
Method Article
Verwendung von Expansionsmikroskopie zur physikalischen Vergrößerung ganzer Drosophila-Embryonen für hochauflösende Bildgebung
In diesem Artikel
Zusammenfassung
In dieser Arbeit wird ein Protokoll für die Implementierung der Expansionsmikroskopie in frühen Drosophila-Embryonen vorgestellt, um eine hochauflösende Bildgebung mit einem konventionellen konfokalen Laser-Scanning-Mikroskop zu erreichen.
Zusammenfassung
Das Arbeitspferd der Entwicklungsbiologie ist das konfokale Mikroskop, mit dem Forscher die dreidimensionale Lokalisierung markierter Moleküle in komplexen biologischen Proben bestimmen können. Während herkömmliche konfokale Mikroskope es ermöglichen, zwei benachbarte Fluoreszenzpunktquellen aufzulösen, die nur wenige hundert Nanometer voneinander entfernt sind, erfordert die Beobachtung der feineren Details der subzellulären Biologie die Fähigkeit, Signale in der Größenordnung von zehn Nanometern aufzulösen. Es wurden zahlreiche hardwarebasierte Methoden für die hochauflösende Mikroskopie entwickelt, die es Forschern ermöglichen, solche Auflösungsgrenzen zu umgehen, obwohl diese Methoden spezielle Mikroskope erfordern, die nicht allen Forschern zur Verfügung stehen. Eine alternative Methode zur Erhöhung des Auflösungsvermögens ist die isotrope Vergrößerung der Probe selbst durch ein Verfahren, das als Expansionsmikroskopie (ExM) bekannt ist und erstmals 2015 von der Boyden-Gruppe beschrieben wurde. ExM ist keine Art der Mikroskopie an sich , sondern vielmehr eine Methode zum Aufquellen einer Probe unter Beibehaltung der relativen räumlichen Organisation ihrer konstituierenden Moleküle. Die expandierte Probe kann dann mit einem herkömmlichen konfokalen Mikroskop mit einer effektiv erhöhten Auflösung betrachtet werden. In dieser Arbeit beschreiben wir ein Protokoll zur Implementierung von ExM in Whole-Mount Drosophila-Embryonen , das verwendet wird, um die Lokalisation von Par-3, Myosin II und Mitochondrien in den Oberflächenepithelzellen zu untersuchen. Dieses Protokoll führt zu einer etwa vierfachen Vergrößerung der Probengröße und ermöglicht die Detektion subzellulärer Details, die mit herkömmlicher konfokaler Mikroskopie nicht sichtbar sind. Als Proof of Principle wird ein Anti-GFP-Antikörper verwendet, um unterschiedliche Myosin-GFP-Pools zwischen benachbarten Zellkortexen zu unterscheiden, und fluoreszenzmarkiertes Streptavidin wird verwendet, um endogene biotinylierte Moleküle zu detektieren, um die feinen Details der mitochondrialen Netzwerkarchitektur aufzudecken. Dieses Protokoll verwendet gängige Antikörper und Reagenzien für die Fluoreszenzmarkierung und sollte mit vielen bestehenden Immunfluoreszenzprotokollen kompatibel sein.
Einleitung
In der Zell- und Entwicklungsbiologie ist Sehen Glauben, und die Fähigkeit, die Lokalisierungsmuster von Proteinen genau zu bestimmen, ist für viele Arten von Experimenten von grundlegender Bedeutung. Die konfokale Laser-Scanning-Mikroskopie ist das Standardwerkzeug für die dreidimensionale Abbildung fluoreszenzmarkierter Proteine in intakten Proben. Herkömmliche konfokale Mikroskope sind nicht in der Lage, benachbarte Fluoreszenzsignale zu unterscheiden (aufzulösen), die weniger als die Hälfte der Wellenlänge des von ihnen emittierten Lichts voneinander entfernt sind1. Mit anderen Worten, zwei Punktquellen müssen in lateraler Richtung mindestens 200-300 nm (500-700 nm in axialer Richtung) voneinander entfernt sein, um sie als zwei unterschiedliche Signale aufzulösen. Diese technische Barriere wird als Beugungsgrenze bezeichnet und stellt eine grundlegende Hürde für die Untersuchung komplexer subzellulärer Strukturen (z. B. des Aktomyosin-Zytoskeletts oder der mitochondrialen Netzwerke) mit räumlichen Merkmalen unterhalb der Beugungsgrenze dar. Daher sind Techniken zur Erhöhung des Auflösungsvermögens herkömmlicher konfokaler Mikroskope von allgemeinem Interesse für die biologische Gemeinschaft.
Um die Beugungsgrenze zu umgehen, wurden eine Reihe verschiedener hochauflösender Mikroskopietechnologien entwickelt, die eine Auflösung in der Größenordnung von zehn Nanometern oder weniger 1,2,3 ermöglichen und eine Welt biologischer Komplexität offenbaren, die bisher nur durch Elektronenmikroskopie zugänglich war. Trotz der offensichtlichen Vorteile dieser hardwarebasierten Methoden haben hochauflösende Mikroskope oft spezifische Anforderungen an die Probenmarkierung und lange Aufnahmezeiten, was ihre Flexibilität einschränkt oder für einige Labore einfach zu teuer ist. Eine Alternative zur mikroskopischen Superauflösung ist die Expansionsmikroskopie (ExM), bei der es sich nicht um eine Art der Mikroskopie an sich handelt, sondern um eine Methode zum Aufquellen einer Probe unter Beibehaltung der relativen räumlichen Organisation ihrer konstituierenden Moleküle4. Die isotrop expandierten Proben können dann mit einem herkömmlichen konfokalen Fluoreszenzmikroskop mit einer effektiv erhöhten Auflösung betrachtet werden. ExM wurde erstmals 2015 von der Boyden-Gruppe beschrieben5, und die grundlegende Technik wurde seitdem für den Einsatz in einer Vielzahl von Experimenten angepasst 6,7,8. ExM wurde auch für den Einsatz in Ganzkörperembryonen angepasst, insbesondere in Drosophila 9,10,11, C. elegans12 und Zebrafisch 13, was es zu einem leistungsstarken Werkzeug für Entwicklungsbiologen macht.
ExM basiert auf zwei verschiedenen Hydrogelchemien: 1) quellfähigen Polyelektrolyt-Hydrogelen, die stark an Größe zunehmen, wenn sie in Wasser eingeweicht werden14, und 2) Polyacrylamid-Hydrogele, die einen extrem kleinen Polymerabstand haben, um eine isotrope Probenexpansion zu ermöglichen15. Es gibt zwar viele veröffentlichte ExM-Protokolle, aber sie haben im Allgemeinen die folgenden Schritte gemeinsam: Probenfixierung, Markierung, Aktivierung, Gelierung, Verdauung und Expansion4. Die Fixierungsbedingungen und Fluoreszenzmarkierungsstrategien variieren natürlich je nach den Anforderungen des Experiments und des Systems, und in einigen Protokollen erfolgt die Markierung nach der Expansion. Die Zielmoleküle in der Probe müssen für die Bindung an das Hydrogel vorbereitet (aktiviert) werden, was mit verschiedenen Chemikalien erreicht werdenkann 4. Während der Gelierungsschritte wird die Probe mit Monomeren des zukünftigen Hydrogels gesättigt (Natriumacrylat, Acrylamid und der Vernetzer Bisacrylamid), und das Hydrogel wird dann durch radikalische Polymerisation gebildet, die durch einen Initiator, wie Ammoniumpersulfat (APS), und einen Beschleuniger, wie Tetramethylendiamin (TEMED)4, katalysiert wird. Nach der Gelierung wird die Probe enzymatisch aufgeschlossen, um die Quellbeständigkeit der Probe zu homogenisieren und die isotrope Ausdehnung des Hydrogels4 zu gewährleisten. Schließlich wird das verdaute Hydrogel in Wasser gelegt, wodurch es sich auf etwa das Vierfache seiner ursprünglichen linearen Größe4 ausdehnt.

