarrollo. Sin embargo, existen numerosas variaciones sobre el concepto básico de ExM 4,5,7 que se pueden utilizar para aumentar la intensidad de la señal, lograr grados aún mayores de expansión y detectar moléculas de ácidos nucleicos y proteínas. En este protocolo, los embriones se incuban con anticuerpos antes de la gelificación y expansión. Alternativamente, las muestras pueden ser tratadas con anticuerpos después de que se expanden 6,30, lo que puede aumentar la intensidad de la señal debido a una mayor accesibilidad a los epítopos y una menor pérdida de anticuerpos unidos durante los pasos de expansión. Además, se pueden utilizar moléculas reticulantes específicas para unir moléculas de ARN al hidrogel para permitir la detección de ARN en geles expandidos utilizando el método de reacción en cadena de hibridación30. Por último, las muestras pueden someterse a múltiples rondas de expansión, como en la microscopía de expansión iterativa (iExM)31, pan-ExM32 y la expansión reveladora (ExR)31, para lograr grados aún más altos de mayor resolución.
Divulgaciones
Los autores no tienen conflictos de intereses que declarar.
Agradecimientos
Nos gustaría agradecer a la Dra. Jennifer Zallen por proporcionar el anticuerpo primario anti-Par-3 para conejillos de indias. Este trabajo contó con el apoyo de una generosa financiación (1R15GM143729-01 y 1P20GM139768-01 5743) del Instituto Nacional de Ciencias Médicas Generales (NIGMS), uno de los miembros de los Institutos Nacionales de Salud (NIH), así como del Instituto de Biociencias de Arkansas (ABI), que proporcionó fondos parciales para la compra de nuestro microscopio confocal.
Materiales
| Name | Company | Catalog Number | Comments |
| acrylamide | Milipore Sigma | 1490-100ML | |
| ammonium persulfate | VWR | BDH9214-500G | |
| anti-GFP rabbit polyclonal antibody | Torrey Pines BioLabs | TP-401 | |
| anti-guinea pig IgG goat polyclonal antibody, Alexa Fluor 568 | Thermo Fisher Scientific | A-11075 | |
| anti-rabbit IgG goat polyclonal antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11008 | |
| bisacrylamide | Research Products International | A11275 | |
| bovine serum albumin (30% solution) | Millipore Sigma | A7284 | |
| conical tubes, 50 mL | fisherscientific | 21008-940 | |
| coverlip glass, square 22 mm | VWR | 48366-227 | |
| coverslip glass, rectangular 40 mm x 24 mm | VWR | 48393-230 | |
| glass capillaries for pulling needles | World Precision Instruments | TW100F-4 | |
| glass microinjection needles (pre-pulled) | World Precision Instruments | TIP10LT | |
| guanidine HCl | VWR | 101970-606 | |
| heptane | VWR | EM-HX0078-1 | |
| latex pipet bulbs | VWR | 82024-554 | |
| methanol | VWR | BDH1135-4LP | |
| methylacylic acid N-hydroxysuccinimidyl ester | VWR | 730300-1G | |
| microfuge tube, 1.5 mL | VWR | 20170-038 | |
| multi-well plate, 6-well | Genesee | 25-100 | |
| paraformaldehyde (16%, EM-grade, methanol-free) | Electron Microscopy Sciences | 509804487 (Fisher) | |
| Pasteur pipet (2 mL, short tip) | VWR | 14673-010 | |
| PDMS kit (Sylgard 184 Kit, base and curing agent) | VWR | 102092-312 | |
| Petri plates | Genesee | 32-107 | |
| phosphate-buffered saline (10x solution) | VWR | 97063-660 | |
| Poly-L-lysine solution (0.1% solution) | VWR | P8920-1ooML | |
| Proteinase K | Thermo Fisher Scientific | E00491 | |
| scintillation vials (30 mL) | VWR | 66022-128 | |
| sodium acrylate | VWR | 101181-226 | |
| sodium azide (powder) | Millipore Sigma | 71289 | make a 1% w/v working stock; acute POISON at this concentration! |
| Streptavidin, Alexa Fluor 488 | Thermo Fisher Scientific | S32354 | |
| TAE (50x) | VWR | 97063-692 | |
| tape (double-sided, 1 inch wide) | Scotch 3M | 665 Scotch double sided 1inch/1296 inches Boxed | |
| TEMED | Thermo Fisher Scientific | PI17919 | |
| TEMPO | VWR | EM8.14681.0005 | catalytic oxidant |
| Tween-20 | VWR | 97063-872 | extremely viscous when pure; make a 10% working stock with water |
| Zeiss LSM 900 | Zeiss | Laser scanning microscope used without AiryScan |
Referencias
- Klar, T. A., Jakobs, S., Dyba, M., Egner, A., Hell, S. W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences of the United States of America. 97 (15), 8206-8210 (2000).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nature Methods. 3 (10), 793-796 (2006).
- Betzig, E. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Wassie, A. T., Zhao, Y., Boyden, E. S. Expansion microscopy: principles and uses in biological research. Nature Methods. 16 (1), 33-41 (2019).
- Chen, F., Tillberg, P. W., Boyden, E. S. Optical imaging. Expansion microscopy. Science. 347 (6221), 543-548 (2015).
- Chen, F., et al. Nanoscale imaging of RNA with expansion microscopy. Nature Methods. 13 (8), 679-684 (2016).
- Tillberg, P. W., et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature Biotechnology. 34 (9), 987-992 (2016).
- Chang, J. -. B. Iterative expansion microscopy. Nature Methods. 14 (6), 593-599 (2017).
- Cahoon, C. K., et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proceedings of the National Academy of Sciences of the United States of America. 114 (33), 6857-6866 (2017).
- Mosca, T. J., Luginbuhl, D. J., Wang, I. E., Luo, L. Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. eLife. 6, e27347 (2017).
- Jiang, N., et al. Superresolution imaging of Drosophila tissues using expansion microscopy. Molecular Biology of the Cell. 29 (12), 1413-1421 (2018).
- Yu, C. -. C. J., et al. Expansion microscopy of C. elegans. eLife. 9, e46249 (2020).
- Freifeld, L., et al. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proceedings of the National Academy of Sciences of the United States of America. 114 (50), E10799-E10808 (2017).
- Tanaka, T., et al. Phase transitions in ionic gels. Physical Review Letters. 45 (20), 1636-1639 (1980).
- Hausen, P., Dreyer, C. The use of polyacrylamide as an embedding medium for immunohistochemical studies of embryonic tissues. Stain Technology. 56 (5), 287-293 (1981).
- Campos-Ortega, J. A., Hartenstein, V. . The Embryonic Development of Drosophila melanogaster. , (2013).
- Chozinski, T. J., et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nature Methods. 13 (6), 485-488 (2016).
- Miller, D. F. B., Holtzman, S. L., Kaufman, T. C. Customized microinjection glass capillary needles for P-element transformations in Drosophila melanogaster. BioTechniques. 33 (2), 366-367 (2002).
- Rothwell, W. F., Sullivan, W. Drosophila embryo dechorionation. CSH Protocols. 2007, (2007).
- Cold Spring Harbor Protocols. . Drosophila apple juice-agar plates. , (2011).
- de Matos Simões, S., et al. Rho-kinase directs bazooka/Par-3 planar polarity during drosophila axis elongation. Developmental Cell. 19 (3), 377-388 (2010).
- Gambarotto, D., et al. Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nature Methods. 16 (1), 71-74 (2019).
- Paré, A. C., Zallen, J. A. Cellular, molecular, and biophysical control of epithelial cell intercalation. Current Topics in Developmental Biology. 136, 167-193 (2020).
- Royou, A., Field, C., Sisson, J. C., Sullivan, W., Karess, R. Reassessing the role and dynamics of nonmuscle myosin II during furrow formation in early Drosophila embryos. Molecular Biology of the Cell. 15 (2), 838-850 (2004).
- Chowdhary, S., Tomer, D., Dubal, D., Sambre, D., Rikhy, R. Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Scientific Reports. 7 (1), 5502 (2017).
- Chowdhary, S., Madan, S., Tomer, D., Mavrakis, M., Rikhy, R. Mitochondrial morphology and activity regulate furrow ingression and contractile ring dynamics in Drosophila cellularization. Molecular Biology of the Cell. 31 (21), 2331-2347 (2020).
- Stelzer, E. H. K., et al. Light sheet fluorescence microscopy. Nature Reviews Methods Primers. 1, 73 (2021).
- Ku, T., et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nature Biotechnology. 34 (9), 973-981 (2016).
- Wen, G., et al. A Universal labeling strategy for nucleic acids in expansion microscopy. Journal of the American Chemical Society. 143 (34), 13782-13789 (2021).
- M'Saad, O., Bewersdorf, J. Light microscopy of proteins in their ultrastructural context. Nature Communications. 11 (1), 3850 (2020).
- Sarkar, D. Expansion revealing: Decrowding proteins to unmask invisible brain nanostructures. bioRxiv. , (2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados