
Method Article
Identification des produits pharmaceutiques dans l’environnement aquatique à l’aide de HPLC-ESI-Q-TOF-MS et élimination d’érythromycine par dégradation Photo
Dans cet article
Résumé
Nous présentons un protocole d’analyse non ciblés à l’aide de temps de vol de spectrométrie de masse comme un outil parfait pour identifier des produits pharmaceutiques dans les eaux. Nous démontrons l’application de l’irradiation UV pour leur élimination. Analyse comportant irradiation, composé isolement, identification et modélisation cinétique des profils dégradation est illustrée.
Résumé
Contrôle des produits pharmaceutiques pendant tout le cycle de l’eau devient de plus en plus important pour le milieu aquatique et finalement pour la santé humaine. Ciblées et analyse non ciblés sont moyens aujourd'hui de choix. Bien que ciblés analyse habituellement réalisée à l’aide d’un triple quadripôle, spectromètre de masse peut-être être plus sensible, seuls composés précédemment sélectionnés peuvent être identifiés. L’analyse plus puissant indifférenciés est effectuée à travers le temps de vol prolongés de spectromètres de masse (TOF-MS) par un analyseur de masse quadripolaire (Q), tel qu’utilisé dans cette étude. Précédé par extraction en phase solide et chromatographie liquide haute performance (HPLC), l’approche non ciblée permet de détecter toutes les substances ionisables avec sélectivité et une grande sensibilité. Profitant pleinement de l’instrument de Q-TOF-MS, des expériences de spectrométrie de masse en tandem (MS/MS) accélérer et facilitent l’identification, alors qu’une méthode ciblée de MS améliore la sensibilité mais s’appuie sur les normes de référence aux fins d’identification. L’identification de quatre produits pharmaceutiques de l’eau du fleuve Rhin est démontrée. Le Rhin est originaire de Toma, Grisons, Suisse et se jette dans la mer du Nord, près de la baie sud, aux Pays-Bas. Sa longueur s’élève à 1232,7 km. Comme il est d’intérêt privilégié pour éliminer efficacement les produits pharmaceutiques à partir du cycle de l’eau, l’irradiation UV-C de l’effet est démontrée à l’échelle laboratoire. Cette méthode permet la dégradation rapide des produits pharmaceutiques, qui apparaît de façon exemplaire pour l’érythromycine antibiotique macrolide. À l’aide de la méthode HPLC-Q-TOF-MS ci-dessus, diagrammes de concentration-temps sont obtenus pour la molécule mère et leurs produits de photodégradation. Après avoir établi les équations des réactions du premier ordre séquentielles, montage numérique permet la détermination des paramètres cinétiques, qui pourrait aider à prédire le temps d’irradiation et conditions lorsqu’il est potentiellement considéré comme quatrième étape au sein traitement des eaux usées.
Introduction
Produits pharmaceutiques figurent régulièrement dans le milieu aquatique1,2,3,4,5. Une source importante sont les effluents des eaux usées traitement plantes (SEEU)6,7,8,9. La présence de produits pharmaceutiques dans la pendant tout le cycle de l’eau a été étudiée exemplairement dans le bassin de la rivière Turia10. Entre autres, les antibiotiques représentent une classe particulière de dangereuse de médicaments, car ils passent souvent l’étape biologique des stations d’épuration inaltéré et peut provoquer des résistances bactériennes dans l’environnement11,12,13 . Macrolides constituent une classe d’antibiotiques qui sont appliqués en homme et en médecine vétérinaire. Leurs représentants ont été trouvés dans la concentration de 1 µg/l dans les effluents14,15,16,17,18,19. L’un d’eux est l’érythromycine (Ery)20,21. Dans les eaux, l’érythromycine est souvent accompagnée d’anhydroerythromycin A (Ery A - H2O), une déshydratation22,23. Élimination de l’eau de l’érythromycine est en raison de l’instabilité acide. Le ratio d’érythromycine vs anhydroerythromycin dépend du pH24,25,26,27.
Chimiquement, les macrolides contiennent une lactone macrocycliques qui sucre différentes fractions sont attachées, par exemple., desosamine, cladinose ou mycaminose. Étant donné que les macrolides sont chimiquement modifiés des produits naturels de procédés de fermentation, elles existent souvent sous forme de mélanges. Les espèces appelées A, B, C, etc.., se distinguent par les substituants de sucre. Les portions de sucre et leur position à la lactone sont responsables pour le mode d’action des macrolides28,29. Afin de minimiser le risque pour l’environnement, il est souhaitable de minéraliser complètement les produits pharmaceutiques avant d’entrer dans le milieu aquatique27,30,31,32.
La première partie de cette étude porte sur la détection de produits pharmaceutiques dans les eaux de surface, qui est important pour la surveillance des effluents et eaux libres. Vous recherchez une variété de substances non identifiées dans le microgramme dans différentes matrices, analyse non ciblée est la méthode de choix20,33,34,35. En particulier, haute performance de chromatographie liquide (HPLC) electrospray ionisation quadrupolaire temps vol de spectrométrie de masse (HPLC-ESI-Q-TOF-MS) a été prouvé de valeur extraordinaire en raison de sa spécificité et de sensibilité. Après l’identification de la substance, la sensibilité peut encore être étendu en utilisant la MS ciblées approche avec le quadripôle fonctionnant en mode select et l’énergie de collision au sein de la cellule de collision à zéro. Par conséquent, les ions arrivent non fragmenté au détecteur TOF.
Le second point de focalisation de ce travail est l’élimination de l’érythromycine. Pour l’élimination des produits pharmaceutiques, procédés d’oxydation avancée dite (NPEA) sont utilisés, par exemple., a commencé par l’irradiation UV lumière36,37,38. Indispensable pour la dégradation est la formation de radicaux hydroxyles de l’eau par VUV / irradiation UVC suite EQ. 1.
H2O + hν(< 200 nm) → H2O * → H. + . OH (1)
Les radicaux hydroxyles possèdent un potentiel d’oxydation élevé de 2,8 V, qui contribue positivement à la dégradation des substances36,37.
La dégradation de l’érythromycine utilisant le vide irradiation UV/UVC dans l’eau est décrit ici, compte tenu de l’influence du pH. La formation de produits encore plus dangereux est censée être un inconvénient de l’utilisation NPEA39,40. Ainsi, il est important d’irradier jusqu'à la minéralisation complète de la pharmacie. Pour mieux estimer la durée d’irradiation, le modèle cinétique de la réaction, les constantes de vitesse de réaction et les demi-vies sont déterminés pour le médicament initial ainsi que pour ses photodegradates. À cette fin, courbe de concentration-temps (c-t) ont été dérivés de mesures HPLC-ESI-Q-TOF-MS et par rapport aux modèles de cinétique chimique à l’aide de MATLAB. La cinétique de la dégradation a procédé selon de premier ordre, et les photodegradates ont été décrits comme produits intermédiaires d’une réaction suivi consécutives ou ultérieure27,41.
Protocole
1. préparation : Extraction en Phase solide
- Prélever environ 1 L d’eau pour la préparation des échantillons.
- Filtrer l’échantillon sur un filtre de bande bleue avec une taille de pore de 2 µm pour éliminer les particules grossières.
- Equilibrer la cartouche SPE en utilisant les 3 mL de méthanol et 3 mL d’eau ultrapure.
- Appliquez le filtrat (1 L) sur la cartouche SPE et augmenter la vitesse d’écoulement à l’aide d’un aspirateur modéré, par exemple., une pompe à membrane.
Remarque : Plusieurs cartouches SPE peuvent être exécutés en parallèle. - Laver l’échantillon avec 3 mL de l’eau ultrapure.
- Éluer les analytes du sorbate de cartouche de 3 mL de méthanol.
- Concentrer l’éluat de 3 mL à sécher à l’aide d’un évaporateur rotatif.
- Dissoudre le résidu dans 1 mL d’eau ultrapure.
- Filtrer la solution sur un filtre de seringue et les stocker dans un flacon d’analyse par HPLC-ESI-Q-TOF-MS indifférenciés.
2. HPLC-ESI-Q-TOF-MS méthode pour l’analyse ciblée et indifférenciés et MS/MS
- Transférer le flacon à l’autosampler HPLC-ESI-Q-TOF-MS.
- Définir tous les paramètres pertinents (tableau 1) pour l’HPLC-ESI-Q-TOF-MS.
Remarque : Si on utilise une énergie de collision finis, i.e., collision énergie (EC) ≠ 0, ions sera fragmenté. Ce mode correspond à la méthode de MS/MS ciblée. - Démarrer la mesure.
- Analyser les chromatogrammes qui en résulte et les spectres de masse.
3. expériences d’Irradiation UV
- Dissoudre le composé antibiotique, par exemple, l’érythromycine (750 mg/L), dans l’eau ultrapure à concentration finale de 20 mg/L.
- Remplir le photoréacteur 1 L, enveloppé dans du papier d’aluminium, avec 750 mL de la solution.
- Introduire la lampe fournissant 15 W de puissance dans le réacteur.
- Appliquer l’agitateur magnétique à 500 tr/min.
- Ajuster le pH à la valeur souhaitée 3-4, 6-7 ou 8 et 9 par goutte à goutte addition de HCl (0,1 M) ou NH3 (0,1 M) si nécessaire. pH 6-7 est utilisé à titre d’exemple.
- Prélever 2 mL de la solution de réaction comme échantillon au temps 0 à l’aide d’une seringue et transférer dans un flacon de verre de 2 mL.
- Allumez la lampe UV et garder une trace de l’époque de filtres.
NOTE : Temps d’Irradiation de 10 min suffisent souvent. Si l’intégralité de la photoréaction est souhaitée, une deuxième série d’expérience devrez peut-être être enregistré en utilisant les résultats de la première série.
ATTENTION : L’irradiation UV peut conduire à la cécité. - Obtenir un échantillon de 2 mL de la solution toutes les 30 s pendant les 5 premières minutes. Ensuite, prenez un échantillon toutes les 60 s jusqu'à la fin de l’expérience. Transférer les échantillons en flacons de 2 mL.
- Conserver les flacons jusqu'à l’analyse HPLC-ESI-Q-TOF-MS à-4 ° C.
- Analyse des 16 échantillons à l’aide de HPLC-ESI-Q-TOF-MS avec les méthodes décrites à l’étape 2.
4. l’analyse cinétique
- Préparer un logiciel approprié comme la boîte à outils de courbe-ajustage de précision de R2016b MATLAB.
- S’adapter à la masse-zone vs chronologiques de la dégradation induite par la photo de l’antibiotique parent composé selon la cinétique de premier ordre, voir EQ 242,43
 (2)
(2)
La concentration se réfère à la concentration initiale de l’aspiration A, cun et Pendant le temps de réaction t avec la vitesse constante k1 la concentration réelle de la première étape de réaction A vers B.
se réfère à la concentration initiale de l’aspiration A, cun et Pendant le temps de réaction t avec la vitesse constante k1 la concentration réelle de la première étape de réaction A vers B. - S’adapter à la masse-zone vs courbes temporelles de l’aussi à l’aide d’EQ. 3 et 4, telles qu’elles peuvent être décrites comme intermédiaires de réaction suivie consécutive ou plue, c’est à dire., modèle de produit B ou C selon la réaction un →B → C → D.
 (3)
(3) (4)
(4)
Les concentrations cB et cC se réfèrent aux intermédiaires B et C ; et k2k3 pour les constantes de vitesse correspondant B à C, C à D. - Utilisez EQ. 5 pour adapter les données, si la durée d’irradiation ne suffisait pas d’observer la dégradation d’un produit photo. Ce degradate peut être traitée comme produit final D avec la concentration CD pour obtenir les constantes de vitesse.
 (5)
(5)- Calculer la concentration de B à l’aide d’EQ. 6 au lieu de l’équation 3, si la réaction se termine par B. Si C est le produit final, calculer la concentration de C selon l’équation 7 au lieu d’EQ. 4.
 (6)
(6) (7)
(7)
- Calculer la concentration de B à l’aide d’EQ. 6 au lieu de l’équation 3, si la réaction se termine par B. Si C est le produit final, calculer la concentration de C selon l’équation 7 au lieu d’EQ. 4.
- Utilisez EQ. 8 pour la détermination des demi-vies t1/2.
 (8)
(8)
Résultats
Comme résultat de l’extraction en phase solide, un jaunâtre à la solution de vert foncée a été obtenue dans tous les cas, qui a révélé la présence de chlorophylle contenant des substances (Figure 1). Produits pharmaceutiques figurant dans cet échantillon d’eau ne conduirait pas à une coloration visible depuis leur concentration et leur absorption serait généralement trop basses. Au lieu de cela, la présence de produits pharmaceutiques doit être analysée à l’aide de HPLC et spectrométrie de masse haute résolution.
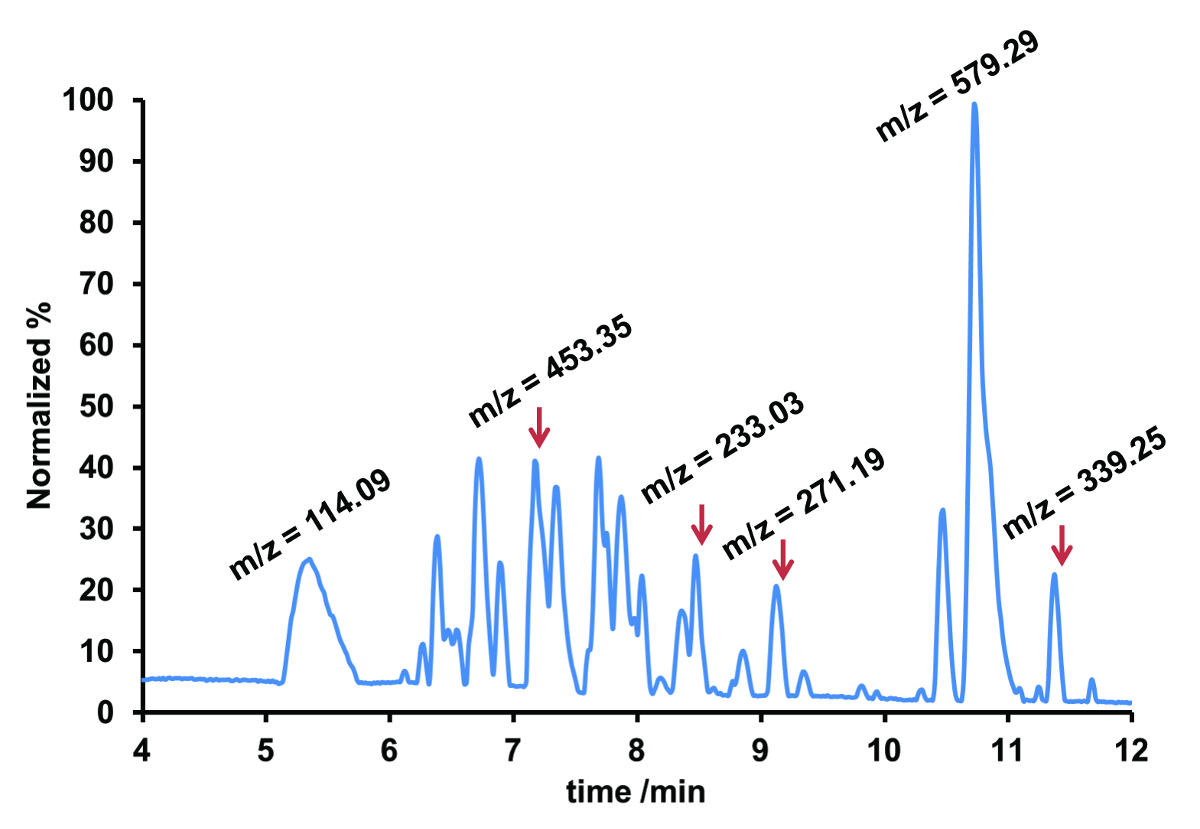
Dans l’analyse non ciblée, un HPLC-ESI-Q-TOF-MS a été utilisé en raison de sa précision exceptionnelle de masse permettant d’obtenir la masse exacte pour chaque composé ionique. Le chromatogramme détecté à la masse de l’analyse effectuée était représenté un chromatogramme pic de base (CPB), qui affiche le pic le plus intense de chaque spectre de masse enregistrée dans le cours de la séparation chromatographique. L’exemple illustré à la Figure 2 présente la CAB d’un échantillon d’eau du fleuve Rhin.
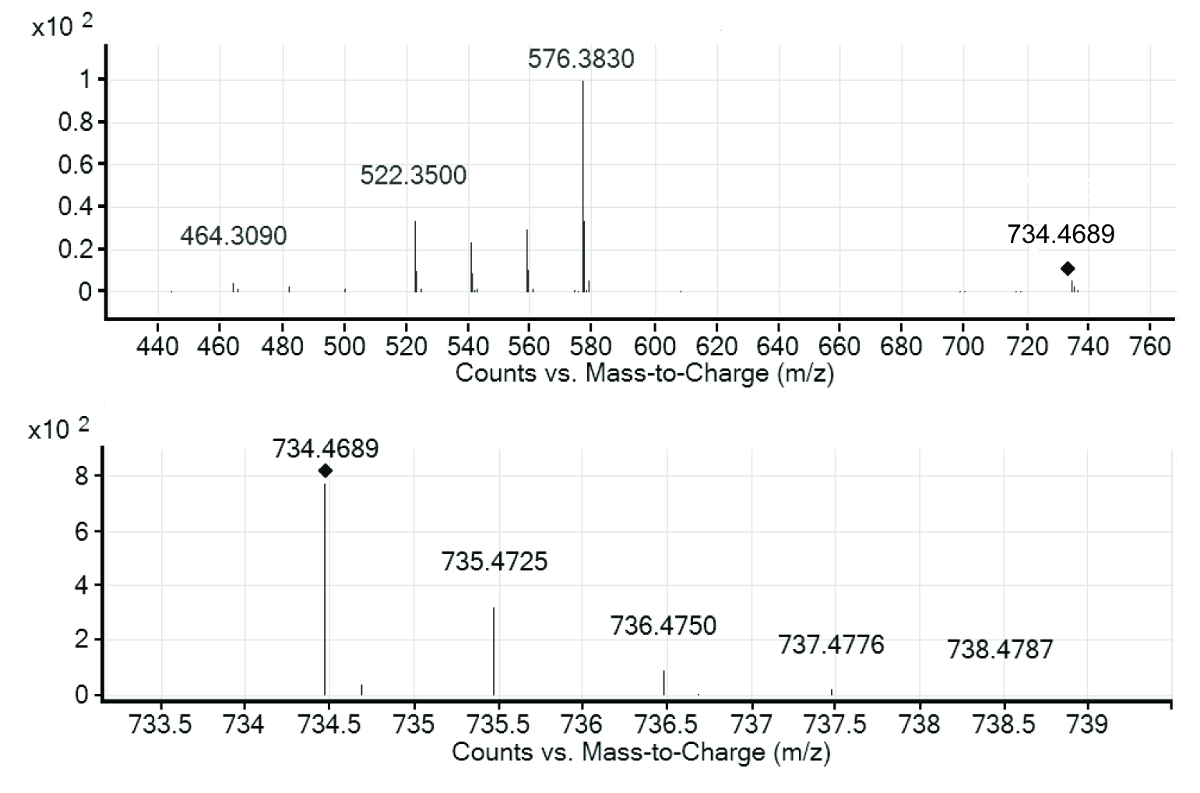
La CAB contenait plus de vingt-cinq pics qui reflète les valeurs différentes de m/z, donc différents composés, dont sept ont été marqués dans la CAB. Étant donné que les substances étaient inconnues a priori, la première étape vers leur identification se compose habituellement de dérivation de la formule moléculaire. Ceci est accompli à travers la masse exacte et isotopique modèle fourni par la détection de la TOF, bien que le modèle isotopique n’est pas observables dans tous les cas en raison de concentrations de l’échantillon faible dans des échantillons environnementaux. Avec l’aide de bases de données publiques, comme les produits pharmaceutiques dans l’environnement de l’Agence de l’environnement allemand (UBA) contenant environ 630 composés, il réussit souvent une identification préliminaire d’un groupe restreint de candidats. Pour une preuve définitive, soit comparaison d’étalons de référence disponible dans le commerce peut-être être effectuée ou patrons de fragmentation MS/MS peuvent être considérées (Figure 3).
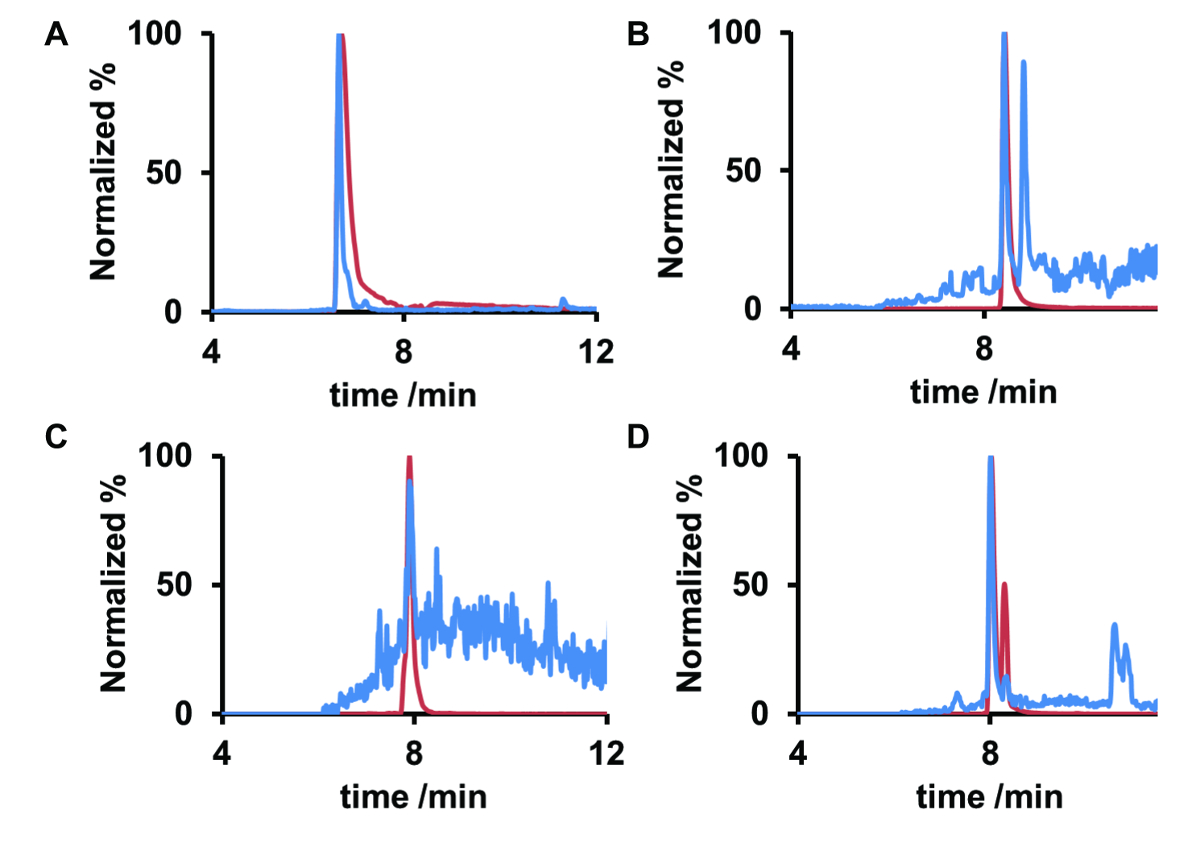
Dans cet ouvrage, comparaison aux normes en ce qui concerne les temps de rétention ont représenté pour l’identification des produits pharmaceutiques très souvent trouvé dans les eaux de surface allemands. Ces substances incluent metoprolol, un β-bloquant, carbamazépine, un analgésique et l’érythromycine antibiotique macrolide A et ses dérivé anhydroerythromycin, A. érythromycine sert d’exemple plus examiné dans cette étude. L’échantillon étudié du fleuve Rhin avait un pH de 7,6 et une température moyenne de 16,5 ° C. À ce pH, anhydroerythromycin devrait également être présent dans l’échantillon d’eau. Pour l’analyse détaillée, les chromatogrammes d’ion extrait (EIC) de l’échantillon ont été comparés avec des étalons de référence (Figure 4).
La comparaison montre la bonne concordance entre le temps de rétention de métoprolol, carbamazépine et anhydroerythromycin et les analytes observées. L’EIC de l’anhydroerythromycin standard de référence affiche deux pics, d'où deux composés où la déshydratation avait eu lieu à deux endroits distincts de l’érythromycine. Pourtant, un seul isomère anhydroerythromycin a été identifié dans l’échantillon du fleuve Rhin. Érythromycine lui-même assistait seulement sous forme de traces. Par conséquent, aucun spectre MS/MS ne pourrait être obtenue. Les masses exactes pour l’antibiotique et sa déshydratation sont donnés dans le tableau 2. À l’aide de CPN, donc temps de valeur et la conservation de m/z, metoprolol, carbamazépine, érythromycine et anhydroerythromycin pourraient être identifiées dans l’échantillon du fleuve Rhin.
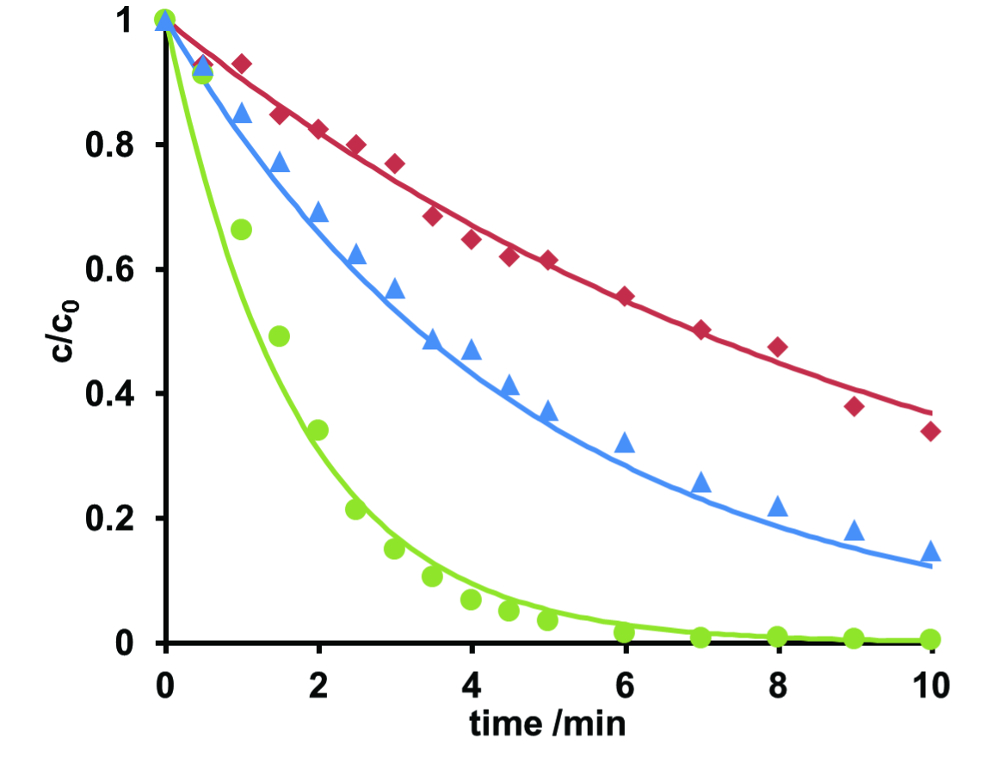
En ce qui concerne le milieu aquatique, il est important d’éviter les produits pharmaceutiques en passant par les usines d’épuration et de pénétrer dans les eaux de surface. Dans la quête d’une élimination efficace, des expériences d’irradiation UV-C à différentes valeurs de pH ont été effectuées pour l’érythromycine comme exemple. Diagrammes de concentration-temps (c-t) ont été enregistrés à l’aide de la région de masse vs temps parcelles dérivées de SCEI. La dégradation a été décrite de l’équation 2. L’érythromycine se compose d’érythromycine A et B anhydroerythromycin A, avec deux isomères de ce dernier. Les courbes de c-t de l’érythromycine A et leurs ajustements calculs sont indiquées dans la Figure 5. À pH 7, dégradation accélérée a été observée. Cela s’applique à tous les quatre composés étudiés, données non présentées. En conséquence, photo-dégradation de l’érythromycine devrait effectuer autour de pH neutre. Dans le cas de l’échantillon de fleuve de Rhin, ajuster le pH n’était pas nécessaire.
Photodegradates de produits pharmaceutiques ont été également identifiés dans les trois valeurs de pH. Tableau 3 donne un aperçu de ces photodegradates avec leurs propositions de structure correspondante. Pour l’analyse de la cinétique de la photodegradates, le produit à m/z = 720 sert à titre d’exemple. Photodegradates peut souvent être décrit comme intermédiaires de réaction. Donc, les photodegradates ont été décrits en termes d’aconsecutive et réaction suivie ultérieure. La décision entre les types qui en résulte des intermédiaires est basée sur la bonté de l’ajustement calculé avec un logiciel adapté, où le coefficient de détermination (R2) et l’erreur résiduelle de quadratique moyenne (EQM) ont servi de critères. Dû au fait que l’érythromycine est acide-instable, aussi qui pourrait se produire lors de l’irradiation étaient présent avant l’irradiation. L’effet qui en résulte sur les équations 3 et 4 était une concentration initiale finie. Par conséquent, un facteur a été ajouté aux équations. La figure 6 montre les données expérimentales et ajustements calculée selon l’équation 3 et 4.
Cet exemple d’un intermédiaire a montré l’augmentation de la concentration avec une sigmoïde montée suivie par une décroissance exponentielle. Ceci est indicatif pour une réaction suivie intermédiaire. Un intermédiaire de réaction consécutive ne montre pas l’augmentation de la sigmoïde. Paramètres de la qualité des statistiques a également indiquent l’accord légèrement supérieure d’ajustement selon le modèle de réaction suivi ultérieur. Le coefficient de détermination R2 de la réaction consécutive a été 0.9898 et donc inférieure à celle de la réaction de suivi ultérieure étant 0.9976. Par conséquent, le photoproduit examiné a été interprété comme intermédiaires de réaction suivie ultérieure. Les valeurs de k résultent de la computational fit aussi bien, la demi-vie a été calculée suivante équation 5. Tous les paramètres cinétiques pertinents sont collectés dans le tableau 3.
La dégradation plus rapide a été observée à pH 7, suivi de pH 9, alors que la dégradation plus lente a été trouvée pour pH 3 (Figure 5). Cette constatation est également appliquée à la formation et la dégradation des photoproduits. Trois photodegradates ont été observés. Leurs valeurs de m/z étaient 750.46 correspondant à Ery F, 720.45 à Ery C et 192.12 à DPEry192, un sucre glycosides dépendant de la structure de l’érythromycine (Figure 7). Aucune dégradation de le photoproduit ne pourrait être observée pour DPEry192 à pH 3 et 9 et pour Ery F à pH 9. Dans ces cas, la durée d’irradiation n’était pas suffisamment longue observer une dégradation totale du produit intermédiaire. Néanmoins, la constante de vitesse de formation puisse être déterminée en utilisant l’équation 5, ce qui correspond à un produit final.

Figure 1 . Comparaison des échantillons provenant du Rhin après SPE (gauche) et traitement water(right) ultrapure. La coloration verte est indicative pour les substances contenant de la chlorophylle. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 . BPC d’un échantillon d’eau après SPE mesuré par HPLC-ESI-Q-TOF-Mme Tous les chromatogrammes ont été normalisées pour le point culminant. M/z-valeurs illustratives obtenu à partir du spectre MS correspondant sont marqués. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 . Q-TOF-MS du spectre de l’érythromycine A (en bas) et le spectre MS/MS de l’ion m/z = 734.4689 (en haut). Les spectres montrent l’ion moléculaire quasi de l’érythromycine A, avec son modèle isotopique et les fragments d’une énergie de collision appliquée de 30 eV. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 . Normalisé SCEI de métoprolol (A), carbamazépine (B), l’érythromycine (C) A et (D) dans un échantillon de fleuve Rhin (bleu) et en eau ultrapure de composés de référence (rouge) anhydroerythromycin A. Les temps de rétention des composés référence et celles de la pharmacie dans l’échantillon d’eau sont les mêmes. Les ratios signal-bruit de métoprolol (A) et anhydroerythromycin (D) sont supérieurs à ceux de la carbamazépine (B) et l’érythromycine (C), ce qui indique que ces derniers étaient présents uniquement dans les traces. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 . Normalisée des courbes concentration-temps de la photodégradation de l’érythromycine A à pH 3 (rouge), pH 7 (vert) et 9 (bleu). Des solutions ont été irradiées pendant 10 min. À pH 7, l’érythromycine a été complètement retiré de l’échantillon. Les courbes concentration-temps pourraient être décrite en utilisant les équations cinétiques de premier ordre. Les constantes cinétiques ont été 0,10 (pH 3), 0,59 (pH 7) = 0,21 (pH 9). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

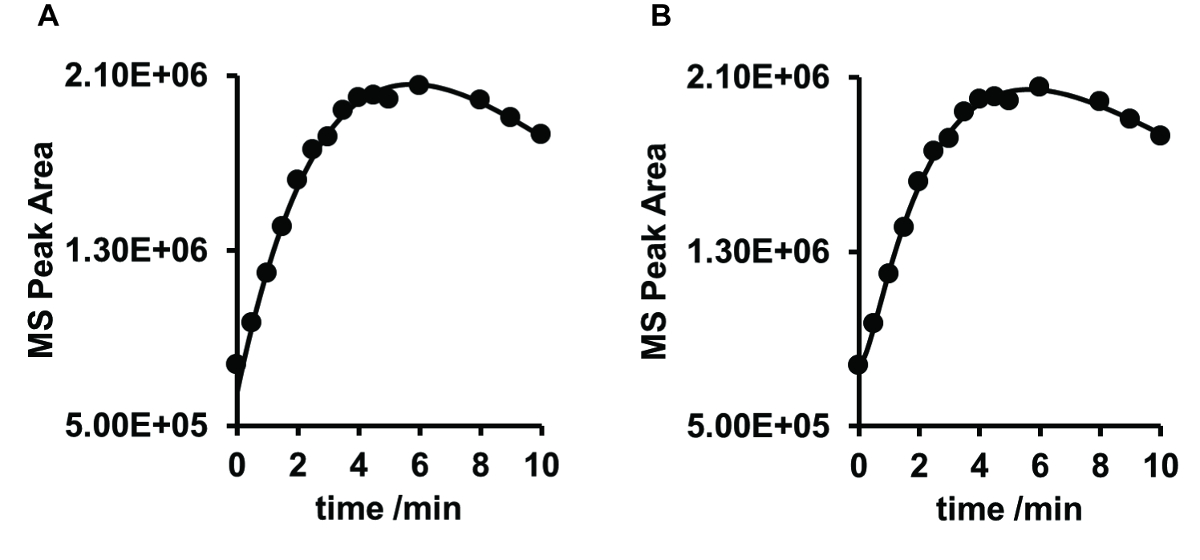
Figure 6 . Comparaison de l’ajustement des courbes concentration-temps de la photoprodegradates d’érythromycine avec m/z = 720 à pH 9 suite d’équations 3 A et 4 B. L’ajustement de la réaction consécutive (A) : R2 = 0.9898, RMSE = 4.645E + 04 et de la réaction de suivi (B) : R2 = 09976, RMSE = 2.366E + 04. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 7 . Structure de l’érythromycine A, érythromycine B et anhydroerythromycin et leurs produits de photdegradation. Ce chiffre a été modifié par Voigt et al. 27. les produits ont été formées après 10 min d’irradiation UVC et identifié à l’aide de HPLC-Q-TOF-MS et MS/MS. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Chromatographie en phase liquide | ||||||
| Colonne : | C-18 de phase inversée | |||||
| Colonne : | CoreShell colonne ; | |||||
| Colonne : | dimensions 50 x 2,1 mm, taille des particules 2.6 μm | |||||
| Température de la colonne | 40 ° C | |||||
| Volume d’injection : | 5 ΜL | |||||
| Débit : | 0,3 mL/min | |||||
| Phase mobile : | Solvant A: eau contenant 0,1 % d’acide formique | |||||
| B: solvant méthanol contenant 0,1 % d’acide formique | ||||||
| Programme de gradient : | ||||||
| Fois/min | 0 | 1 | 10 | 11.1 | 11.2 | 12 |
| Ratio de solvant a : b | 99 : 1 | 70 : 30 | 25 : 75 | 1 : 99 | 1 : 99 | 99 : 1 |
| Spectrométrie de masse | ||||||
| Source : | Double AJS ESI (mode positif) | |||||
| Gaz et source | ||||||
| Température des gaz : | 300 ° C | |||||
| Séchage des gaz : | 8,0 L/min | |||||
| Nébuliseur : | 14 lb/po2 | |||||
| Température de gaz de la gaine : | 300 ° C | |||||
| Débit de gaz de gaine : | 8 L/min | |||||
| Gamme de masse : | 100 - 1000 m/z | |||||
| Fréquence d’acquisition : | 1. spectre/s | |||||
| Temps d’acquisition : | 1000 ms/spectre | |||||
| Transient / spectre | 10014 | |||||
| Pour la méthode de MS ciblé | ||||||
| Énergie de collision (CE) : | 0 eV | |||||
| Préféré de masse - Table | 734.4685 | |||||
| Pour MS/MS (typiquement le mode auto MS/MS) | ||||||
| Énergie de collision (CE) : | 30 eV | |||||
| Seuil absolu | comtes de 3000 | |||||
| Seuil de relative | 0,01 % | |||||
| Gamme de masse : | 100 - 100 m/z | |||||
| Fréquence d’acquisition : | 1. spectre/s | |||||
| Temps d’acquisition : | 1000 ms/spectre | |||||
| Transient / spectre | 9964 | |||||
| Pour la méthode ciblée de MS/MS | ||||||
| Préféré de masse - Table | 734.4685 | |||||
Tableau 1. Conditions et paramètres utilisés pour l’analyse HPLC-ESI-Q-TOF-MS de produits pharmaceutiques dans les matrices eau. Il est conseillé d’introduire une étape de rinçage entre les pistes chromatographiques en roulant un échantillon d’eau ultrapure pur entre deux analyses ou par le biais de prolongement au moment de l’exécution de la méthode chromatographique pour éluer toutes les substances.

Le tableau 2. Produits pharmaceutiques trouvées dans l’échantillon de fleuve de Rhin avec leurs temps de rétention, théoriques et observés [M + H]+ et leur structure. Le mode de l’ESI a défini à positif, afin que [M + H]+-ions ont été détectés. Le temps de rétention peuvent varier au minimum pour des raisons connues expérimentales habituelles.
| pH 3 | pH 3 | pH 7 | pH 7 | pH 7 | pH 7 | pH 7 | pH 7 | pH 9 | pH 9 | pH 9 | pH 9 | |
| Produit | k1 [min-1] | t1/2 [min] (k1) | k1 [min-1] | k2 [min-1] | k3 [min-1] | t1/2 [min] (k1) | t1/2 [min] (m2) | t1/2 [min] (m3) | k1 [min-1] | k2 [min-1] | t1/2 [min] (k1) | t1/2 [min] (m2) |
| Ery A | 0,1 | 6.81 | 0,59 | - | - | 1.18 | - | - | 0,21 | - | 3.37 | - |
| Ery B | 0.05 | 14.23 | 0,66 | - | - | 1.04 | - | - | 0,22 | - | 3.21 | - |
| Ery A-Oa de2H | 0,11 | 6.53 | 0,59 | - | - | 1.17 | - | - | 0,19 | - | 3.72 | - |
| Ery A – Ob de2H | 0,15 | 4.76 | 1.11 | - | - | 0,63 | - | - | 0,21 | - | 3.35 | - |
| Ery F | non observée | - | 0,89 | 0,35 | - | 0,78 | 1.98 | - | 1.09* | - | 0,64 | - |
| Ery C | non déterminée | - | 0,74 | 5.27 | 0,78 | 0,94 | 0,13 | 0,89 | 0,17 | 0,18 | 4.04 | 3.92 |
| DPEry192 | 0.35* | 1,97 | non observée | - | - | - | - | - | 0.30* | - | 2.34 | - |
| * Aucune autre dégradation observée | ||||||||||||
Tableau 3. Constantes cinétiques et demi-vie correspondante de la dégradation de l’érythromycine et ses photodegradates adapté de Voigt et al. 27 . L’érythromycine se compose de l’érythromycine A, érythromycine B et deux formes d’anhydroerythromycin. Trois photodegradates ont été observés. Il sont appelés Ery F, Ery C et DEry192.
Discussion
L’exemple d’une analyse non ciblées présentée dans ce rapport ont démontré l’identification des produits pharmaceutiques dans les eaux de surface à l’aide de HPLC-ESI-Q-TOF-MS, MS/MS et comparaison avec les normes de référence comme la preuve définitive. La force de l’analyse non ciblés à l’aide de TOF-MS repose sur la détection de tous les ions présentes à un temps de rétention donné et la haute précision de masse qui mène à la prédiction de la formule moléculaire est indicative. Comme alternative à un spectromètre de masse de TOF, l’application d’un piège à ions orbital a été décrite pour l’analyse des contaminants dans l’eau,44. La prédiction de formule moléculaire a été utilisée comme point de départ pour sélectionner rapidement les étalons de référence. L’application de la méthode de MS ciblée de l’instrument de Q-TOF-MS a permis la détection de composés spécifiques, puisque seuls les ions présélectionnées passent le filtre quadripolaire. En général l’analyse ciblée est effectuée en utilisant le spectromètre de masse quadripolaire triple également dans l’analyse de l’eau45. Pour compenser l’écart par rapport à la masse théorique dues aux imperfections instrumentales, une comparaison par chromatographie en phase avec un étalon de référence peut être exécutée. La méthode MS/MS ciblée peut-être également être retenue pour l’analyse de l’identification. Ici, les ions sont sélectionnées, fragmenté et leurs fragments détectés. MS/MS étant moins sensibles que les MS, la concentration de la pharmacie dans les échantillons d’eau étudiés était trop faible pour produire des fragments significatifs. Toutefois, si les fragments sont détectés, composés peuvent être identifiés avec confiance plus élevé. La sensibilité insuffisante pourrait être surmontée en concentrant un plus grand volume d’échantillon de l’eau initiale. En outre, la mesure doit être effectuée dès que possible après le prélèvement en raison du potentiel de biodégradation46,47,48,49. Dans le cas contraire, les échantillons doivent être stockés à-20 ° C d’exclure composée dégradation ou réaction.
Parfois, les mêmes valeurs de m/z apparaissent à des moments différents de rétention. C’est peut-être à cause que les isomères exigent différentes techniques d’analyse. Il peut arriver également qu’aucun composé ne peut-être être détectées à tous, qui ne prouve pas nécessairement leur absence. Ils pourraient tout simplement pas former des ions ou se produisent sous le seuil de détection. Le type d’eau exerce également une influence sur la présence de produits pharmaceutiques. Produits pharmaceutiques rarement entrer dans l’eau de source et des eaux souterraines par rapport aux eaux usées et les effluents des eaux usées traitement plantes48,50,51,52,53.
Pour les expériences de dégradation, la source d’irradiation doit être caractérisée à l’avance, étant donné que le flux de photons ou taux de fluence des photons de la lampe contribue de manière significative à la dégradation et le mécanisme de dégradation. Pour les premiers essais, une lampe VUV/UVC, probablement une lampe de mercure à basse pression est suffisante. En règle générale, l’ajout de peroxyde d’hydrogène, H2O2, accélère la dégradation27,36,37,54. Lorsqu’une lampe différente, par exemple., une lampe UVA, est utilisée, la formation des radicaux hydroxyles devrait être assurée, par exemple., grâce à l’ajout de dioxyde de titane 23,24,30, 31. pour de nombreux composés, comme l’érythromycine, radicaux OH plutôt que photoréactivité du produit pharmaceutique lui-même27sont les espèces induisant la dégradation.
Pour la détermination des paramètres cinétiques, la zone des signaux dans les chromatogrammes de masse détectés, ce qui représente la concentration, est tracée en fonction du temps d’irradiation. Pour ajuster les données, il est conseillé d’utiliser un logiciel adapté. Ici, la sertir courbe de MATLAB a servi, ce qui a permis à rapidement calculer et ajuster les données avec les équations correctes. La cinétique des intermédiaires est déterminée par des équations plus complexes. Les paramètres de qualité de l’ajustement, c’est à dire., R2 et RMSE, on obtient facilement aussi bien.
Cette étude a démontré l’analyse de l’eau de la rivière pour détecter et identifier les polluants pharmaceutiques et la photodégradation d’érythromycine en eau ultrapure. Dans les eaux environnementales, telles que l’eau de surface, les vitesses de dégradation des différents et des constantes de vitesse obtiendrait due à la lumière d’absorber des substances, telles que humines. Selon l’expérience des auteurs, souvent la dégradation se déroule plus lentement, mais parfois à des taux comparables41,56.
Le problème dans le monde entier des produits pharmaceutiques, notamment des antibiotiques, dans le milieu aquatique et les dangers qui en résultent continuent de croître de1. En raison de la variété et la diversité des produits chimiques, les métabolites et aussi, l’analyse indifférenciés deviendra la principale arme analytique pour leur découverte de l' environnement57. Pour l’élimination effective, nouveaux stades dans les usines de traitement des eaux usées devront être conçus basés sur les procédés d’oxydation avancée, dont l’irradiation UV pourrait faire partie de.
Déclarations de divulgation
Les auteurs déclarent sans intérêts financiers concurrents.
Remerciements
Melanie Voigt est reconnaissante pour une allocation de formation de la Promotionskolleg des Niederrhein University of Applied Sciences. Les auteurs remercient leur institution pour une nouvelle aide financière.
matériels
| Name | Company | Catalog Number | Comments |
| Methanol for liquid chromatography LiChrosolv | Merck | 1060181000 | |
| formic acid | Fluka | 94318 | |
| HCl | Riedel-de Haen | ||
| NH3 | Riedel-de Haen | ||
| Simplicity 185 Water Purification System | EMD Millipore | for producing MilliQ-water | |
| Erythromycin | BioChemica AppliChem | A2275,0005 | |
| Filter Rotilabo-filter, Typ 113A | Roth | AP78.1 | |
| SPE-Cartridges Oasis HLB 3cc (60mg) | Waters | WAT094226 | |
| BAKER SPE-12G | J.T. Baker | ||
| membrane pump PC3001 VarioPro | Vacuubrand | ||
| rotary evaporator; Laborota 4000 efficient | Heidolph Instruments | ||
| syringe, 2 mL | Terumo | ||
| Nylon Syringe Filters Target2 | Thermo Scientific | 10301345 | |
| C-18 CoreShell column 50 mm x 2.1 mm dimensions, 2.6 μm particle size | Thermo Scientific | ||
| HPLC 1200 | Agilent | ||
| ESI-Q-ToF-MS 6530 | Agilent | ||
| photoreactor, UV Labor Reactor System 3 | Peschl Utraviolet GmbH | ||
| VUV/UVC-lamp, TNN 15/32, 15 W | Heraeus | ||
| pH-meter, pHenomenal pH 1100L | vwr | 662-1657 | |
| magnetic stirrer | Heidolph Instruments | ||
| MassHunter Workstation B.06.00 | Agilent | ||
| MATLAB R2016b | Mathworks |
Références
- Kümmerer, K. Antibiotics in the aquatic environment - a review - part I. Chemosphere. 75 (4), 417-434 (2009).
- Tijani, J. O., Fatoba, O. O., Petrik, L. F. A review of pharmaceuticals and endocrine-disrupting compounds: Sources, effects, removal, and detections. Water, Air, and Soil Pollution. 224 (11), (2013).
- Li, W. C. Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environmental Pollution. 187, 193-201 (2014).
- Jones, O., Voulvoulis, N., Lester, J. N. Human pharmaceuticals in the aquatic environment a review. Environmental technology. 22 (12), 1383-1394 (2001).
- Carmona, E., Andreu, V., Picó, Y. Multi-residue determination of 47 organic compounds in water, soil, sediment and fish-Turia River as case study. Journal of Pharmaceutical and Biomedical Analysis. 146, 117-125 (2017).
- Kostich, M. S., Batt, A. L., Lazorchak, J. M. Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation. Environmental Pollution. 184, 354-359 (2014).
- Chiffre, A., Degiorgi, F., Buleté, A., Spinner, L., Badot, P. -M. Occurrence of pharmaceuticals in WWTP effluents and their impact in a karstic rural catchment of Eastern France. Environmental Science and Pollution Research. 23 (24), 25427-25441 (2016).
- Gros, M., Petrovic, M., Barceló, D. Wastewater treatment plants as a pathway for aquatic contamination by pharmaceuticals in the Ebro river basin (northeast spain). Environmental Toxicology and Chemistry. 26 (8), 1553-1562 (2007).
- Ibáñez, M., Borova, V., et al. UHPLC-QTOF MS screening of pharmaceuticals and their metabolites in treated wastewater samples from Athens. Journal of Hazardous Materials. 323, 26-35 (2017).
- Carmona, E., Andreu, V., Picó, Y. Occurrence of acidic pharmaceuticals and personal care products in Turia River Basin: From waste to drinking water. Science of the Total Environment. 484 (1), 53-63 (2014).
- Martínez, J. L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science Mag. 321, 365-368 (2008).
- World Health Organization Antimicrobial resistance - Global Report on Surveillance. Bulletin of the World Health Organization. World Health Organization. 61 (3), 383-394 (2014).
- Proia, L., Von Schiller, D., Alexandre, S., Balc, L. Occurrence and persistence of antibiotic resistance genes in river bio fi lms after wastewater inputs in small rivers. Environmental Pollution. 210, 121-128 (2016).
- Karthikeyan, K. G., Meyer, M. T. Occurrence of antibiotics in wastewater treatment facilities in Wisconsin, USA. Science of the Total Environment. 361 (1-3), 196-207 (2006).
- Prieto-Rodriguez, L., Miralles-Cuevas, S., Oller, I., Agüera, A., Puma, G. L., Malato, S. Treatment of emerging contaminants in wastewater treatment plants (WWTP) effluents by solar photocatalysis using low TiO2 concentrations. Journal of Hazardous Materials. 211, 131-137 (2012).
- Dela Cruz, N., Giménez, J., Esplugas, S., Grandjean, D., de Alencastro, L. F., Pulgarín, C. Degradation of 32 emergent contaminants by UV and neutral photo-fenton in domestic wastewater effluent previously treated by activated sludge. Water research. 46 (6), 1947-1957 (2012).
- Zuccato, E., Castiglioni, S., Bagnati, R., Melis, M., Fanelli, R. Source, occurrence and fate of antibiotics in the Italian aquatic environment. Journal of Hazardous Materials. 179 (1-3), 1042-1048 (2010).
- Castiglioni, S., Bagnati, R., Fanelli, R., Pomati, F., Calamari, D. Removal of Pharmaceuticals in Sewage Treatment Plants in Italy. Environmental Science and Technology. 40 (1), 357-363 (2006).
- Watkinson, J., Murby, E. J., Costanzo, S. D. Removal of antibiotics in conventional and advanced wastewater treatment: implications for environmental discharge and wastewater recycling. Water research. 41 (18), 4164-4176 (2007).
- López-Serna, R., Petrović, M., Barceló, D. Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)-tandem mass spectrometry (MS/MS). Chemosphere. 85 (8), 1390-1399 (2011).
- Christian, T., Schneider, R. J., Färber, H. A., Skutlarek, D., Meyer, M. T., Goldbach, H. E. Determination of Antibiotic Residues in Manure, Soil, and Surface Waters. Acta hydrochimica et hydrobiologica. 31, 36-44 (2003).
- Sacher, F., Thomas, F. Pharmaceuticals in groundwaters Analytical methods and results of a monitoring program in Baden-Württemberg, Germany. Journal of Chromatography. 938, 199-210 (2001).
- Kasprzyk-Hordern, B., Dinsdale, R. M., Guwy, J. Multi-residue method for the determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-phase extraction and ultra performance liquid chromatography-positive electrospray ionisation tandem mass spectrometry. Journal of chromatography. A. 1161 (1-2), 132-145 (2007).
- Zuckerman, J. M. Macrolides and ketolides: azithromycin, clarithromycin, telithromycin. Infectious Disease Clinics of North America. 18 (3), 621-649 (2004).
- Hassanzadeh, A., Helliwell, M., Barber, J. Determination of the stereochemistry of anhydroerythromycin A, the principal degradation product of the antibiotic erythromycin A. Organic & biomolecular chemistry. 4 (6), 1014-1019 (2006).
- Hassanzadeh, A., Barber, J., Morris, G., Gorry, P. Mechanism for the degradation of erythromycin A and erythromycin A 2'-ethyl succinate in acidic aqueous solution. Journal of Physical Chemistry A. 111 (4), 10098-10104 (2007).
- Voigt, M., Jaeger, M. On the photodegradation of azithromycin, erythromycin and tylosin and their transformation products - A kinetic study. Sustainable Chemistry and Pharmacy. 5, 131-140 (2017).
- Delaforge, M., Jaouen, M., Mansuy, D. Dual effects of macrolide antibiotics on rat liver cytochrome P-450. Biochemical Pharmacology. 32 (15), 2309-2318 (1983).
- Hansen, J. L., Ippolito, J., Ban, N., Nissen, P., Moore, P. B., Steitz, T. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Molecular Cell. 10 (1), 117-128 (2002).
- Xekoukoulotakis, N. P., Xinidis, N., et al. UV-A/TiO2 photocatalytic decomposition of erythromycin in water: Factors affecting mineralization and antibiotic activity. Catalysis Today. 151 (1-2), 29-33 (2010).
- Yuan, F., Hu, C., Hu, X., Wei, D., Chen, Y., Qu, J. Photodegradation and toxicity changes of antibiotics in UV and UV/H(2)O(2) process. Journal of hazardous materials. 185 (2-3), 1256-1263 (2011).
- Monteagudo, J. M., Durán, A., San Martín, I. Mineralization of wastewater from the pharmaceutical industry containing chloride ions by UV photolysis of H2O2/Fe(II) and ultrasonic irradiation. Journal of Environmental Management. 141, 61-69 (2014).
- Malik, A. K., Blasco, C., Picó, Y. Liquid chromatography-mass spectrometry in food safety. Journal of chromatography. A. 1217 (25), 4018-4040 (2010).
- Hu, C., Xu, G. Mass-spectrometry-based metabolomics analysis for foodomics. TrAC Trends in Analytical Chemistry. 52, 36-46 (2013).
- Castro-Puyana, M., Herrero, M. Metabolomics approaches based on mass spectrometry for food safety, quality and traceability. TrAC Trends in Analytical Chemistry. 52, 74-87 (2013).
- Parsons, S. Advanced Oxidation Processes for Water and Wastewater Treatment. , IWA Publishing. London. (2004).
- Oppenländer, T. Photochemical Purification of Water and Air: Advanced Oxidation Processes (AOPs): Principles, Reaction Mechanisms, Reactor Concepts (Chemistry). , Wiley-Vch Verlag. Weinheim. (2003).
- Giannakis, S., Gamarra Vives, F. A., Grandjean, D., Magnet, A., De Alencastro, L. F., Pulgarin, C. Effect of advanced oxidation processes on the micropollutants and the effluent organic matter contained in municipal wastewater previously treated by three different secondary methods. Water Research. 84, 295-306 (2015).
- Fatta-Kassinos, D., Vasquez, M. I., Kümmerer, K. Transformation products of pharmaceuticals in surface waters and wastewater formed during photolysis and advanced oxidation processes - degradation, elucidation of byproducts and assessment of their biological potency. Chemosphere. 85 (5), 693-709 (2011).
- Vasconcelos, T. G., Henriques, D. M., König, A., Martins, A. F., Kümmerer, K. Photo-degradation of the antimicrobial ciprofloxacin at high pH: Identification and biodegradability assessment of the primary by-products. Chemosphere. 76 (4), 487-493 (2009).
- Voigt, M., Savelsberg, C., Jaeger, M. Photodegradation of the antibiotic spiramycin studied by high-performance liquid chromatography-electrospray ionization-quadrupole time-of-flight mass spectrometry. Toxicological & Environmental Chemistry. 99 (4), 624-640 (2017).
- Mauser, H. Formale Kinetik. Experimentelle Methoden der Physik und der Chemie. , Bertelsmann-UniversitĂtsverlag. Düsseldorf. (1974).
- Connors, K. A. Chemical Kinetics The Study of Reaction Rates in Solution. , VCH Verlagsgesellschaft. (1990).
- Comtois-Marotte, S., Chappuis, T., et al. Analysis of emerging contaminants in water and solid samples using high resolution mass spectrometry with a Q Exactive orbital ion trap and estrogenic activity with YES-assay. Chemosphere. 166, 400-411 (2017).
- Gago-Ferrero, P., Borova, V., Dasenaki, M. E., Thomaidis, N. S. Simultaneous determination of 148 pharmaceuticals and illicit drugs in sewage sludge based on ultrasound-assisted extraction and liquid chromatography-tandem mass spectrometry. Analytical and bioanalytical chemistry. 407 (15), 4287-4297 (2015).
- Yang, C., Hsiao, W., Chang, B. Chemosphere Biodegradation of sulfonamide antibiotics in sludge. Chemosphere. 150, 559-565 (2016).
- Gartiser, S., Urich, E., Alexy, R., Kümmerer, K. Ultimate biodegradation and elimination of antibiotics in inherent tests. Chemosphere. 67 (3), 604-613 (2007).
- Guerra, P., Kim, M., Shah, a, Alaee, M., Smyth, S. Occurrence and fate of antibiotic, analgesic/anti-inflammatory, and antifungal compounds in five wastewater treatment processes. The Science of the total environment. 473, 235-243 (2014).
- Jelic, A., Gros, M., et al. Occurrence, partition and removal of pharmaceuticals in sewage water and sludge during wastewater treatment. Water Research. 45 (3), 1165-1176 (2011).
- Lin, A. Y. -C., Tsai, Y. -T. Occurrence of pharmaceuticals in Taiwan's surface waters: Impact of waste streams from hospitals and pharmaceutical production facilities. Science of The Total Environment. 407 (12), 3793-3802 (2009).
- Sun, J., Luo, Q., Wang, D., Wang, Z. Occurrences of pharmaceuticals in drinking water sources of major river watersheds, China. Ecotoxicology and Environmental Safety. 117, 132-140 (2015).
- Nikolaou, A., Meric, S., Fatta, D. Occurrence patterns of pharmaceuticals in water and wastewater environments. Analytical and Bioanalytical Chemistry. 387 (4), 1225-1234 (2007).
- Gao, P., Ding, Y., Li, H., Xagoraraki, I. Occurrence of pharmaceuticals in a municipal wastewater treatment plant: Mass balance and removal processes. Chemosphere. 88 (1), 17-24 (2012).
- Andreozzi, R., Caprio, V., Insola, A., Marotta, R. Advanced oxidation processes (AOP) for water purification and recovery. Catalysis Today. 53, 51-59 (1999).
- Fernández, C., Callao, M. P., Larrechi, M. S. Kinetic analysis of C.I. Acid Yellow 9 photooxidative decolorization by UV-visible and chemometrics. Journal of hazardous materials. 190 (1-3), 986-992 (2011).
- Voigt, M., Bartels, I., Nickisch-Hartfiel, A., Jaeger, M. Photoinduced degradation of sulfonamides, kinetic, and structural characterization of transformation products and assessment of environmental toxicity. Toxicological & Environmental Chemistry. 99 (9-10), 1304-1327 (2017).
- Hoff, R., Mara, T., Diaz-Cruz, M. Trends in Environmental Analytical Chemistry Trends in sulfonamides and their by-products analysis in environmental samples using mass spectrometry techniques. Trends in Environmental Analytical Chemistry. 9, 24-36 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.