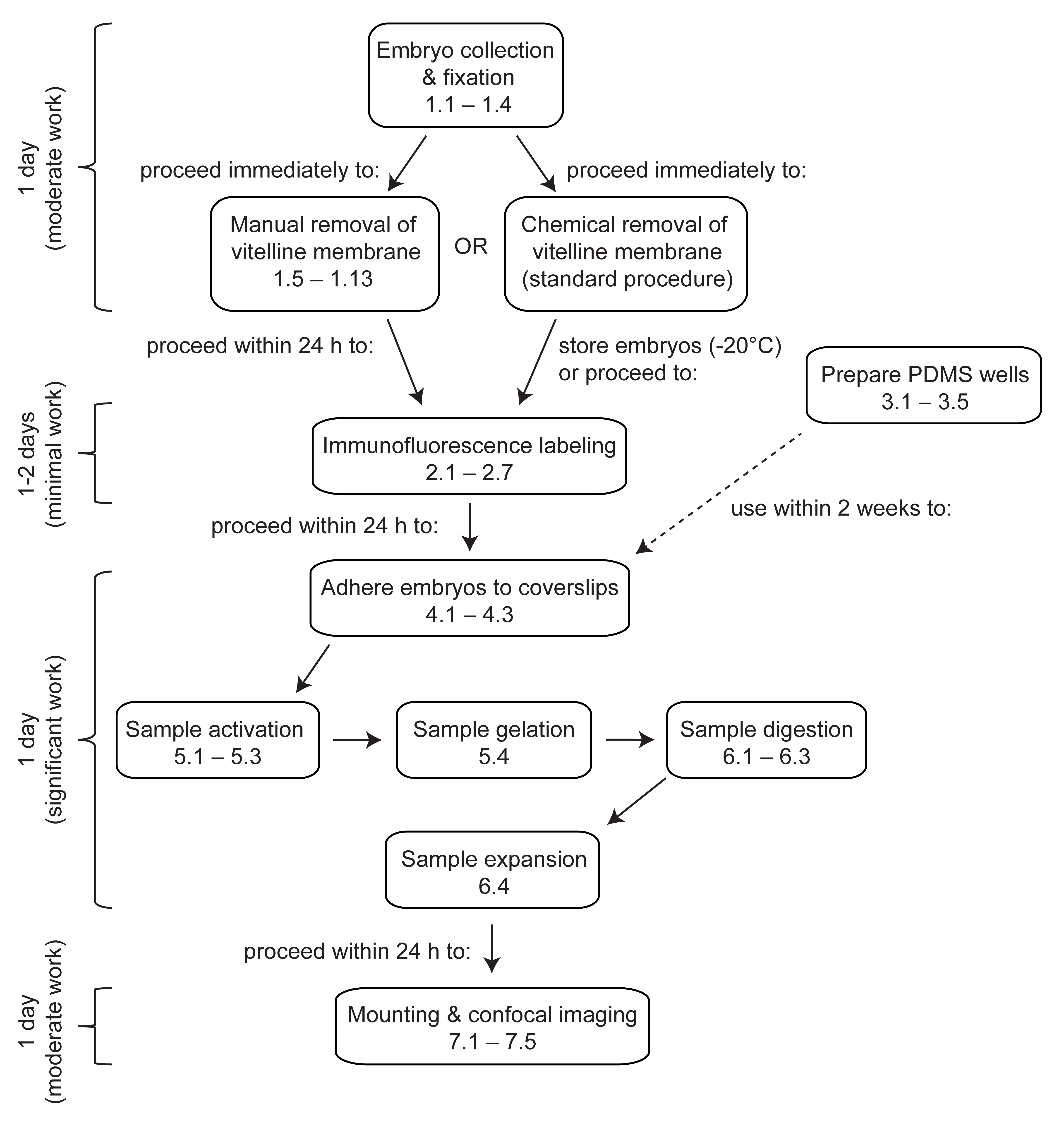
Abbildung 1: Überblick über die Expansionsmikroskopie in Drosophila-Embryonen . ExM ist ein mehrstufiges Protokoll, das mindestens 4 Tage in Anspruch nimmt. Die Entnahme, Fixierung und Devitellinisierung von Embryonen dauert 1 Tag oder länger, je nachdem, ob Embryonen aus mehreren Sammlungen gepoolt werden. Die Immunfluoreszenzmarkierung dauert 1 Tag oder 2 Tage, je nachdem, ob die Embryonen über Nacht mit den primären Antikörpern inkubiert werden. Die Aktivierung, Gelierung, Verdauung und Expansion des Embryos kann an einem einzigen Tag durchgeführt werden. Die Gele können sofort nach der Expansion montiert und abgebildet werden, obwohl es aus praktischen Gründen oft wünschenswert ist, am nächsten Tag mit der Bildgebung zu beginnen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Dieses Protokoll beschreibt, wie ExM an Drosophila-Embryonen im frühen bis mittleren Stadium durchgeführt wird16 , um subzelluläre Proteinlokalisierungsmuster mit Superauflösung zu visualisieren (Abbildung 1). Diese Methode verwendet die Chemie des Methylacrylsäure-N-Hydroxysuccinimidylesters (MA-NHS), um Proteinmoleküle zu aktivieren und an dem Hydrogel zu verankern17, und es handelt sich um eine Modifikation eines zuvor veröffentlichten ExM-Protokolls für die Verwendung in Drosophila-Embryonen und -Geweben im späten Stadium11. Dieses Protokoll verwendet Polydimethylsiloxan (PDMS)-Wells, um die Hydrogele zu formen und den Lösungsaustausch während der Aktivierung und Gelierung zu erleichtern. Ein alternatives Verfahren, das die Erzeugung von PDMS-Vertiefungen nicht erfordert, beinhaltet das Absenken von Embryonen, die an Deckgläsern befestigt sind, in Tropfen der Monomerlösung, die auf einem Stück Laborversiegelungsfolie22 sitzen. Darüber hinaus beschreibt dieses Protokoll eine Methode zur manuellen Entfernung der undurchlässigen Vitellinmembran, die Drosophila-Embryonen umgibt, was eine Voraussetzung für die Immunfluoreszenzfärbung ist. Wichtig ist, dass diese Methode des manuellen Schälens von Embryonen verwendet werden kann, um vor der Probenmarkierung nur richtig stadifizierte Drosophila-Embryonen auszuwählen, was die Wahrscheinlichkeit, dass am Ende erweiterte Proben des richtigen Stadiums und der richtigen Ausrichtung erhalten, erheblich erhöht und somit die nachgelagerte Datenerfassung viel effizienter macht.
Protokoll
Dieses Protokoll folgt den Richtlinien der University of Arkansas (UARK) für die Forschung an wirbellosen Tieren, wie z. B. Drosophila melanogaster, und wurde vom UARK Institutional Biosafety Committee genehmigt (Protokoll #20001).
1. Fixierung und Devitellinisierung von Drosophila-Embryonen
HINWEIS: Schritt 1 beschreibt ein Verfahren (Handpeeling) zur manuellen Entfernung der Vitellinmembran, einer transparenten, undurchlässigen Membran, die den Embryo umgibt. Wichtig ist, dass das Peeling von Hand die Auswahl von Embryonen in einem ordnungsgemäßen Stadium zu Beginn des ExM-Protokolls ermöglicht, wodurch die Wahrscheinlichkeit, am Ende des ExM-Protokolls Embryonen in einer brauchbaren Ausrichtung zu erhalten, erheblich erhöht wird. Dieses ExM-Protokoll ist jedoch vollständig kompatibel mit der Entnahme von Embryonen in großen Mengen und den Standardverfahren für die Entfernung der Vitellinmembran auf Methanolbasis, in diesem Fall kann man direkt zu Schritt 2 (Immunfluoreszenzmarkierung) übergehen.
- Bereiten Sie eine Anzahl feiner Glasnadeln vor oder kaufen Sie sie. Die tatsächlichen Abmessungen der Nadelspitze sind nicht entscheidend, stellen aber sicher, dass die Nadeln starr und scharf genug sind, um die Vitellinmembranen von fixierten Embryonen zu durchdringen. Herstellung von Nadeln aus Glaskapillarröhrchen (1 mm Außendurchmesser, 0,75 mm Innendurchmesser) mit einem Mikropipettenzieher, wie man Nadeln für die Mikroinjektion von Embryonen vorbereitenwürde 18; Alternativ können Sie auch vorgezogene Nadeln kaufen.
- Embryonen werden mit Standard-Drosophila-Techniken19 entnommen, indem >100 erwachsene Drosophila in einen belüfteten Plastikbecher gegeben werden, der mit einer Fruchtsaft-/Agarplatte20 verschlossen ist. Verwenden Sie zeitgesteuerte Entnahmefenster, um Embryonen des richtigen Stadiums16 anzureichern. Um zum Beispiel für die Phase der Gastrulation (Stadium 6) und der konvergenten Extension (Stadium 7) anzureichern, wechseln Sie die Fruchtsaftplatte, sammeln Sie die Embryonen für 2 Stunden bei 25 °C, entfernen Sie dann die Platte und lassen Sie sie weitere 2 Stunden bei 25 °C altern, um Embryonen zu erhalten, die ~2-4 Stunden alt sind.
- Um die eierschalenartige Chorion von den Embryonen zu entfernen, bedecken Sie die Oberfläche der Fruchtsaftplatten mit 50%igem Bleichmittel (Tabelle 1), lösen Sie die Embryonen von der Oberfläche des Agars, indem Sie sie mit einem kleinen Pinsel bewegen, und warten Sie 3 Minuten, bis sich die Chorion aufgelöst hat.
- Übertragen Sie die dechorionierten Embryonen mit einem Pinsel in ein 30-ml-Szintillationsfläschchen, das 4 ml Heptan (organische Oberphase) und 4 ml Fixierungspuffer (wässrige Unterphase; Tabelle 1). Verdünnen Sie das Formaldehyd frisch aus einer frisch zubereiteten oder kürzlich geöffneten Brühe und mischen Sie es mit 10x PBS und deionisiertem Wasser unmittelbar vor der Zugabe der Embryonen.
HINWEIS: Bereiten Sie das Formaldehyd aus Paraformaldehydkraft oder 16% EM-Formaldehyd in Glasampullen vor. Vorräte an konzentriertem Formaldehyd (z. B. 37 % Formaldehyd) können verwendet werden, obwohl die Ergebnisse möglicherweise weniger konsistent sind. - Die Embryonen sammeln sich an der Schnittstelle zwischen der organischen und der wässrigen Phase an. Fügen Sie so viele Embryonen hinzu, wie eine einzige Schicht an der Schnittstelle bildet. Wenn zu viele Embryonen in ein Fläschchen gegeben werden, werden sie nicht so gut fixiert.
- Fixieren Sie die Szintillationsfläschchen mit starkem Klebeband auf einem Tischschüttler auf der Seite und schütteln Sie sie 20 Minuten lang bei 220 U/min. Für eine optimale Fixierung ist während der gesamten Fixierung eine kräftige Emulsion zwischen der organischen und der wässrigen Phase beizubehalten.
- Bereiten Sie während der Fixierungszeit für jede der Proben eine der folgenden Präparate vor.
- Nimm einen 6 cm großen Petrischalenboden aus Kunststoff, der zur Hälfte mit 3%igem Agar gefüllt ist, und ritze mit einer Rasierklinge oder einem Skalpell ein Rechteck von ~5 cm x 3 cm in den Agar. Hierfür können auch Fruchtsaft-/Agarplatten verwendet werden.
- Mit einem kleinen Laborspatel die Agar-Agarplatte entfernen. Drehen Sie den Boden der Petrischale um und stellen Sie sie auf die Bank. Legen Sie die Agarplatte auf die umgedrehte Schale (Abbildung 2A).
- Nehmen Sie den Deckel der Petrischale und stellen Sie sicher, dass er trocken ist. Tragen Sie Handschuhe und legen Sie ein Stück doppelseitiges Klebeband in den Deckel (das Stück Klebeband sollte etwas größer sein als die Agar-Platte; Abbildung 2B).
- Nehmen Sie die Fläschchen aus dem Shaker, stellen Sie sie aufrecht auf die Bank und lassen Sie die organische und die wässrige Phase trennen. Richtig fixierte Embryonen verbleiben an der Schnittstelle zwischen den beiden Phasen.
- Die fixierten Embryonen werden mit einer Pasteur-Glaspipette mit einer Latexbirne auf die Agarplatte übertragen. Um zu verhindern, dass die Embryonen an der Innenseite der Pipette haften, versuchen Sie, die Embryonen innerhalb des schmalen Halses der Pipette zu halten, und übertragen Sie die Embryonen in mehreren kleinen Chargen und nicht alle auf einmal. Sobald sich alle Embryonen auf der Agarplatte befinden, entfernen Sie den größten Teil des restlichen Heptans mit einer P200-Pipette aus der Umgebung der Embryonen. Führen Sie diesen Schritt so schnell wie möglich durch (<3 Minuten), um ein Austrocknen der fixierten Embryonen zu vermeiden, was sich negativ auf die Morphologie auswirken kann.
- Aus einer Höhe von ~2 cm lassen Sie den Deckel mit dem doppelseitigen Klebeband auf die Agarplatte fallen, um die Embryonen auf das Klebeband zu kleben (Abbildung 2C). Nehmen Sie vorsichtig den Deckel von der Agarplatte ab, legen Sie sie kopfüber auf die Bank und fügen Sie dann genügend PBS-Tween hinzu (Tabelle 1), um die Embryonen im Deckel zu bedecken.
- Identifizieren Sie mit einem Stereo-Präpariermikroskop bei etwa 100-facher Vergrößerung und indirekter Beleuchtung ordnungsgemäß stadifizierte Embryonen anhand morphologischer Marker. Verwenden Sie für Embryonen im Stadium 6 Marker wie eine sichtbare Kopffurche und ein einstülptes Mesoderm; Verwenden Sie für Embryonen im Stadium 7 Marker wie ein verlängertes Keimband; Verwenden Sie für Embryonen im Stadium 11 Marker wie einen vollständig verlängerten Kemmand und eine sichtbare Segmentierung entlang der Kopf-Schwanz-Achse16.
- Um die gewünschten Embryonen zu sammeln, stechen Sie zunächst mit einer feinen Glasnadel in die Vitellinmembran (eine transparente ovale Membran um den Embryo) in der Nähe des vorderen oder hinteren Endes des Embryos; Die Membran entleert sich ein wenig, wenn der Druck nachlässt. Schieben Sie dann mit einer feinen Pinzette oder einer Metallsonde den Embryo am anderen Ende vorsichtig durch das Loch. Die Vitelline-Membran bleibt auf dem doppelseitigen Klebeband haften. Lassen Sie unerwünschte Embryonen auf dem Klebeband kleben.
- Entnehmen Sie regelmäßig die schwimmenden devitellinisierten Embryonen mit einer Pasteurpipette aus Glas und bringen Sie sie in ein 1,5-ml-Mikrofugenröhrchen.
- Führen Sie an dieser Stelle einen der folgenden Schritte aus.
- Fahren Sie direkt mit den Schritten zur Immunfluoreszenzmarkierung fort. Die devitellinisierten Embryonen können direkt in die Blockierungslösung überführt werden (Schritt 2.2). Lassen Sie die Embryonen nicht länger als 16 Stunden in PBS-Tween oder Blocklösung (Tabelle 1) verbleiben, bevor Sie mit dem nächsten Schritt fortfahren.
- Bringen Sie die Embryonen zur Lagerung in Methanol. Entfernen Sie so viel PBS-Tween wie möglich und fügen Sie dann 1 ml Methanol hinzu. Sobald sich die Embryonen beruhigt haben, entfernen Sie so viel Methanol wie möglich und fügen Sie 1 ml frisches Methanol hinzu. Lagern Sie die Embryonen auf unbestimmte Zeit bei −20 °C. Die Lagerung von Methanol ermöglicht auch das Poolen von Embryonen aus mehreren Sammlungen.
2. Immunfluoreszenz-Markierung
HINWEIS: Abgesehen von den Inkubationsschritten der Antikörper sind die genauen Flüssigkeitsmengen und -zeiten in diesem Abschnitt nicht entscheidend. Um eine Spülung oder Waschung durchzuführen, lassen Sie die Embryonen sich am Boden des Röhrchens absetzen, entfernen Sie so viel Flüssigkeit wie möglich, ohne die Embryonen aufzusaugen, und fügen Sie dann ~1 ml neue Flüssigkeit hinzu. Verwenden Sie eine Pasteurpipette aus Glas, die mit einem Latexkolben ausgestattet ist, um optimale Klarheit und Kontrolle zu gewährleisten. Für den Spülschritt werden die Embryonen nicht geschaukelt, sondern nur gelassen; Für den Waschschritt werden die Embryonen für die angegebene Zeit auf einem Nutator geschaukelt und dann gelassen.
- Wenn die Embryonen nicht in Methanol gelagert wurden, fahren Sie mit Schritt 2.2 fort. Wenn die Embryonen in Methanol gelagert wurden, spülen Sie sie zweimal mit PBS-Tween und waschen Sie sie dann zweimal 20 Minuten lang mit PBS-Tween.
- Waschen Sie die Embryonen 30-60 Minuten lang in 1 ml Blockierungslösung.
- Die Embryonen werden mit in Antikörperlösung verdünnten Primärantikörpern (Tabelle 1) für 2 h bei Raumtemperatur oder vorzugsweise über Nacht bei 4 °C inkubiert. Führen Sie diesen Schritt in einem möglichst kleinen Volumen (50-300 μl) durch, um die Primärantikörper zu konservieren. Das Schaukeln auf einem Nutator ist nicht unbedingt erforderlich.
- Erhöhen Sie die Menge des Primärantikörpers, die in einem typischen Immunfluoreszenzexperiment verwendet wird, um mindestens 50 % für ExM. Verwenden Sie die folgenden Primärantikörperkonzentrationen: 1:200 für polyklonales Anti-Par-3-Meerschweinchen21 und 1:100 für polyklonales Anti-GFP-Kaninchen.
- Die Primärantikörperlösung entfernen (auf Wunsch bei 4 °C aufbewahren), zweimal mit PBS-Tween spülen und dann viermal 15 Minuten lang mit PBS-Tween waschen.
- Die Embryonen werden mit fluoreszierenden Sekundärantikörpern in einem Endvolumen von 300 μl (verdünnt in Antikörperlösung) für 1 h bei Raumtemperatur auf einem Nutierer inkubiert. In diesem Schritt kann fluoreszenzmarkiertes Streptavidin zugegeben werden. Schützen Sie die Embryonen ab diesem Schritt nach Möglichkeit vor übermäßiger und längerer Lichteinwirkung, indem Sie die Röhrchen mit einem undurchsichtigen Kastendeckel abdecken oder die Proben in einer Schublade aufbewahren.
- Verwenden Sie die folgenden Konzentrationen: 1:500 für polyklonales Anti-Kaninchen-IgG-Ziegenfleisch, das mit Alexa Fluor 488 fusioniert ist; 1:500 für polyklonales Anti-Meerschweinchen-IgG-Ziegenfleisch, das mit Alexa Fluor 568 fusioniert wurde; und 1:1000 für Streptavidin-Alexa Fluor 488.
- Entfernen und entsorgen Sie die sekundäre Antikörperlösung. Spülen Sie die Embryonen zweimal mit PBS-Tween und waschen Sie sie viermal 15 Minuten lang mit PBS-Tween.
- Zu diesem Zeitpunkt können die Embryonen bei 4 °C im Dunkeln gelagert werden, aber die Proben werden so schnell wie möglich (<24 h) verarbeitet.
3. Vorbereiten von PDMS-Vertiefungen
HINWEIS: Die PDMS-Wells können bis zu 2 Wochen im Voraus hergestellt werden.
- Stellen Sie einen Inkubator oder eine Heizplatte auf 55 °C ein und stellen Sie eine Zentrifuge ein, die konische Röhrchen auf 15 °C schleudern kann.
- Zur Herstellung der PDMS-Lösung (Tabelle 1) wird ein konisches 50-ml-Röhrchen in einen sekundären Behälter auf einer Waage gelegt und mit einer Spritze 10 g Silikonelastomerbasis in das Röhrchen gegeben. Fügen Sie dann 1 g Silikonelastomerhärter hinzu und drehen Sie das Rohr mehrmals um, um es zu mischen.
- Stellen Sie ein Gleichgewichtsröhrchen her, indem Sie eine geeignete Menge Wasser in ein zweites konisches 50-ml-Röhrchen geben. Zentrifugieren Sie die PDMS-Lösung bei 500 x g für 3 min bei 15 °C und gießen Sie sie dann in eine 10 cm Petrischale bis zu einer Tiefe von ~1 mm. Falls erforderlich, entfernen Sie Blasen, indem Sie die Lösung mit einem Luftschlauch vorsichtig anpusten. Lassen Sie die PDMS-Lösung über Nacht bei 55 °C erstarren.
- Sobald die PDMS-Platte verfestigt ist, ritzen Sie mit einem Skalpell quadratische Bereiche, die etwas kleiner sind als ein Deckglas von 22 mm x 22 mm. In jedem Quadrat eine ~8 mm breite quadratische Vertiefung einritzen und entfernen.
- Übertragen Sie jede quadratische PDMS-Vertiefung auf ein Deckglas von 22 mm x 22 mm und kleben Sie es fest auf (Abbildung 2D). Die Anfertigung von sechs oder mehr Deckgläsern sollte eine gute Anzahl von vergrößerten Embryonen für das Bild ergeben.
4. Aufkleben der Embryonen auf die Deckgläser
- Tragen Sie so viel 0,1 % Poly-L-Lysin auf, dass die Deckglasoberfläche in jeder Vertiefung bedeckt ist (~50 μl), und legen Sie sie zum Trocknen in einen 55 °C warmen Inkubator. Wiederholen Sie diesen Schritt, um die Haftung zu erhöhen.
- Spülen Sie die Embryonen einmal in 1x PBS kurz ab, um das Tween-Detergens zu entfernen, und übertragen Sie dann >10 Embryonen in jede der mit Poly-L-Lysin beschichteten Vertiefungen.
- Lassen Sie die Embryonen sich auf dem Boden der Vertiefungen niederlassen. Entfernen Sie die überschüssige Flüssigkeit aus den anhaftenden Embryonen mit einer Pasteurpipette. Fahren Sie sofort mit dem nächsten Schritt fort.
5. Aktivierung und Gelierung
HINWEIS: Aktivierung bezieht sich auf die Zugabe von MA-NHS zu den Embryonen, wodurch die Probenproteine und Antikörper so modifiziert werden, dass sie an das Hydrogel binden können. Die Gelierung bezieht sich auf die Erzeugung eines Hydrogels in und um die Embryonen in jeder Vertiefung. Bei der Gelierung werden die Embryonen mit einer Monomerlösung durchdrungen und dann mit einer Gelierungslösung behandelt, um das Hydrogel zu bilden.
- Aktivieren Sie die Embryonen für 1 h bei Raumtemperatur, indem Sie die Vertiefungen mit Aktivierungslösung füllen (1 mM MA-NHS frisch verdünnt in 1x PBS; Tabelle 1). Wechseln Sie diese Lösung etwa alle 10 Minuten im Laufe von 1 Stunde.
- Spülen Sie die Embryonen dreimal mit 1x PBS. Die Embryonen werden 45 Minuten lang bei 4 °C in Monomerlösung (Tabelle 1) inkubiert.
- Während die Embryonen in der Monomerlösung sitzen, wird die Gelierlösung vorbereitet (Tabelle 1). Die Zubereitung von ~2 ml Gelierlösung reicht aus, um die PDMS-Vertiefungen aus einer ganzen 10-cm-Petrischale abzudecken. Achten Sie darauf, das APS zuletzt hinzuzufügen, da es die Polymerisation einleitet und mit der Gelierung beginnt.
- Verdünnen Sie das katalytische Oxidationsmittel frisch aus dem Pulver (z. B. 1 % TEMPO w/v in Wasser). Kombinieren Sie 1.960 μl Monomerlösung mit 30 μl 10 % TEMED und 10 μl 1 % TEMPO.
- Um zu vermeiden, dass die gesamte Charge der Gelierlösung auf einmal polymerisiert wird, arbeiten Sie in kleinen Chargen. Die Gelierlösung (ohne APS) wird zwischen den acht Röhrchen eines PCR-Streifens in 125 μl Aliquots aufgeteilt.
- Entfernen Sie die Monomerlösung mit Hilfe eines Vakuums aus den drei PDMS-Vertiefungen, wobei Sie darauf achten müssen, die Embryonen nicht zu stören. Geben Sie 5 μl APS in eines der PCR-Röhrchen mit der Gelierlösung, um die Polymerisation einzuleiten. Verteilen Sie die polymerisierende Gelierungslösung schnell auf die Vertiefungen (~40 μl pro Vertiefung). Wiederholen Sie dies, bis alle Vertiefungen und Embryonen bedeckt sind.
- Lassen Sie die Proben 1,5-2,5 h bei 37 °C gelieren. Rühren Sie die Hydrogele von Zeit zu Zeit, um die Polymerisation zu überwachen. Verfestigte Hydrogele wackeln nicht. Dickere Hydrogele brauchen länger, um die Polymerisation abzuschließen und sich zu verfestigen.
6. Verdauung und Expansion
HINWEIS: Bei dickeren und größeren Gelen dauert es länger, bis sie sich ausdehnen, und es kann mehrere Stunden dauern, bis sich das Zentrum der Gele vollständig ausdehnt. Dies kann beschleunigt werden, indem die Ränder des Gels abgeschnitten werden. Wenn sich die Gele ausdehnen, wird ihr Brechungsindex fast identisch mit dem von Wasser, und sie werden sehr schwer zu sehen sein.
- Nachdem die Gelierung abgeschlossen ist, ziehen Sie die PDMS-Vertiefungen aus dem Deckglas ab und versuchen Sie, die Hydrogele nicht zu stören. Schneiden Sie überschüssiges Hydrogel-Material ab, falls gewünscht.
- Übertragen Sie die Hydrogele (die noch an den Deckgläsern befestigt sind) einzeln in die Vertiefungen einer Platte mit sechs Vertiefungen (Abbildung 2E). Beachten Sie, dass sich die Hydrogele während der Verdauung leicht ausdehnen können.
- Die Gele werden für 1 h bei 37 °C vollständig mit Aufschlusspuffer (Tabelle 1) bedeckt. Im Allgemeinen reichen 30 ml Aufschlusspuffer aus, um die Gele in einer 6-Well-Platte zu bedecken.
- Nach dem Aufschluss geben Sie jedes Hydrogel einzeln in eine 6 cm große Petrischale, indem Sie sie vom Deckglas abschieben. Es kann notwendig sein, ein zweites Deckglas zu verwenden, um das Gel zu entfernen. Füllen Sie jede Petrischale mit deionisiertem Wasser, um das Gel zu erweitern. Wechseln Sie das Wasser drei- bis viermal im Laufe von 1-2 Stunden, bis sich die Gele vollständig entfaltet haben (rechnen Sie mit einer ungefähren Vervierfachung der Breite).
7. Montage und Bildgebung
HINWEIS: Expandierte Hydrogele bestehen fast ausschließlich aus Wasser, wodurch sie nahezu transparent und extrem zerbrechlich sind. Die Gele können mit langen Deckgläsern manipuliert werden, um sie zu bewegen und aufzunehmen. Montieren und fotografieren Sie jeweils nur ein oder zwei Gele, da die Gele nach und nach Wasser abgeben und beginnen, um das Deckglas zu gleiten.
- Entfernen Sie mit einer Pasteurpipette so viel überschüssiges Wasser wie möglich aus der Petrischale, um die Bewegung der Gele bei der Handhabung zu minimieren.
- Manövrieren Sie jedes expandierte Gel mit den Embryonen auf der Unterseite auf ein großes Deckglas (z. B. 24 mm x 40 mm) für die Bildgebung.
- Montieren Sie jedes Deckglas mit Gel über das Objektiv eines inversen konfokalen Laser-Scanning-Mikroskops. Lokalisieren Sie korrekt inszenierte und ausgerichtete Proben mit den Epifluoreszenz- oder Hellfeldmikroskopie-Modi am Mikroskop mit Luftobjektiven mit niedriger Vergrößerung (5x oder 10x) oder mittlerer Vergrößerung (20x).
- Um Bilder mit hoher Auflösung zu erstellen, wechseln Sie zu einem Öl- oder Wasserimmersionsobjektiv mit hoher Vergrößerung (60-fach, 63-fach oder 100-fach). Die Oberfläche der Embryonen muss sich innerhalb des Fokussierbereichs des Objektivs befinden (<300 μm vom Deckglas für ein 63x-Objektiv), um abgebildet werden zu können.
- Erfassen Sie Daten von Proben mit dem konfokalen Laserscanning-Modus des Mikroskops. Stellen Sie sicher, dass Sie nicht gesättigte Bilder mit einem guten Dynamikbereich sammeln, und verwenden Sie eine angemessene Anzahl von Pixeln pro Bild, um die maximal möglichen Informationen über die Probe23 zu erfassen.

Abbildung 2: Manuelle Devitellinisierung und Arbeiten mit Hydrogelen . (A) Schneiden einer Agarplatte von einer Agar-/Fruchtsaftplatte. (B) Doppelseitiges Klebeband in den Deckel einer 6 cm langen Petrischale legen. (C) Aufkleben von Embryonen auf den abgeklebten Deckel. (D) Eine PDMS-Platte mit einer quadratischen Vertiefung, die auf ein Deckglas von 22 mm x 22 mm geklebt ist. (E) Deckglas mit einer PDMS-Vertiefung in einer 6-Well-Platte. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergebnisse
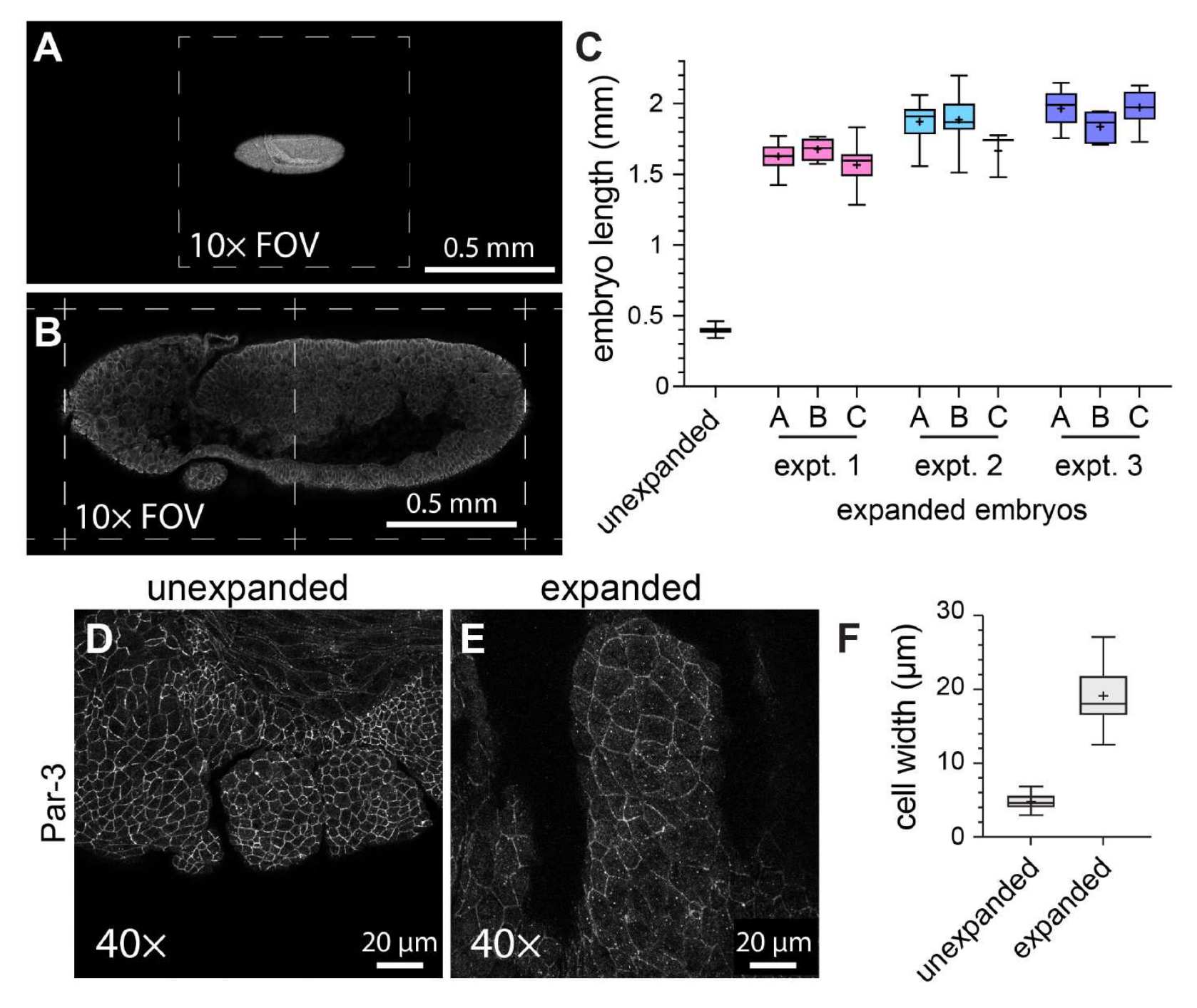
Um die allgemeine Wirksamkeit von ExM in Whole-Mount-Drosophila-Embryonen zu charakterisieren, wurde die Embryolänge entlang der Kopf-zu-Schwanz-Achse in nicht expandierten Kontrollembryonen im Vergleich zu expandierten Embryonen gemessen (Abbildung 3A-C). Die nicht expandierten Kontrollembryonen wurden den gleichen Fixierungsbedingungen und Immunfluoreszenz-Markierungsschritten unterzogen wie die expandierten Embryonen, mit der Ausnahme, dass sie vor der Bildgebung mit einem verfestigten Eindeckmedium montiert wurden. Die einzelnen nicht expandierten Embryonen erstreckten sich über etwa die Hälfte eines Gesichtsfeldes, wenn ein 10x-Objektiv verwendet wurde (Abbildung 3A). Im Gegensatz dazu erstreckten sich die expandierten Embryonen über etwa zwei volle Sichtfelder, wenn sie das gleiche 10-fache Objektiv verwendeten (Abbildung 3B). Um zu beurteilen, wie der Grad der Ausdehnung sowohl innerhalb als auch zwischen den Experimenten variierte, wurde das gleiche ExM-Protokoll bei drei verschiedenen Gelegenheiten durchgeführt, und die Embryonallänge wurde in drei verschiedenen Gelen innerhalb jedes einzelnen Experiments gemessen. Die durchschnittliche Kopf-zu-Schwanz-Länge der nicht expandierten Kontrollembryonen betrug 398,8 μm (Standardabweichung [SD] = 22,93 μm; n = 74; Abbildung 3C). Für Experiment 1, Experiment 2 und Experiment 3 betrugen die durchschnittlichen Embryonenlängen 1.596 μm (SD = 159,9 μm; n = 57), 1.868 μm (SD = 150,5 μm; n = 51) bzw. 1.954 μm (SD = 120,3 μm; n = 44), was Expansionsfaktoren von 4,0-fach, 4,7-fach bzw. 4,9-fach entspricht (Abbildung 3C). Die intraexperimentelle Variation zwischen den Gelen war viel weniger auffällig als die interexperimentelle Variation, die etwa 20 % betrug (Abbildung 3C). Um die Auswirkungen von ExM auf die Zell- und Embryomorphologie zu beurteilen, wurde ein Antikörper gegen die adhärenzköpfige Verbindungskomponente Par-3 (Bazooka)21 verwendet, um die apikalen Zellmembranen zu markieren, und wir bildeten die sich entwickelnden Mundsegmente von Drosophila-Embryonen im Stadium 11 ab – einem Stadium mit einer komplexen segmentierten Struktur (Abbildung 3D-F). In der Kontrollprobe wiesen die Zellen im Oberkiefersegment eine durchschnittliche Breite von 4,76 μm auf (SD = 1,053 μm, n = 25; Abbildung 3D,F). In den erweiterten Proben, die mit dem gleichen 40-fachen Objektiv und Zoomfaktor (1x) abgebildet wurden, hatten die Zellen im Oberkiefersegment eine durchschnittliche Breite von 19,10 μm (SD = 3,966 μm, n = 18; Abbildung 3E,F), die eine 4,0-fache Ausdehnung darstellt. Daher waren wir in Übereinstimmung mit früheren Berichten11 in der Lage, Drosophila-Embryonen mit Hilfe von ExM in linearen Dimensionen um etwa das Vierfache zu erweitern, ohne dass die Proben zerrissen oder offensichtliche Verzerrungen in der Zell- oder Gewebemorphologie auftraten.

Abbildung 3: Vierfache Expansion von Drosophila-Embryonen. (A) Unexpandierte und (B) expandierte Drosophila-Embryonen, aufgenommen mit einem 10-fachen Objektiv (0,3 NA) bei 1-fachem Zoom. Einzelne Sichtfelder (FOV) sind mit gestrichelten Linien gekennzeichnet. Die Embryonen exprimierten eine GFP-markierte Version der Myosin-Leichtkette und wurden mit einem Anti-GFP-Antikörper gefärbt. (C) Quantifizierung der Embryonenlänge (entlang der Kopf-zu-Schwanz-Achse) in drei Hydrogelen pro Experiment und aus drei separaten ExM-Experimenten im Vergleich zu nicht expandierten Kontrollen. (D,E) Oberkiefersegmente von (D) unexpandierten und (E) expandierten Drosophila-Embryonen im Stadium 11, aufgenommen mit einem 40-fachen Objektiv (1,3 NA) bei 1-fachem Zoom. Die Zellumrisse (adhärens junctions) wurden mit einem Anti-Par-3/Bazooka-Antikörper (weiß) detektiert. (F) Quantifizierung der Zellbreite (Längsachse) aus äquivalenten Zellgruppen aus (D) und (E). Die Boxplots in (C) und (F) zeigen den Bereich des 25., 50. und 75. Perzentils. die Schnurrhaare zeigen die Minimal- und Maximalwerte an; Die "+"-Symbole geben den Mittelwert an. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Um zu zeigen, dass ExM verwendet werden kann, um subzelluläre Details unterhalb der typischen Beugungsgrenze aufzulösen, wurde das Actomyosin-Zytoskelett in unexpandierter Kontrolle im Vergleich zu expandierten Embryonen in konvergenter Extension (Stadium 7) abgebildet. Die Gewebeumbauereignisse der Gastrulation und konvergenten Extension werden weitgehend durch Veränderungen in der Lokalisation des Motorproteins Myosin II II24 gesteuert. Im dicht gepackten säulenförmigen Epithel des frühen Drosophila-Ektoderms ist es jedoch schwierig, viele feine Details des Myosin-II-Lokalisierungsmusters zu beobachten, selbst wenn es mit 158-facher Vergrößerung (63-faches Objektiv mit einem 2,5-fachen optischen Zoom) aufgenommen wird – ein typisches maximales Auflösungsvermögen für ein konfokales Laser-Scanning-Mikroskop. Da es sich bei Myosin II beispielsweise um ein kortikales Protein handelt (das sich direkt unter der Plasmamembran befindet), waren Pools von Myosin II25 , die sich auf beiden Seiten der Zell-Zell-Kontakte befanden, in Embryonen im Stadium 7 nicht auflösbar, und sie erschienen als eine einzige Linie, in der benachbarte Zellen aufeinandertrafen (Abbildung 4A). Im Gegensatz dazu konnten in expandierten Embryonen im Stadium 7 parallele Linien von Myosin II an Zell-Zell-Verbindungen beobachtet werden, die kortikale Proteinpools in benachbarten Zellen darstellen (Abbildung 4B). Der Abstand zwischen parallelen Myosin-II-Linien in expandierten Proben betrug 892,7 nm (SD = 0,171 nm, n = 12); geteilt durch vier ergibt dies einen vorhergesagten Abstand von ~220 nm zwischen den Myosinlinien in benachbarten Zellen in unexpandierten Embryonen, was in der Tat knapp unter der Beugungsgrenze für ein mit Alexa 488 detektiertes Signal liegt (maximale Emission von ~520 nm/2 = 260 nm).
Darüber hinaus haben wir auch getestet, ob ExM verwendet werden kann, um die mitochondriale Netzwerkarchitektur in dicht gepackten Zellen von gastrulierenden Drosophila-Embryonen (Stadium 6) aufzulösen. Die mitochondriale Funktion ist eng mit der Netzwerkstruktur verbunden (d. h. fusionierte vs. fragmentierte Organellen), aber die Details der mitochondrialen Netzwerkorganisation sind mit konventioneller konfokaler Mikroskopie in Zelltypen, die nicht flach und/oder dünn sind, schwer zu visualisieren. Mitochondrien sind von Natur aus reich an biotinylierten Molekülen, und daher können Mitochondrien im frühen Drosophila-Embryo mit fluoreszenzmarkiertem Streptavidinmarkiert werden 26. Bei den nicht expandierten Embryonen im Stadium 6, die mit Streptavidin-Alexa 488 markiert waren, erschien das Signal als zytoplasmatische Punkte, die sich oft überlappten und schwer aufzulösen waren (Abbildung 4C). Im Gegensatz dazu waren in den erweiterten Embryonen im Stadium 6 viel mehr feine Details des mitochondrialen Netzwerks sichtbar und die Puncta waren leichter auflösbar (Abbildung 4D)26,27. Diese Ergebnisse deuten darauf hin, dass ExM verwendet werden kann, um die mitochondriale Netzwerkorganisation in Zelltypen zu untersuchen, die traditionell nicht für die mitochondriale Analyse geeignet sind.

Abbildung 4: Details des Actomyosin-Zytoskeletts und der Mitochondrien, die durch Expansionsmikroskopie enthüllt wurden. (A,B) Myosin-II-Lokalisation in Neuroektodermzellen (Keimband), aufgenommen mit einem 63-fachen Objektiv (1,4 NA) bei 2,5-fachem Zoom in Stadium 7 (A) unexpandierten und (B) expandierten Embryonen. Myosin II wurde in den Embryonen nachgewiesen, die eine transgene GFP-markierte Version der Myosin II regulatorischen Leichtkette (sqh-GFP) exprimierten, die mit einem Anti-GFP-Antikörper (rot) nachgewiesen wurde. Unterschiedliche Pools von kortikalem Myosin, die sich in benachbarten Zellen befinden, können im expandierten Embryo aufgelöst werden (weiße Pfeile). (C,D) Mitochondriale Netzwerke in Neuroektodermzellen, die mit einem 63× Objektiv (1,4 NA) bei 2,5-fachem Zoom in unexpandierten (C) und expandierten (D) Embryonen im Stadium 6 abgebildet wurden. Die Mitochondrien wurden mit Streptavidin-Alexa 488 (grün) und die Zellumrisse mit einem Anti-Par-3/Bazooka-Antikörper (magenta) detektiert. Die Experimente wurden mit einem konfokalen Laser-Scanning-Mikroskop durchgeführt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Tabelle 1: Rezepte für Lösungen. Zusammensetzung der in diesem Protokoll verwendeten Lösungen in der Reihenfolge ihres Auftretens. Alle Aktien sind, sofern nicht anders angegeben, liquid. Die Chemikalien wurden, sofern nicht anders angegeben, in autoklaviertem gefiltertem Wasser resuspendiert oder verdünnt. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Manuelle Devitellinisierung
Die meisten Protokolle zur Fixierung von Drosophila-Embryonen beinhalten die Entfernung der Vitellinmembran durch Schütteln fixierter Embryonen in einer Emulsion aus Methanol und Heptan, wodurch die Membranen durch osmotischen Bruch platzen26. Während die Methanol-basierte Devitellinisierung (Methanol-Popping) effektiv und für viele Anwendungen geeignet ist, bietet die manuelle Devitellinisierung (Handpeeling) einige wesentliche Vorteile. Erstens ermöglicht das manuelle Peeling die Auswahl von genau inszenierten Embryonen, die devitellinisiert und gesammelt werden sollen, was die Wahrscheinlichkeit, am Ende des Experiments vergrößerte Embryonen in einer brauchbaren Ausrichtung zu erhalten, erheblich erhöht. Diese Anreicherung ist von entscheidender Bedeutung, wenn bestimmte Aspekte schneller Entwicklungsprozesse (z. B. Mesoderm-Invagination oder konvergente Extension) untersucht werden, bei denen entsprechend inszenierte Embryonen nur wenige Prozent aller Embryonen ausmachen können, selbst innerhalb eines eng getakteten Zeitfensters. Natürlich ist für viele Anwendungen das traditionellere Methanol aus einem zeitgesteuerten Entnahmefenster ausreichend, und das Schälen von Hand ist den zusätzlichen Aufwand möglicherweise nicht wert. Zweitens wird die Bindung bestimmter primärer Antikörper und Farbstoffe durch eine vorherige Exposition der Probe gegenüber Methanol negativ beeinflusst. Aus diesem Grund kann das Schälen von Hand im Vergleich zu mit Methanol gepoppten Proben zu einer signifikanten Verbesserung der Immunfluoreszenzsignalqualität führen, was es zu einer nützlichen allgemeinen Technik für Drosophila-Entwicklungsbiologen macht.
Hochauflösende konfokale Mikroskopie in expandierten Drosophila-Embryonen
Während die Durchführung der hochauflösenden konfokalen Mikroskopie an expandierten Proben konzeptionell die gleiche ist wie an unexpandierten Proben, bringt ExM einige technische Hürden mit sich. Insbesondere die Embryonenorientierung, die zufällig ist, wird mit zunehmender Stichprobengröße noch wichtiger, da Objektive mit hoher Vergrößerung und hoher NA nur in der Lage sind, Licht von Probenbereichen zu fokussieren, die sich sehr nahe am Deckglas27 befinden. Daher ist es in der Regel nur möglich, sich auf die Zellen an oder nahe der Oberfläche des Embryos zu konzentrieren, die bei der Bildung des Gels neben dem Deckglas gelandet sind. Der beste Weg, um sicherzustellen, dass am Ende Proben mit der richtigen Ausrichtung vorhanden sind, besteht darin, das ExM-Protokoll mit einer eng gestaffelten Entnahme von fixierten Embryonen zu beginnen (z. B. durch Schälen von Hand) und viele Embryonen in jede Vertiefung zu säen (>10). Um Zellen tief im Inneren des Embryos sichtbar zu machen, kann es notwendig sein, spezialisiertere Bildgebungsaufbauten zu verwenden, wie z. B. Lichtblattmikroskopie28. Darüber hinaus stellen wir fest, dass die Bildqualität verbessert werden kann, indem die konfokale Lochblende auf eine Größe von mehr als einer luftigen Einheit geöffnet wird. Natürlich geht eine größere Lochblende auf Kosten einer verringerten maximalen Auflösung, aber in der Praxis können selbst kleine Erhöhungen der Lochblendengröße die Signalintensität erheblich erhöhen (Daten nicht gezeigt). Zukünftige Studien sollten sich systematisch mit der Größe der Lochblenden und der effektiven Auflösung in ExM-Proben befassen.
Variationen des Basis-ExM
Das hier beschriebene Protokoll ist ein relativ einfaches Beispiel für ExM, das für viele Anwendungen funktionieren und in den meisten entwicklungsbiologischen Laboren leicht zu implementieren sein sollte. Es gibt jedoch zahlreiche Variationen des Grundkonzepts von ExM 4,5,7, mit denen die Signalintensität erhöht, noch weitere Expansionsgrade erreicht und Nukleinsäuremoleküle sowie Proteine detektiert werden können. In diesem Protokoll werden die Embryonen vor der Gelierung und Expansion mit Antikörpern inkubiert. Alternativ können die Proben mit Antikörpern behandelt werden, nachdem sie expandiert wurden 6,30, was die Signalintensität aufgrund der erhöhten Epitopzugänglichkeit und des verringerten Verlusts gebundener Antikörper während der Expansionsschritte erhöhen kann. Darüber hinaus können spezifische Vernetzermoleküle verwendet werden, um RNA-Moleküle an das Hydrogel zu binden, um den Nachweis von RNA in expandierten Gelen unter Verwendung des Hybridisierungskettenreaktionsverfahrens30 zu ermöglichen. Schließlich können die Proben mehreren Expansionsrunden unterzogen werden, wie z. B. bei der iterativen Expansionsmikroskopie (iExM)31, der pan-ExM32 und der Expansionsenthüllung (ExR)31, um noch höhere Grade an erhöhter Auflösung zu erreichen.
Offenlegungen
Die Autoren haben keine Interessenkonflikte zu deklarieren.
Danksagungen
Wir danken Dr. Jennifer Zallen für die Bereitstellung des Meerschweinchen-Anti-Par-3-Primärantikörpers. Diese Arbeit wurde durch großzügige Mittel (1R15GM143729-01 und 1P20GM139768-01 5743) vom National Institute of General Medical Science (NIGMS), einem der Mitglieder der National Institutes of Health (NIH), sowie dem Arkansas Biosciences Institute (ABI) unterstützt, das den Kauf unseres konfokalen Mikroskops teilweise finanzierte.
Materialien
| Name | Company | Catalog Number | Comments |
| acrylamide | Milipore Sigma | 1490-100ML | |
| ammonium persulfate | VWR | BDH9214-500G | |
| anti-GFP rabbit polyclonal antibody | Torrey Pines BioLabs | TP-401 | |
| anti-guinea pig IgG goat polyclonal antibody, Alexa Fluor 568 | Thermo Fisher Scientific | A-11075 | |
| anti-rabbit IgG goat polyclonal antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11008 | |
| bisacrylamide | Research Products International | A11275 | |
| bovine serum albumin (30% solution) | Millipore Sigma | A7284 | |
| conical tubes, 50 mL | fisherscientific | 21008-940 | |
| coverlip glass, square 22 mm | VWR | 48366-227 | |
| coverslip glass, rectangular 40 mm x 24 mm | VWR | 48393-230 | |
| glass capillaries for pulling needles | World Precision Instruments | TW100F-4 | |
| glass microinjection needles (pre-pulled) | World Precision Instruments | TIP10LT | |
| guanidine HCl | VWR | 101970-606 | |
| heptane | VWR | EM-HX0078-1 | |
| latex pipet bulbs | VWR | 82024-554 | |
| methanol | VWR | BDH1135-4LP | |
| methylacylic acid N-hydroxysuccinimidyl ester | VWR | 730300-1G | |
| microfuge tube, 1.5 mL | VWR | 20170-038 | |
| multi-well plate, 6-well | Genesee | 25-100 | |
| paraformaldehyde (16%, EM-grade, methanol-free) | Electron Microscopy Sciences | 509804487 (Fisher) | |
| Pasteur pipet (2 mL, short tip) | VWR | 14673-010 | |
| PDMS kit (Sylgard 184 Kit, base and curing agent) | VWR | 102092-312 | |
| Petri plates | Genesee | 32-107 | |
| phosphate-buffered saline (10x solution) | VWR | 97063-660 | |
| Poly-L-lysine solution (0.1% solution) | VWR | P8920-1ooML | |
| Proteinase K | Thermo Fisher Scientific | E00491 | |
| scintillation vials (30 mL) | VWR | 66022-128 | |
| sodium acrylate | VWR | 101181-226 | |
| sodium azide (powder) | Millipore Sigma | 71289 | make a 1% w/v working stock; acute POISON at this concentration! |
| Streptavidin, Alexa Fluor 488 | Thermo Fisher Scientific | S32354 | |
| TAE (50x) | VWR | 97063-692 | |
| tape (double-sided, 1 inch wide) | Scotch 3M | 665 Scotch double sided 1inch/1296 inches Boxed | |
| TEMED | Thermo Fisher Scientific | PI17919 | |
| TEMPO | VWR | EM8.14681.0005 | catalytic oxidant |
| Tween-20 | VWR | 97063-872 | extremely viscous when pure; make a 10% working stock with water |
| Zeiss LSM 900 | Zeiss | Laser scanning microscope used without AiryScan |
Referenzen
- Klar, T. A., Jakobs, S., Dyba, M., Egner, A., Hell, S. W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences of the United States of America. 97 (15), 8206-8210 (2000).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nature Methods. 3 (10), 793-796 (2006).
- Betzig, E. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Wassie, A. T., Zhao, Y., Boyden, E. S. Expansion microscopy: principles and uses in biological research. Nature Methods. 16 (1), 33-41 (2019).
- Chen, F., Tillberg, P. W., Boyden, E. S. Optical imaging. Expansion microscopy. Science. 347 (6221), 543-548 (2015).
- Chen, F., et al. Nanoscale imaging of RNA with expansion microscopy. Nature Methods. 13 (8), 679-684 (2016).
- Tillberg, P. W., et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature Biotechnology. 34 (9), 987-992 (2016).
- Chang, J. -. B. Iterative expansion microscopy. Nature Methods. 14 (6), 593-599 (2017).
- Cahoon, C. K., et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proceedings of the National Academy of Sciences of the United States of America. 114 (33), 6857-6866 (2017).
- Mosca, T. J., Luginbuhl, D. J., Wang, I. E., Luo, L. Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. eLife. 6, e27347 (2017).
- Jiang, N., et al. Superresolution imaging of Drosophila tissues using expansion microscopy. Molecular Biology of the Cell. 29 (12), 1413-1421 (2018).
- Yu, C. -. C. J., et al. Expansion microscopy of C. elegans. eLife. 9, e46249 (2020).
- Freifeld, L., et al. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proceedings of the National Academy of Sciences of the United States of America. 114 (50), E10799-E10808 (2017).
- Tanaka, T., et al. Phase transitions in ionic gels. Physical Review Letters. 45 (20), 1636-1639 (1980).
- Hausen, P., Dreyer, C. The use of polyacrylamide as an embedding medium for immunohistochemical studies of embryonic tissues. Stain Technology. 56 (5), 287-293 (1981).
- Campos-Ortega, J. A., Hartenstein, V. . The Embryonic Development of Drosophila melanogaster. , (2013).
- Chozinski, T. J., et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nature Methods. 13 (6), 485-488 (2016).
- Miller, D. F. B., Holtzman, S. L., Kaufman, T. C. Customized microinjection glass capillary needles for P-element transformations in Drosophila melanogaster. BioTechniques. 33 (2), 366-367 (2002).
- Rothwell, W. F., Sullivan, W. Drosophila embryo dechorionation. CSH Protocols. 2007, (2007).
- Cold Spring Harbor Protocols. . Drosophila apple juice-agar plates. , (2011).
- de Matos Simões, S., et al. Rho-kinase directs bazooka/Par-3 planar polarity during drosophila axis elongation. Developmental Cell. 19 (3), 377-388 (2010).
- Gambarotto, D., et al. Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nature Methods. 16 (1), 71-74 (2019).
- Paré, A. C., Zallen, J. A. Cellular, molecular, and biophysical control of epithelial cell intercalation. Current Topics in Developmental Biology. 136, 167-193 (2020).
- Royou, A., Field, C., Sisson, J. C., Sullivan, W., Karess, R. Reassessing the role and dynamics of nonmuscle myosin II during furrow formation in early Drosophila embryos. Molecular Biology of the Cell. 15 (2), 838-850 (2004).
- Chowdhary, S., Tomer, D., Dubal, D., Sambre, D., Rikhy, R. Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Scientific Reports. 7 (1), 5502 (2017).
- Chowdhary, S., Madan, S., Tomer, D., Mavrakis, M., Rikhy, R. Mitochondrial morphology and activity regulate furrow ingression and contractile ring dynamics in Drosophila cellularization. Molecular Biology of the Cell. 31 (21), 2331-2347 (2020).
- Stelzer, E. H. K., et al. Light sheet fluorescence microscopy. Nature Reviews Methods Primers. 1, 73 (2021).
- Ku, T., et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nature Biotechnology. 34 (9), 973-981 (2016).
- Wen, G., et al. A Universal labeling strategy for nucleic acids in expansion microscopy. Journal of the American Chemical Society. 143 (34), 13782-13789 (2021).
- M'Saad, O., Bewersdorf, J. Light microscopy of proteins in their ultrastructural context. Nature Communications. 11 (1), 3850 (2020).
- Sarkar, D. Expansion revealing: Decrowding proteins to unmask invisible brain nanostructures. bioRxiv. , (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten