
Method Article
Tecniche di micromanipolazione che consente analisi delle dinamiche morfogenetiche e fatturato dei regolatori del citoscheletro
In questo articolo
Riepilogo
Descriviamo come tecniche di micro - e photomanipulation come FRAP e fotoattivazione permettono la determinazione dei parametri di motilità e la dinamica spazio-temporale delle proteine all'interno di migrazione delle cellule. Letture sperimentali includono subcellulare dinamiche e fatturato dei regolatori della motilità o del citoscheletro di actina sottostante.
Abstract
Esaminando la dinamica spazio-temporale delle proteine può rivelare loro importanza funzionale in vari contesti. In questo articolo, è discusso come fluorescente recupero dopo photobleaching (FRAP) e tecniche di fotoattivazione può essere utilizzato per studiare la dinamica spazio-temporale delle proteine nelle posizioni sottocellulari. Mostriamo anche come queste tecniche consentono la semplice determinazione di vari parametri collegati all'actina del citoscheletro regolamento cellulare motilità e. Inoltre, il microinjection delle cellule inoltre è descritto come un trattamento alternativo (potenzialmente precede o integrando le tecniche suddette photomanipulation) a grilletto effetti istantanei delle proteine spostati sulla cella morfologia e funzione. Micromanipolazione come proteina iniezione o l'applicazione locale di farmaci membrana plasmatica-permeabile o inibitori del citoscheletro può servire come potente strumento per registrare le conseguenze immediate di un dato trattamento il comportamento delle cellule presso la singola cella e subcellulare livello. Questo è esemplificato qui da induzione immediata della sporgenza bordo di lamellipodial delle cellule tramite l'iniezione di proteine ricombinanti di Rac1, come stabilito un quarto di secolo fa. Inoltre, mettiamo a disposizione un protocollo per la determinazione del fatturato di migliorata della proteina fluorescente verde (EGFP)-VASP, una polimerasi del filamento di actina prominente accumulando alle punte di lamellipodial delle cellule B16-F1, impiegando FRAP e compresi dati associati analisi e adattamento della curva. Siamo presenti anche linee guida per la stima dei tassi di polimerizzazione di rete lamellipodial dell'actina, come esemplificato dalle cellule che esprimono EGFP-Tag β-actina. Infine, le istruzioni sono date per come analizzare i tassi di mobilità di monomero di actina all'interno del citoplasma delle cellule, seguito dall'incorporazione di actina in siti di Assemblea del filamento rapida, ad esempio le punte sporgenti lamellipodi, utilizzando fotoattivazione si avvicina. Nessuno di questi protocolli è limitata ai componenti o regolatori del citoscheletro, ma può essere facilmente esteso per esplorare in moda analoga la dinamica spazio-temporale e funzione delle proteine in varie strutture subcellulari differenti o funzionale contesti.
Introduzione
Monitoraggio la dinamica spazio-temporale delle proteine e altre molecole nelle cellule viventi è diventato uno strumento essenziale in molti campi della biologia cellulare e molecolare. Tecniche di microscopia compreso trasferimento di energia di risonanza di fluorescenza (FRET) e la durata di FRET-fluorescenza (FRET-FLIM) di imaging avanzate di fluorescenza, o consentire FRAP, perdita di fluorescenza in photobleaching (FLIP) e fotoattivazione, così come molti altri per il temporale e spaziale di rilevamento delle interazioni proteina-proteina, cambiamenti conformazionali, nonché su come determinare la cinetica di diffusione e localizzazione delle proteine differenti in cella1,2. FRAP e fotoattivazione tecniche, in particolare, sono ampiamente applicabili per l'esame i regolatori della migrazione delle cellule e del citoscheletro di actina. Queste tecniche possono essere applicate da solo o in combinazione con tecniche di micromanipolazione aggiuntive quali microiniezione3e coinvolgono l'espressione delle proteine fluorescente etichettati. Consentono la valutazione della cinetica di associazione di proteine actina ricche strutture coinvolte nella migrazione cellulare, ad esempio filopodi o lamellipodi, il fatturato delle proteine in adesioni focali4, o ramificata actina reti5. Essi consentono anche la determinazione dei tassi di polimerizzazione dell'actina lamellipodial, la valutazione della dispersione di actina monomerica nel citosol, il tasso di traslocazione monomero subcellulare actina polimerizzazione filamenti dell'actina in sporgenti lamellipodi6e altri parametri.
FRAP è un metodo per visualizzare e quantificare la mobilità delle proteine all'interno di una cellula vivente, originariamente sviluppato nel 1970 da Axelrod7. Una regione di interesse (ROI) all'interno di una cella, popolata con proteine fluorescente identificate, transitoriamente è esposto ad un laser di alta intensità, sufficiente a causare lo sbiancamento delle molecole fluoroforo presenti in questa regione durante un dato periodo breve di tempo. La greggia, fluorescente etichettato proteine localizzate all'esterno il ROI durante lo sbiancamento, diffusa e infiltrarsi regione sbiancata in funzione della loro dinamica spazio-temporale, causando lo spostamento di molecole photobleached nel tempo. Il tasso di recupero di fluorescenza in regioni sbiancate dipende da vari fattori, tra cui le dimensioni e il tasso di diffusione di una data molecola, e naturalmente suo tasso di fatturato entro il presunto associato struttura sbiancato. Così, proteine solubili medierà il recupero della fluorescenza all'interno il ROI sbiancato rapidamente attraverso la diffusione, mentre proteine strettamente associate a strutture, quali adesioni focali, avrà tempi più lunghi di fatturato, così come loro recupero di fluorescenza dipendono entrambi sulla diffusione della frazione solubile della proteina e dissociazione-associazione cinetica della frazione struttura-collegata. Recupero di fluorescenza è solitamente acquisita e quantificato fino a quando non viene raggiunto il livello iniziale di pre-candeggina intensità di fluorescenza. Tuttavia, questo non si verifica se una parte dell'intensità di fluorescenza iniziale appartiene alla cosiddetta frazione immobile, che è in grado di essere rifornito tramite diffusione o è reintegro al prezzo molto lento rispetto la maggior parte delle molecole che comprende il mobile frazione. Per determinare il tasso di turnover delle proteine, FRAP curve sono generate, che rappresenta il grado di fluorescenza recupero nel corso del tempo. Da queste curve di recupero, metà-tempi medi di recupero della proteina possono essere calcolati. Con la creazione si adatta curva del medio FRAP dati e analisi quindi matematiche, è anche possibile dedurre se il tasso di turnover medio della frazione mobile costituisce un composito di una popolazione omogenea di molecole, o se è composto da due o più sottopopolazioni di molecole girarsi al differenziale di prezzo. Oltre alla stima di tassi di turnover delle proteine dopo approcci quantitativi, il recupero delle regioni photobleached lamellipodi di rilevamento può anche permettere per quantificazione accurata dei parametri di motilità di lamellipodial come flusso retrogrado, protrusione, e tassi di polimerizzazione dell'actina. Così, FRAP costituisce uno strumento versatile per essere applicato per valutare diversi parametri all'interno delle strutture delle cellule viventi.
Fotoattivazione è un metodo utilizzato per rilevare la diffusione e la mobilità delle proteine o molecole provenienti da una posizione cellulare designata. La tecnica utilizza, per esempio, una variante del selvaggio-tipo proteina fluorescente verde (GFP), inizialmente sviluppata da Patterson e Lippincott-Schwartz8, che è mutato in un modo che permette la sua fluorescenza altamente essere aumentato sopra l'esposizione ad luce ultravioletta (UV) (circa 400 nm; qui, 405 nm). Come descritto da Patterson et al., cromofori GFP wild type esistano come una popolazione mista di fenoli neutri e phenolates anionici, che producono un picco di assorbanza principali a circa 397 nm e una minore a 475 nm, rispettivamente. All'irradiazione della proteina con luce UV, la popolazione subisce fotoconversione, spostando verso la forma anionica. Quando sono eccitati da 488 nm, la proteina di photoconverted/fotoattivazione esibisce un aumento di 3 volte in fluorescenza, insufficiente in pratica per distinguere tra non attivati e GFP dovuto la fluorescenza di fondo intrinseco elevato. Tuttavia, una diminuzione nell'intensità di sfondo è stata realizzata con l'introduzione di una mutazione di singolo amminoacido nella sequenza di GFP (sostituzione dell'istidina alla posizione 203). Il mutante di T203H risultante, anche conosciuto come fuse-GFP (PA-GFP) è caratterizzata da una riduzione significativa di assorbanza del picco minore, che al momento di irradiazione con luce UV è aumentato quasi 100 volte quando successivamente eccitato dalla luce di 488 nm. Quindi, l'iperespressione delle proteine PA-GFP-etichettato è un approccio ampiamente utilizzato, che permette la determinazione di diffusione e la motilità delle molecole all'interno delle cellule. In precedenza abbiamo applicato PA-GFP-etichettato di actina per determinare il tasso di dispersione di monomeri di actina via dalle regioni citosoliche, permettendo non solo l'esplorazione della loro mobilità all'interno del citosol, ma anche il loro tasso di incorporazione in sporgente l'actina lamellipodial rete6. Letteratura più recente descrive anche proteine romanzo, foto-convertibile che possono essere utilizzate in maniera analoga, in linea di principio ma che harboring il vantaggio potenziale per essere visibili già prima della conversione di foto. Gli esempi per questo gruppo di proteine fluorescenti includono Dendra2 e mEos29,10,11,12.
In questo articolo, spiegheremo la metodologia delle cellule microinjecting con proteine. Abbiamo ulteriormente spiegare come questa tecnica può essere combinata con FRAP, di photobleaching proteine coinvolte nella regolazione del citoscheletro di actina e la motilità, e come può essere derivati FRAP curve e metà-tempo di recupero delle frazioni mobile. Inoltre, forniamo un esempio di come la tecnica FRAP può essere utilizzata per determinare i tassi di polimerizzazione dell'actina di lamellipodial reti. Forniamo anche le istruzioni e suggerimenti su come eseguire esperimenti di fotoattivazione, che possono essere utilizzati per determinare la mobilità citosolico di actina monomerica e tassi di incorporazione dell'actina lamellipodi. Queste tecniche, naturalmente, non sono solo limitati a rilevamento dei componenti del citoscheletro di actina, ma al momento potenzialmente richiesto adattamento moderato o ottimizzazione, può essere ampiamente applicato ad altri tipi di cella o per indagare le diverse proteine, strutture, e parametri.
Protocollo
1. vetrino coprioggetto lavaggio e sterilizzazione
- Immergere i vetri di coperchio di 15 mm (diametro) (n ° 1) in un pallone da 500 mL contenente una miscela di 40 mL 37% HCl e 60 mL 100% EtOH (non più di 100 vetrini coprioggetto per 100ml soluzione di lavaggio).
Nota: anche se appena acquistato, le lamelle devono essere rigorosamente pulite prima della semina di cellule sulle loro superfici. Ciò è perché possono contenere film sottili di grasso, che non sono visibili in modo macroscopico, ma può interferire in modo efficiente con adesione e diffusione appropriata delle cellule vive. Considerando che tali pellicole possono essere rimossi in modo efficiente con soluzioni contenenti acidi o basici (Vedi Fischer et al. 13), usiamo ordinariamente la miscela di acido/alcool sopra descritta. - Agitare il matraccio contenente i vetri di copertura per 30 minuti su un agitatore di rotazione. Scegliere una velocità che consente i vetri di copertura essere roteato liberamente, ma abbastanza per evitare la frequente rottura lento. La soluzione per rimuovere i pezzi di vetro rotto se riutilizzando del filtrato.
- Trasferire i vetri di copertura in un pallone contenente almeno 200 mL di acqua sterile e incubare su un agitatore di rotazione, mentre la sostituzione ripetutamente l'acqua fino a quando l'odore acidulo è scomparso. Diversi lavaggi sopra parecchie ore sono raccomandati per la completa eliminazione di tracce di HCL-EtOH.
- Asciugare i singoli vetri di coperchio su un foglio di carta da filtro.
- Posto i vetri di copertura nella parte inferiore di una capsula di Petri 10 cm (diametro) ricoperta di carta da filtro e calore secco-sterilizzare. Evitare di sterilizzazione in autoclave ciò causerebbe coprioggetto stare insieme.
2. il trattamento delle cellule, la transfezione e semina su vetrini coprioggetti
- Crescere le cellule di melanoma B16-F1 del mouse secondo le condizioni di coltura standard celle in DMEM (4,5 g/L glucosio) contenente 10% siero fetale del vitello, la glutamina 2mm e 1% di penicillina-streptomicina a 37 ° C, 7% CO2.
- Crescere le cellule dei fibroblasti NIH3T3 per microiniezioni secondo le condizioni di coltura cellulare standard (coltura tissutale incubatore a 37 ° C, 7% CO2) in DMEM (4,5 g/L glucosio) contenente 10% di siero bovino fetale, piruvato di sodio di 1 mM, 1 x MEM non essenziali aminoacidi... , la glutamina 2mm e 1% di penicillina-streptomicina.
- Per transfezioni, crescere cellule B16-F1 alla confluenza del 100% in un piatto di 10 cm e il passaggio ad un rapporto di 1:5 in un piatto di plastica di 3 cm (diametro).
- Lo stesso giorno, dopo che le cellule B16-F1 furono permesso di aderire per almeno 6 h, transfect con 500 ng/piatto di fuse PA-GFP-actina o DNA plasmidico di EGFP-Tag β-actina. Per co-transfezioni di PA-GFP-actina con vettori mCherry-codifica, mescolare un totale di 1 µ g di DNA plasmidico per ogni piatto di 3 cm.
- Transfect le cellule B16-F1 con il reagente di transfezione (Tabella materiali). Per un piatto di 3cm, mescolare 200 µ l di 150 mM NaCl contenente 500 ng di DNA costruire con 200 µ l di 150 mM NaCl contenente 1 µ l di reagente di transfezione (cioè, DNA (µ g): reagente (µ l) rapporto di 1:2 è stato usato).
- Incubare la miscela di transfezione per 20 min a temperatura ambiente (TA) e dispensare drop-wise sul piatto da portata 3 cm contenente le celle. Agitare delicatamente il piatto per miscelare e incubare per una notte a 37 ° C, 7% CO2.
- Preparare il tampone di rivestimento laminin contenente 50 mM Tris, pH 7,4 e 150 mM NaCl.
- Per le celle di B16-F1, cappotto 15 mm copertura vetri diffondendo 150 µ l di laminina (25 µ g/mL in tampone di rivestimento di laminina) e incubare per 1 h a TA. Per le cellule NIH3T3, rivestire i vetri di copertura con soluzione di fibronectina (25 µ g/mL in tampone fosfato salino (PBS)) e incubare per 1 h a TA.
- Lavare vetri di copertura laminina e fibronectina incubati con PBS, poi aspirare il PBS e aggiungere 2 mL di cellule trasfettate.
- Le cellule trasfettate B16-F1 di semi (a 01:30 rapporto da un piatto confluente), il giorno dopo la trasfezione in lamelle rivestite con laminina. I fibroblasti NIH3T3 di semi (a 01:20 rapporto da un piatto confluente) su vetrini coprioggetti rivestite di fibronectina.
- Permettono alle cellule di diffondere il coprioggetto fibronectin-rivestito o di laminina - pernottamento in un coltura tissutale incubatore a 37 ° C prima della microscopia. In alternativa, esperimenti di microscopia possono essere avviati nello stesso giorno, dato che le cellule sono autorizzate a diffondere per almeno 2 – 3 h.
3. montaggio della camera di formazione immagine di microscopia
- Utilizzare un calore conduttivo RC-26 in alluminio imaging camera per microscopia (Figura 1a). Spalmare il grasso di silicone intorno al contorno dell'apertura di plastica isolante usando una siringa (Figura 1b).
- Mettere il vetro di copertura con il lato di cellule-up sulla camera (Figura 1c).
- Inserire il sigillante di plastica sopra il vetro di copertura per fare un sigillo sicuro tra il vetrino coprioggetto e camera. Fissare la plastica isolante (diagonalmente per evitare la rottura del vetrino coprioggetto) avvitando i morsetti scorrevoli sulla camera per evitare il mezzo colatura (Figura 1d).
- Medio-microscopia preriscaldato pipetta 37 ° C in zona centrale. Per medie e ridotto in autofluorescenza e così ottimizzata per microscopia, utilizzare la stessa ricetta come descritto sopra, ma con F12-prosciutto invece DMEM, inoltre contenente 20 mM HEPES per la coltura delle cellule in assenza di CO2 (figura del terreno di coltura 1e).
- Inserire il rilevatore di calore nello slot designato dell'alloggiamento e collegare gli elettrodi della camera di un regolatore di temperatura automatico TC-324B mantenendo una temperatura costante di 37 ° C (Figura 1f).
- Mettere una piccola goccia di olio per immersione sull'obiettivo e posizionare la camera in cima.
- Incubare la camera con le cellule per almeno 10-30 min consentire loro di recuperare il calo della temperatura durante il montaggio e per adattarsi al mezzo di microscopia.
- Prima di microscopia è avviata, sostituire il mezzo di coltura nel serbatoio centrale della camera (circa 800 µ l) per evitare inadeguata concentrazione di componenti medi e del siero a causa dell'evaporazione media. Sessioni di microscopia prolungata con chambers aperta richiederà cambiando ordinaria del mezzo d'evaporazione.
4. microinjection procedura
- Rivestire i coprioggetti, preparare le cellule e montare la camera di imaging, come descritto in precedenza.
- Scongelare un'aliquota di proteina purificata da iniettare (tipicamente 10 µ l o meno) e diluirlo con il buffer di microiniezione appropriato.
Nota: Composizione di Buffer può variare secondo il tipo delle cellule e proteine, ma fare attenzione a utilizzare un pH compreso tra 6.95 e 8.00 ed evitare PBS, come la maggior parte delle cellule non piace essere iniettato con PBS. - Per il microinjection di Rac1, preparare tampone contenente NaCl 100 mM, 50 mM Tris-HCl a pH 7.5, 5 mM MgCl2, 1mm DTT. Gli ioni Mg2 + sono essenziali per piccole GTPasi stabilità.
Nota: Le concentrazioni di proteina normalmente variano tra 0,1 – 1 mg/mL (massimo 2 mg/mL), a seconda della proteina, il tipo di esperimento e cella tipo. - Se applicabile, è possibile aggiungere colorante fluorescente, come destrano inerte (0,5 µ g/mL, 70 kDa) per la soluzione della proteina, che può confermare la presenza di flusso dell'ago prima di iniezioni e permette la documentazione delle iniezioni successo dopo l'esperimento.
Nota: Non è puntato su l'esperimento qui seguendo la dinamica di Rac1 iniettato, che sarebbe solo possibile su fluorescenza diretta etichettatura della proteina. Accoppiamento delle proteine con coloranti fluorescenti o fusione di una proteina fluorescente è possibile, ma evitare qui come nasconde il rischio di interferire con la funzione di segnalazione, in particolare di piccole proteine come la famiglia Rho GTPase Rac1 (20 kDa). - Centrifugare la soluzione della proteina a 10.000 x g per almeno 30 min per rimuovere aggregati proteici che possono condurre ai ferri intasamento se presente nella microiniezione capillare.
- Caricare un ago per microiniezione (microiniezione capillare) con 1 µ l di miscela di iniezione dal lato posteriore utilizzando un puntale flessibile di punta/microloader.
- Se le bolle d'aria presenti nella punta dell'ago, picchiettare delicatamente la base dell'ago per rimuoverli. Procedere rapidamente per evitare la disidratazione della punta dell'ago, che potrebbe causare intasamento dell'ago.
- Regolare con cura il supporto dell'ago del dispositivo di micromanipolazione. Se si utilizza un microscopio invertito per l'imaging di contrasto di fase, prima del caricamento dell'ago, assicurarsi c'è abbastanza spazio per spostare l'ago su e giù senza ostruire il condensatore del microscopio.
- Al momento di avvitamento microcapillary sul supporto dell'ago, applicare pressione (pressione di base di 20 – 50 hPa) l'ago usando un dispositivo di pressione di microiniezione prima dispostamento la punta dell'ago nel terreno di coltura delle cellule.
Nota: Attivando la pressione quando l'ago è nel mezzo verrà causare il mezzo risucchiato dalla forza capillare e così proibire l'iniezione della soluzione di interesse. - Posizionare l'ago nel campo visivo (facilitato usando gli obiettivi a basso ingrandimento). Un secco obiettivo 40x per esperimenti di microiniezione è stata usata qui.
- Posizionare la punta dell'ago in modo macroscopico in posizione verticale rispetto al centro della lente dell'obiettivo (questo accelererà trovando la punta dell'ago). Utilizzare il microscopio con ottica a contrasto di fase per spostare la punta dell'ago sul piano orizzontale riguardante il campo di vista, a un piano ottico bene-sopra lo strato delle cellule.
Nota: L'ago inizialmente apparirà come un'ombra nel campo visivo e il piano di messa a fuoco può essere regolato per visualizzare il suggerimento. Una volta che la punta dell'ago è trovata, abbassare gradualmente sul piano ottico, seguito dalla punta dell'ago verso il basso per una posizione vicino lo strato delle cellule. - Controllare il flusso dell'ago passando a un canale fluorescente quando si utilizza il destrano fluorescente e sintonizzare il flusso utilizzando il dispositivo di pressione per ottenere un flusso costante "background".
Nota: In questo articolo, descriviamo iniezione manuale, che è mediata dalla rottura attraverso la membrana plasmatica tramite toccare la cella in superficie e delicato movimento della punta dell'ago durante il flusso costante dell'ago. Questo deve essere distinto da dispositivi di iniezione automatica accompagnati da aumento di pressione di abbassamento e l'ago ago programmato durante eventi di iniezione, che sono più appropriati per l'iniezione di più alti numeri di cellulare seguiti da una popolazione di cellule successiva analisi. Il metodo qui descritto è ottimizzato per analisi unicellulare di microscopia time-lapse, prima, durante e dopo la microiniezione. - Trovare una cella di interesse e ridurre gradualmente l'ago sopra la cella.
- Quando si è pronti a microinject, abbassare l'ago gradualmente verso la regione perinucleare di cella utilizzando il pignone bene del joystick micromanipolatore, mantenendo le cellule a fuoco.
- Per il microinjection, toccare delicatamente la membrana plasmatica della cellula, che può essere sufficiente per penetrare la cellula, o la rottura della membrana transitoria di aiuto da un tocco molto delicato sul setup microscopio.
Nota: Un punto bianco sulla punta dell'ago indicherà il tempo di contatto con la membrana plasmatica; Dopo la rottura della membrana, la punta dell'ago sarà reseal, accompagnato da un delicato flusso di soluzione dell'iniezione nella cella. - Interrompere il processo di iniezione, non appena è visibile il flusso nella cellula (idealmente entro 0,3 s) spostando la punta dell'ago nel mezzo. Quando si utilizza il destrano fluorescente, iniezioni di successo possono essere documentate immediatamente dalla fluorescenza.
- Se lo si desidera, Avvia acquisizione immagine time-lapse prima o dopo la microiniezione.
Nota: L'applicazione locale di farmaci o inibitori può essere eseguito in tutte le fasi qui, tranne per il microinjection evento per sé. Per applicazioni locali, la diffusione della molecola attiva può essere controllata dalla pressione di flusso e documentata di fluorescenza e la punta dell'ago può essere posizionata all'altezza desiderata. Per esempi di esperimenti di applicazione locale, vedere ad esempio, piccole e Rottner14 o Kaverina et al. 15 - In seguito la microiniezione, attendere che si verifichi l'effetto della proteina. Per proteine differenti e a seconda dei risultati attesi, tempi di incubazione possono variare. Per il piccolo GTPase Rac1, la risposta del lamellipodio formazione può essere avviato entro 1 min o meno, ma dura circa 10-15 min in media a sviluppare pienamente (Figura 1g, h).
- Giudicare l'attuabilità delle cellule dopo la microiniezione.
Nota: Iniezioni Inappropriate o dannose possono causare danni alle cellule, che è frequentemente accompagnata da retrazione del bordo di cella non specifici o rottura della membrana del plasma.- Evitare che spuntavano attraverso la membrana plasmatica sia la parte superiore e inferiore, che può verificarsi per iniezioni nelle regioni piatto cellulare.
Nota: I volumi di iniezione devono essere mantenute al minimo (idealmente < 5% del volume cellulare) e sarà solitamente nella gamma femtoliter. Necessari volumi di iniezione possono anche essere controllati da cambiamenti nella concentrazione, ma si noti che per le proteine, le concentrazioni > 2 mg/mL può diventare impraticabile a causa di intasamento dell'ago di frequente. Tuttavia, questo dipende anche la qualità e il comportamento della proteina purificata; ad esempio, iniezione di actina fluorescente-accoppiato è complicata dalla polimerizzazione di concentrazione-dipendente e inevitabile nella punta dell'ago, e così viene raramente eseguita oggi (Vedi piccolo et al. 16).
- Evitare che spuntavano attraverso la membrana plasmatica sia la parte superiore e inferiore, che può verificarsi per iniezioni nelle regioni piatto cellulare.
- Prima, durante o dopo l'effetto della microiniezione, FRAP o fotoattivazione può essere eseguita sulla stessa cellula (veda la sezione 5 e 6).
5. procedura di FRAP
- Transfect il tipo di cella di interesse (B16-F1 celle qui) con il plasmide DNA codifica per una proteina fluorescente-tag di interesse (qui, è stata usata una versione di EGFP-etichetta di β-actina). Seme le cellule sul lamelle rivestite con laminina (punto 2.10).
- Assemblare la camera imaging (sezione 3).
- Utilizzare le seguenti impostazioni per lamellipodial regione photobleaching: potenza di 65 mW laser (variabile secondo la fonte di laser e installazione sperimentale); diametro del raggio laser 10 pixel; 1 ms candeggina dwell tempo/pixel; tempo di esposizione di 500 ms GFP; intervallo di tempo di 1.500 ms. In questa carta i risultati sperimentali sono stati effettuati con un obiettivo apocromatico 100 X 1.4NA.
- Eseguire la calibrazione del laser per garantire precisione nelle dimensioni della regione photobleached. Prima della calibrazione, spostare il campo di vista in un'area priva di qualsiasi segnale di cellule/fluorescenza e osservare l'immagine sul display.
- Selezionare l'ingrandimento dell'obiettivo facendo clic sul pulsante di ingrandimento rispettivi e ridurre la potenza del laser (3 – 5 mW) nel "pannello | Menu di intensità". Per avviare la calibrazione manuale sul software Visiview (v 2.1.4), selezionare il "Configura | FRAP"menu e fare clic sul" Calibra | Menu regolazione manuale". Assicurarsi che il laser può essere distinto come un punto forte. Se non, o riorientare o regolare l'hardware laser.
- Eseguire la calibrazione manuale guidando il laser al pre-determinato software X-Y coordinate. Questo indica che il software come bersaglio specificamente designate al laser di una regione definita dall'utente per l'ingrandimento corrente.
- Prima di attivare il laser, passare al canale GFP e Avvia acquisizione di lasso di tempo di immagine.
- Disegnare manualmente la regione per essere photobleached sul canale GFP, durante la visualizzazione del display.
- Avviare photobleaching da un trigger manuale del laser 405 nm, almeno 3 – 4 fotogrammi dopo l'inizio dell'acquisizione immagine. L'acquisizione di fotogrammi prima del photobleaching è richiesto per la normalizzazione dell'immagine in successiva analisi dei dati.
6. fotoattivazione procedura
Nota: Software, installazione di microscopio e impostazioni, fatta eccezione per la potenza del laser, sono simili a quelli per FRAP. In fotoattivazione, un'importante differenza rispetto ai FRAP, che è una potenza laser 405nm significativamente inferiore a quello impiegato per photobleaching deve essere utilizzato, per attivare PA-GFP senza contemporaneamente photobleaching esso.
- Co-transfect il tipo di cella di interesse (B16-F1 celle qui; Vedi punto 2.5) con il DNA del plasmide codifica PA-GFP-actina e un'altra proteina fluorescente etichettati (ad es., mCherry o mCherry-Lifeact).
Nota: Nella maggior parte dei casi, le cellule mCherry-positive anche sarà positive per il vettore di PA-GFP-actina, quest'ultimo normalmente non visto sul canale GFP prima di fotoattivazione. Per aumentare la possibilità che le cellule mCherry-positive sono anche positive per PA-GFP, utilizzare un rapporto di transfezione di 1:2 di mCherry:PA-GFP-actina. Seguendo questo protocollo, oltre il 90% delle cellule che esprimono mCherry visualizzato avvenuta attivazione dell'actina-GFP-PA. - Cellule di seme il B16-F1 sui vetrini coprioggetti rivestite con laminina (punto 2.10).
- Assemblare la camera imaging (sezione 3).
- Prima di iniziare gli esperimenti di fotoattivazione, se necessario, eseguire la calibrazione del laser per l'obiettivo selezionato (punto 5.4 – 5.6).
- Impostare l'acquisizione di immagini GFP/488 nm a intervallo di tempo di esposizione e 1.500 ms 500 ms (secondo il disegno sperimentale).
-
Regolare le impostazioni del software per l'acquisizione di filmati time-lapse doppio canale o a triplo canale marcatura la piazza "Serie di lunghezza d'onda" e selezionando il numero desiderato di canali nella "Acquire | Menu di lunghezza d'onda". È consigliabile che i film di lasso di tempo vengono acquisiti con contrasto di fase e canali GFP.
- Facoltativamente, includere anche il canale di mCherry; Tuttavia, esponendo le cellule con troppa luce potrebbe indurre photodamage. Questo potrebbe essere evitato con organismi saprofagi dell'ossigeno come Oxyrase17, anche se un trattamento efficace richiede cella camera di tenuta.
- Trovare le celle transfected sul canale mCherry.
- Prima di attivare il laser, Avvia acquisizione di lasso di tempo di immagine e disegnare manualmente la regione per essere fotoattivazione sul canale di contrasto di fase, durante la visualizzazione del display.
- Avviare la fotoattivazione di un trigger manuale del laser 405 nm (intensità impostata tra 5 – 15 mW dal "pannello | Menu di intensità"), almeno 3 – 4 fotogrammi dopo l'inizio di acquisizione immagine.
7. analisi dei dati e presentazione dei risultati FRAP
Nota: Il metodo presentato viene utilizzato per indagare il fatturato di una proteina accumulando presso siti di actina dinamico, in questo caso VASP, che associa con siti di adesione e le punte sporgenti lamellipodi. Stiamo analizzando il fatturato sulla punta di lamellipodio, ma gli stessi principi di analisi possono essere applicati per indagare il fatturato di VASP o qualsiasi altra proteina e altri compartimenti subcellulari.
- Aprire i filmati time-lapse, derivati da Visiview sul software Metamorph. In questo articolo, è stato usato Metamorph v7.8.10.
- Ricavare i valori di intensità per le regioni photobleached manualmente delineando rispettive regioni su Metamorph. Disegnare una forma sulla punta del lamellipodio che copre l'intera o parte della zona di photobleached e regola manualmente la posizione su fotogrammi successivi se necessario (ad esempio, se il bordo è sporgente), in ordine per tenere traccia delle modifiche in lamellipodial intensità di il rispettivo componente durante lo spostamento di punta.
- Per la correzione di sfondo e photobleaching acquisizione, analizzare le regioni all'interno e all'esterno della cella. Vedere Figura 2una per regioni rappresentative delle intensità misurata.
- Mentre è selezionato un ROI, estrarre i valori di intensità su Metamorph utilizzando il menu "misura | Misurazioni di regione". Assicurarsi "Elapsed time" e "Media intensità" opzioni siano selezionate nel menu "Configurazione". Fare clic su "Open log" e selezionare "Dynamic Data Exchange". Fare clic su "OK" per aprire un foglio di calcolo di Excel e fare clic sul pulsante "Aprire il registro" per incollare i valori di Metamorph in Excel.
Nota: Questi valori vengono utilizzati per generare le curve di recupero di fluorescenza. - Per la generazione di curve di recupero fluorescenza sulla punta di lamellipodio delle regioni photobleached (normalizzato all'intensità della regione prima photobleaching), applicare la seguente equazione:
 Equazione 1
Equazione 1
Dove: FRAPTn è l'intensità della regione di photobleached per ogni frame di interesse seguendo photobleaching; FuoriTn è un intensità di regione prese all'esterno della cella (sfondo) per ogni frame di interesse seguendo photobleaching; InsTn è due in media all'interno della regione di intensità per ogni frame di interesse seguendo photobleaching (utilizzato per normalizzare per acquisizione photobleaching nel tempo); FRAPT-1 è l'intensità di regione photobleached prima photobleaching; T-1 è un intensità di regione prese all'esterno della cella (sfondo) prima photobleaching; e InsT-1 sono due media all'interno della regione di intensità per ogni frame di interesse prima di photobleaching. - Per ogni intervallo di tempo di interesse, è possibile utilizzare equazione 1 per ottenere una curva di recupero fluorescenza contenente tutti i fotogrammi di tempo per essere studiato. La lunghezza del tempo è strettamente dipendente della proteina in esame. Quando sconosciuto, eseguire esperimenti preliminari per acquisire il tasso di turnover della proteina.
- Per calcolare il tempo di recupero, incollare i valori della curva di recupero fluorescenza con il corrispondente tempo (in secondi) in un appezzamento di Sigma (v. 12) ed eseguire una curva utilizzando il "Dynamic Fit guidata | Strumento di ascesa esponenziale al massimo". Selezionare mono-esponenziale (single, 3 parametri) o bi-esponenziale (double, 4 parametri) funzioni, a seconda la curva migliore.
- Utilizzare la seguente formula per funzione mono-esponenziale:
 Equazione 2
Equazione 2
- Utilizzare la seguente formula per funzione bi-esponenziale:
 Equazione 3
Equazione 3
- Incollare i parametri "b" e "d", derivato da Sigma Plot (equazione 2 o equazione 3) in Excel per calcolare l'intervallo di recupero. Si applicano le seguenti equazioni:
 Equazione 4
Equazione 4
o Equazione 5
Equazione 5
- Quando la funzione mono-esponenziale si traduce in una curva precisa, applicare solo equazione 4.
- Quando la funzione mono-esponenziale non comporta una buona curva, è possibile applicare la formula bi-esponenziale risolvendo sia equazione 4 e 5 di equazione. Considerare la risultante due volte a mezzo di recupero come che rappresenta due frazioni differenti della proteina: una rapida e una frazione lentamente lo scambio, rispettivamente.
8. determinazione del tasso di polimerizzazione dell'actina Lamellipodial di FRAP
- Per determinare il tasso di polimerizzazione dell'actina lamellipodial, transfect cellule B16-F1 con etichetta EGFP β-actina, e photobleach della regione lamellipodial (passo 5,9) utilizzando 1,5 s intervallo di tempo e l'esposizione GFP di 500 ms.
- In Metamorph, aprire i filmati time-lapse acquisiti da Visiview e calibrare il rapporto pixel/µm secondo l'obiettivo utilizzato dalla "misura | Strumento di calibrare le distanze".
- Riprodurre il filmato time-lapse e fermarlo sul telaio quando il recupero di fluorescenza di lamellipodial, che scorre all'indietro verso la lamella come una linea, ha raggiunto la lamella e nessun ulteriore flusso all'indietro può essere rintracciato.
- Misurare la distanza in µm tra la punta del lamellipodio e il retro della fluorescenza recuperato. Questa distanza corrisponde alla somma del flusso retrogrado e distanze di protrusione.
- In alternativa, per separare la protrusione dal flusso retrogrado, contrassegnare la punta di lamellipodial con un telaio di una linea prima il photobleaching. Utilizzare la linea come riferimento punto in frame successivi per riferirsi alla posizione originale della punta del lamellipodio al momento del photobleaching; il punto di riferimento può essere utilizzato per misurare la distanza di protrusione e flusso retrogrado.
- Si noti il tempo (in secondi) necessario per il ripristino di fluorescenza dopo photobleaching si verifichi. Il tempo può essere calcolato manualmente dal frame rate o visualizzato da Metamorph attraverso la "misura | Strumento di misura di regione".
- Derivare il tasso di polimerizzazione dell'actina utilizzando la seguente equazione (con alcuni parametri di equazione sulla base di misurazioni di Metamorph da passaggi 8.4 e 8.6):
 Equazione 6
Equazione 6
dove tasso di polimerizzazione dell'actina è in µm/min, distanza di flusso retrogrado è in µm, lamellipodial protrusione distanza è in µm e tempo è espresso in secondi.
9. analisi della proteina diffusione e della mobilità su fotoattivazione
Nota: Il metodo presentato qui descrive l'analisi della mobilità del monomero di actina impiegando fotoattivazione di actina fusa a PA-GFP, come illustrato dalla visualizzazione e quantificazione della diffusione della proteina attraverso il citosol.
- Per la misura della diffusione dell'actina fuse da una regione citosolica, nonché l'accumulo all'interno di una regione di lamellipodial, utilizzare Metamorph per determinare l'intensità nel corso del tempo nelle seguenti regioni (illustrato nella Figura 3un ): una regione citosolica fotoattivazione (PA); una regione di lamellipodial, in cui fotoattivazione proteine prevedibilmente verranno accumulati nel tempo (Lam); una regione di fuori della cella utilizzata per la normalizzazione di fluorescenza di fondo (fuori).
- Quando si determina la mobilità di actina all'interno del citosol, misurare distinte regioni citosoliche (vedere la Figura 3un, regioni R1-R5). Si noti che l'acquisizione di photobleaching non può essere determinato in modo simile a FRAP, dovuto un aumento nella fluorescenza focale - e alla fine tutto il cellulare al momento dell'attivazione.
- Trasferire i valori di intensità per tutte le regioni da Metamorph in un foglio di Excel, come descritto al punto 7.4.
- Per l'esame della velocità di spostamento di actina fuse dalla regione citosolica di fotoattivazione o il tasso di incorporazione all'interno di una regione di lamellipodial (entrambi rappresentati come percentuali intensità della regione citosolica fotoattivazione al tempo 0) , generare le curve di fluorescenza dai dati nel passaggio 9.3. Si applicano le seguenti equazioni:
 Equazione 7
Equazione 7 Equazione 8
Equazione 8
Dove: PATn è l'intensità di una regione citosolica fotoattivazione per ogni frame di interesse seguendo fotoattivazione; LAMTn è l'intensità di una regione di lamellipodial per ogni frame di interesse seguendo fotoattivazione; FUORITn è l'intensità di una regione presa all'esterno della cella (sfondo) per ogni frame di interesse seguendo fotoattivazione; PAT-1 è l'intensità di una regione citosolica fotoattivazione prima fotoattivazione; LAMT-1 è l'intensità di una regione di lamellipodial prima di fotoattivazione; T-1 è l'intensità di una regione presa all'esterno della cella (sfondo) prima di fotoattivazione; PAT0 è l'intensità di una regione citosolica fotoattivazione al tempo 0 (cioè, primo fotogramma dopo fotoattivazione); e fuoriT0 è l'intensità di una regione presa all'esterno della cella (sfondo) al tempo 0 (cioè, primo fotogramma dopo fotoattivazione). - Facoltativamente, per una migliore visualizzazione dei dati, normalizzare le curve di intensità da 0 sottraendo l'intensità del primo fotogramma dopo fotoattivazione da ogni fotogramma successivo.
Nota: Il seguente metodo di analisi (passaggi 9,6-9,8) consente inoltre di calcolare la dispersione citosolica di fotoattivazione actina nel citosol. - Misurare l'intensità per regioni citosoliche, che consecutivamente vengono posizionate distalmente dalla regione di attivazione.
- Per rappresentare le intensità di queste regioni come percentuale intensità della regione fotoattivazione al tempo 0, applicare equazione 8, dove lamellipodial intensità vengono sostituiti con intensità per ogni ROI citosolico. La dimensione e il numero delle regioni possono variare a seconda della distanza di dimensione e dispersione delle cellule deve essere misurata.
- Per derivare un valore quantificabile per il tasso di proteine di fotoattivazione ogni regione citosolica di infiltrazione, incollare i valori della curva di aumento di intensità di fluorescenza per ogni regione e tempo di trama Sigma (simile all'analisi di FRAP nella sezione 7), utilizzare Equazione 2 ed equazione 4 per derivare la metà tempo dell'intensità di fluorescenza raggiunge un plateau. Confrontare i valori di t1/2 tra differenti gruppi sperimentali.
Risultati
Figura 1 g h Visualizza immagini di contrasto di fase di una cellula di fibroblasti NIH3T3 preventiva e il post-microinjection 10 min di Rac1, che è una piccola GTPasi Rho-famiglia in grado di indurre la formazione di lamellipodi attraverso la sua interazione con l'onda complessa. La cella è in primo luogo visualizzata prima la microiniezione (Figura 1g), per confermare la sua vitalità e la morfologia, per esempio, mancanza di lamellipodi. A 10 min post-microiniezione, la cella è cambiato chiaramente la sua morfologia, che ci si aspetta da questo trattamento e indica un successo iniezione (Figura 1h).
Per semplicità e chiarezza, forniamo successiva Risultati esemplare per analisi FRAP e fotoattivazione nelle cellule, che non sono stati inoltre microiniettati.
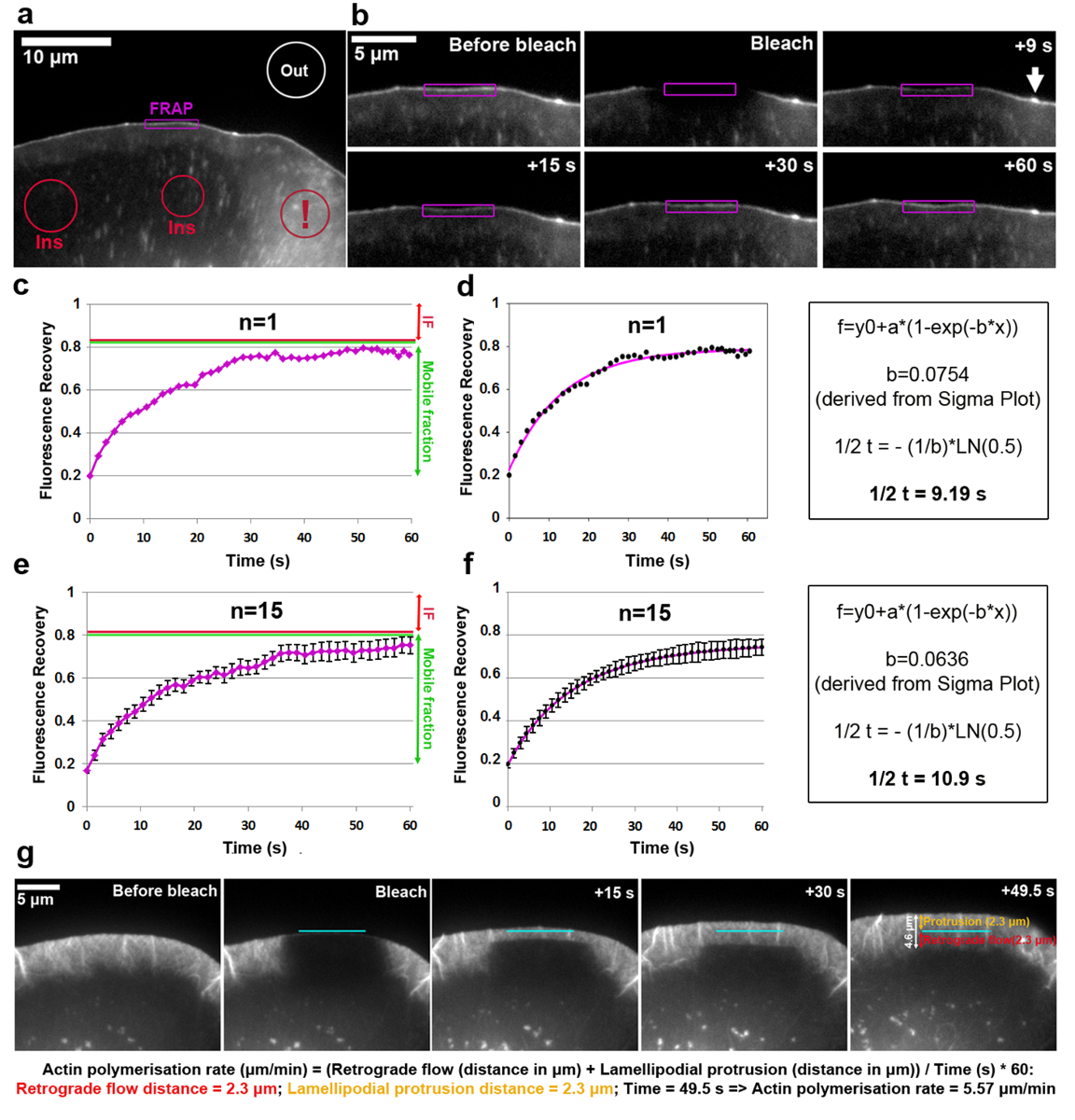
Analisi del fatturato di EGFP-etichetta VASP sulla punta di lamellipodio sono illustrato nella Figura 2a e f. Si noti che VASP mira inoltre alla nascente e focale aderenze, puntini piccoli e allungati a cella interni18,19. L'intensità di fluorescenza di una regione di lamellipodial con un accumulo di VASP chiaro sulla punta era sbiancato e misurato per ogni intervallo di tempo, seguendo il contorno del ROI, prima, durante e dopo lo sbiancamento come il lamellipodio sporge in avanti. Come sbiancato EGFP-VASP proteine sono riciclati da molecole non sbiancato in questi siti, recupero graduale della fluorescenza è osservato (Figura 2b). La curva di recupero FRAP ottenute in questo modo e normalizzato per l'intensità di sbianca preliminare (espressa come 1) può essere visto in Figura 2c. Photobleaching efficienza può variare ed è stato di circa il 20% del valore prima dello sbiancamento in questo esempio, come determinato dal valore a t0 (primo fotogramma dopo photobleaching). L'aumento della fluorescenza raggiunge un plateau nell'esempio mostrato a circa il 80% della fluorescenza prima dello sbiancamento. In una struttura statica durante il corso di tempo dell'esperimento, ad esempio un'adesione focale, la differenza tra l'intensità di sbianca preliminare e la fluorescenza di altopiano raggiunto dopo recupero è definito come la frazione immobile (IF, freccia rossa in Figura 2 c, e), mentre la quantità di fluorescenza recuperato tra il momento dell'imbianchimento e pieno recupero è definito come la frazione mobile (verde doppia freccia in Figura 2c, e). Si noti che in una struttura dinamicamente mutevole come la punta di lamellipodio qui analizzata, nella misura del se potrebbe non solo rappresentano molecole immobile ma anche derivare da una riduzione della velocità di sporgenza, come intensità di EGFP-VASP è conosciuto per dipendere da questo parametro18. Per calcolare il tempo di recupero, una curva che è stata creata sulla trama Sigma (Figura 2d). In questo caso, il valore del parametro "b" Estratto dalla soluzione di equazione 2 è uguale a 0.0754, che una volta applicato alla funzione logaritmica (equazione 4) risultati in un metà-tempo stimato del recupero di 9,19 s (Figura 2d , pannello di estrema destra), che è relativamente veloce in questa cella particolare rispetto alla media pubblicati precedentemente5. Si deve constatare che metà-tempi di recupero possono a volte variano significativamente tra le celle all'interno della popolazione stessa. Per ottenere risultati rappresentativi, è pertanto consigliabile determinare questo parametro come una media da almeno 15-20 cellule. Per illustrare il grado di varianza, mezzi aritmetici di EGFP-VASP recupero in media da 15 celle per ogni punto di tempo sono stati generati (Figura 2e), e curva media adatta creato e visualizzato in maniera analoga (Figura 2 f).
Il tasso di polimerizzazione della rete actina lamellipodial comprende la somma di protrusione rete avanti e flusso retrogrado. FRAP può essere applicato per misurare il tasso di polimerizzazione dell'actina trasfezione le cellule (in questo caso B16-F1) con etichetta EGFP β-actina e photobleaching una regione lamellipodial sporgente (Figura 2g). Per analisi di polimerizzazione di rete lamellipodial dell'actina, il recupero di fluorescenza con lo sbiancamento di EGFP-Tag β-actina viene valutato nel corso del tempo. Come la polimerizzazione dell'actina monomeri progredisce alle estremità pungenti dei filamenti dell'actina di lamellipodial (che tutti puntano verso il frontale20), la rete è costantemente spostati all'indietro e procedendo in avanti, i tassi di cui possono essere facilmente ottenute attraverso il recupero polarizzato di fluorescenza su photobleaching. Recupero di fluorescenza del lamellipodio è completo, non appena la zona sbiancata ha raggiunto la zona di transizione tra la parte posteriore del lamellipodio e la lamella, che è caratterizzata da una densità più bassa di altri fasci di filamenti disposti orizzontalmente gira molto più lentamente di quanto si osserva nel lamellipodio. Come illustrato nella Figura 2g, recupero di fluorescenza può essere visualizzato come una linea orizzontale a bordo e che scorre all'indietro verso la lamella, che permette di misurare le distanze della protrusione e flusso retrogrado (rappresentato individualmente nel pannello di destro della Figura 2g come frecce a due punte arancione e rosse, rispettivamente).
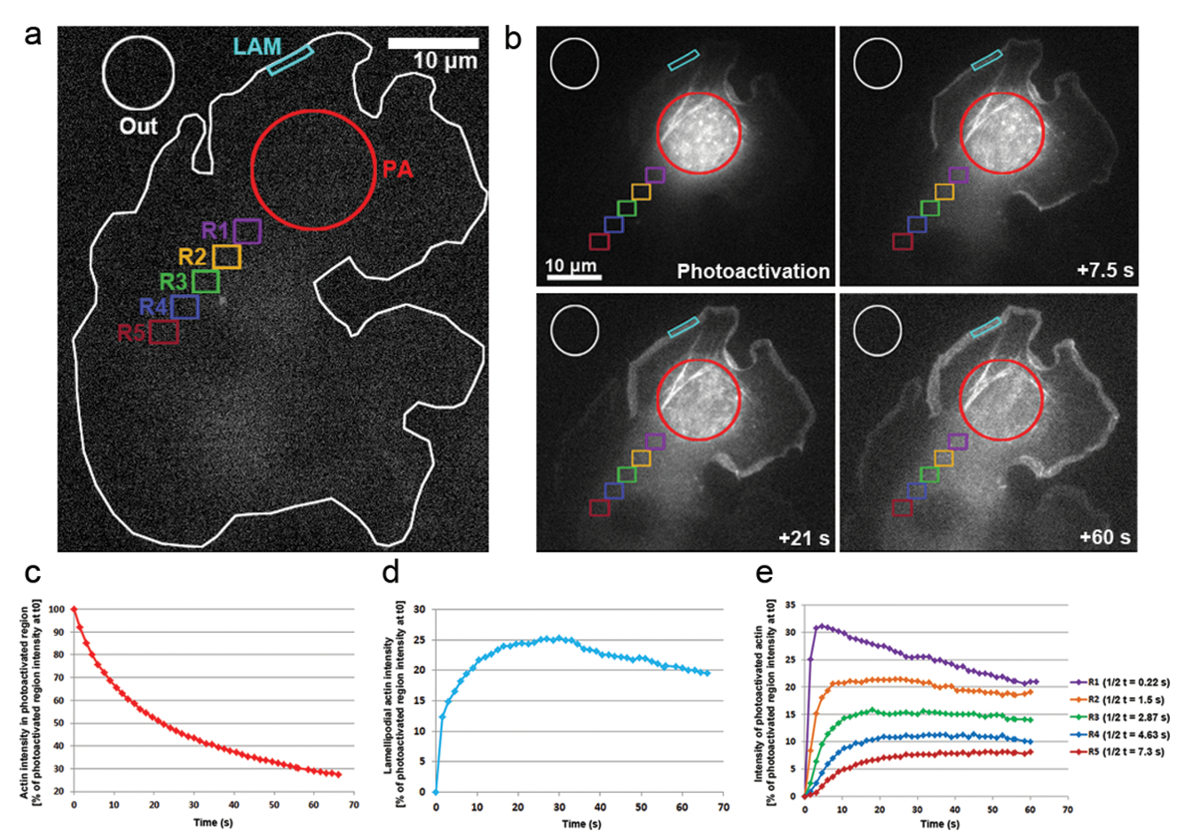
Inoltre abbiamo applicato fotoattivazione in B16-F1 cellule trasfettate con PA-GFP-actina per monitorare la mobilità dei monomeri di actina all'interno del citosol e il tasso della loro incorporazione all'interno sporgente lamellipodi. Come illustrato nella Figura 3a, b, una regione citosolica era fotoattivazione tramite l'esposizione a un laser di 405 nm, mentre le immagini sono state acquisite sulla GFP canale ogni 1,5 s per visualizzare la distribuzione di GFP-etichettato, l'actina di fotoattivazione. Photoactivated GFP-actina può essere visto diffondere fuori dalla regione citosolica in Figura 3b. Il tasso di diminuzione di intensità di fluorescenza nella regione citosolica fotoattivazione è rappresentato come la percentuale dell'intensità iniziale t0 (primo fotogramma dopo fotoattivazione; Figura 3 c). l'actina fotoattivazione integra anche alle punte dei lamellipodi, dove nuovi monomeri di actina sono aggiunte all'estremità pungenti crescente di allungando i filamenti dell'actina durante la protrusione. Per stimare il tasso di incorporazione di lamellipodial, abbiamo misurato l'intensità della fluorescenza nel tempo di un contorno o una regione bidimensionale di circa 5 µm di larghezza e 1 µm in altezza; la regione è stata costantemente ri-posizionata sulla punta del lamellipodio come esso ha sporto. Incorporazione di actina è stata rappresentata come la percentuale di intensità di fluorescenza della regione citosolica fotoattivazione a t0 (Figura 3d). Come allungamento dei filamenti dell'actina progredito, nuovi monomeri di actina sono stati incorporati nella parte anteriore di lamellipodial. Una frazione di questi monomeri di actina casualmente è stata derivata dal pool citosolico dove monomeri erano fotoattivazione. In questo modo il rapido aumento della fluorescenza in lamellipodi nel primo 20 s dopo fotoattivazione. Come nuovi monomeri vengono aggiunti alla parte anteriore di lamellipodial, flusso di monomeri di actina precedentemente incorporato con filamenti verso la lamella di flusso retrogrado. Nel corso del tempo, il ROI è completamente riempito con monomeri fluorescente e un plateau in fluorescenza è raggiunto (Figura 3d). Un calo graduale di fluorescenza è quindi osservato quando, a seguito di diffusione di fotoattivazione monomeri in tutta la cellula, non fotoattivazione monomeri di actina sono sempre più essere ri-aggiunto alla parte anteriore di lamellipodial. Questa diminuzione nella fluorescenza troveranno una nuova piattaforma, che sarà raggiunto, non appena è raggiungere un equilibrio nella cella intera tra fotoattivazione e non-fotoattivazione monomeri (dati non mostrati).
La mobilità dei monomeri di actina durante il cytosol è stata derivata misurando intensità di fluorescenza nelle regioni di uguali dimensioni posizionato distalmente dalla fotoattivazione regione (esemplificata in Figura 3un di regioni di colore codificato con l'etichetta R1-R5). Come illustrato nella Figura 3e, intensità di fluorescenza in ciascuna di queste regioni è diminuendo gradualmente lontano dalla cytosolically regione di fotoattivazione, come la frazione di monomeri di actina di fotoattivazione diventa sempre più diluito con non attivato monomeri (cioè, non fluorescenti). Inoltre, il picco di fluorescenza è raggiunto più tardi: più lontane della regione misurata si trova dalla regione fotoattivazione, più il tempo che è richiesto per i monomeri di actina diffondere in queste regioni. Un valore rappresentativo per il grado di infiltrazione di monomero di actina in ogni regione può essere derivato da quantificare il tempo di raggiungere l'altopiano di fluorescenza. Più lontane della regione, più tempo ci vuole per l'actina di fotoattivazione di diffondere in esso, e così è necessario più tempo per il plateau fluorescente essere raggiunto, infine conducendo su un valore maggiore di1/2 t (Figura 3e).

Figura 1 : Imaging procedura di assemblaggio e microiniezione camera. (un) Imaging camera componenti. (b) Silicone grasso è accuratamente spalmato intorno all'apertura di un sigillante di plastica. (c) il coprioggetto è posizionato con il lato di cella rivolto verso l'alto al centro della camera di formazione immagine di apertura. guarnizione (d) un sicuro è stabilito posizionando la plastica isolante sopra il vetrino coprioggetto e stringendo i morsetti laterali. (e), microscopia medio è dispensato nel vano di alloggiamento. (f), la camera di imaging è posizionato sul palco microscopio, elettrodi e rivelatore di calore sono collegati ad un'unità di riscaldamento pre-impostato a 37 ° C e le cellule sono permesso di adattare per almeno 30 min prima di microscopia è iniziata. In questo esempio, il tavolino del microscopio è anche dotato di un micromanipolatore per l'esecuzione di microiniezioni, e l'ago per microiniezione è immerso nel mezzo che copre lo strato delle cellule nella camera di imaging. (g), An NIH3T3 cellula del fibroblasto è visualizzato prima microiniezione di microscopia di contrasto di fase. La croce rossa nel compartimento perinucleare indica la posizione della microiniezione futuri, che corrisponde a una regione citoplasmatica alta a causa della vicinanza al nucleo ingombrante. (h) 10 min dopo microiniezione con Rac1, la cellula reagisce prominente formazione di lamellipodi intorno alla periferia di intera cella (indicata dalle frecce verdi). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: FRAP consente di determinare i tassi di polimerizzazione di actina proteina fatturato o lamellipodial. (una) rappresentante esempio di cella B16-F1 che esprimono EGFP-VASP prima photobleaching di una regione di lamellipodial, come indicato. Contorni/forme diversamente colorate sono etichettati per indicare quali regioni sono state considerate per misure di intensità di fluorescenza nel corso del tempo. Nota il contorno rosso contrassegnati con un punto esclamativo, che etichette una regione citosolica posizionata in un'area che racchiude molteplici vescicole e increspature superficiali delle cellule. Aree dinamiche come questo dovrebbero essere evitati per selezionare le regioni di riferimento di fluorescenza, come essi sono caratterizzati da forti fluttuazioni a breve termine della fluorescenza, causando risultati non accurati. (b) Lamellipodial regione della cellula che esprimono EGFP-VASP prima e dopo photobleaching. Il recupero del segnale fluorescente dopo photobleaching all'interno della regione contrassegnato in viola è visualizzato nel corso del tempo. Freccia indica la punta di un microspike, arricchita per VASP probabilmente a causa dell'alta densità dei filamenti dell'actina polimerizzazione là19. curva di recupero (c), un esempio di un FRAP come derivato da quantificare l'intensità di fluorescenza del lamellipodio photobleached (contorno viola) in b. rosso e linee verdi sulla destra indicano, rispettivamente, le frazioni di immobile e mobile. (d), A forma di curva di recupero del FRAP in c (pannello sinistro) e un esempio del metodo di calcolo utilizzato per ricavare il tempo di recupero (pannello di destra). (e), un esempio di un recupero FRAP curva derivata da una media le curve di recupero di fluorescenza di 15 cellule, con barre di SEM che indicano il grado di variabilità all'interno della popolazione campione. (f) una curva misura derivata da in media gli accoppiamenti di curva FRAP recupero di 15 celle (pannello sinistro) e un esempio del metodo di calcolo utilizzato per ricavare il tempo di recupero (pannello di destra). (g) time-lapse pannelli di sporgenti lamellipodio di una cella B16-F1 che esprimono EGFP-Tag β-actina, prima e dopo lo sbiancamento di una regione di lamellipodial come indicato, seguita dal recupero di fluorescenza nel lamellipodio nel corso del tempo. Sul pannello di destro, i valori misurati per protrusione e retrograde distanze sono forniti (in arancione e rosso, rispettivamente). I calcoli sotto i riquadri dell'immagine rivelano come la somma di protrusione e retrograde distanze vengono utilizzati per derivare il tasso di polimerizzazione della rete actina lamellipodial. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Fotoattivazione di PA-GFP-actina per monomero di monitoraggio in tutta la cellula. (un) A esempio rappresentativo di una cella B16-F1 esprimendo PA-GFP-actina prima di far scattare la fotoattivazione in una regione citosolica come indicato dal cerchio rosso (PA). Diversamente colorati contorni sono etichettati per indicare quali regioni sono state considerate per misure di intensità di fluorescenza nel corso del tempo. (b) un'illustrazione della distribuzione temporale dei seguenti PA-GFP-actina fotoattivazione. Si noti la riduzione graduale della fluorescenza nella fotoattivazione, regione citosolica (cerchio rosso), come l'actina di fotoattivazione diffonde lontano da esso. A causa della loro diffusione alla parte anteriore e l'Assemblea nella rete, monomeri di actina di fotoattivazione sono gradualmente migliorati in lamellipodi (ciano regione) e nel citosol (diverse regioni color-coded) in un modo dipendente dal tempo e della distanza. (c) rappresentante, temporale declino della fluorescenza all'interno della regione citosolica di fotoattivazione (contorno rosso in b). (d) temporale cambiamenti nell'intensità di fluorescenza nella regione lamellipodial (ciano contorno in b). (e) curve rappresentative dei cambiamenti temporali nell'intensità di fluorescenza delle regioni citosoliche (color-coded in b) a causa di posizionamento a distanza variabile dalla zona di fotoattivazione. Nota come altopiano volte mezzo di raggiungere la fluorescenza (indicato nella legenda a destra) aumenta con la distanza del dato regione nell'area di fotoattivazione, probabilmente correla con l'aumento tempi di diffusione di monomeri di actina nella rispettiva regione. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Qui discutiamo passaggi critici nelle tecniche descritte in questo articolo, e come possono essere ottimizzati per l'applicazione in differenti condizioni sperimentali.
Microiniezione è un metodo che può essere applicato per monitorare in cellule effetti istantanei da introdurre proteine esogene, inibitori o farmaci. Può essere particolarmente vantaggioso per determinare le funzioni delle proteine nelle difficili a transfect tipi cellulari o nelle situazioni in cui espressione a lungo termine non è desiderato. Si deve constatare che la sopravvivenza di alcuni tipi di cellule varia a seconda della matrice extracellulare che sono seminati su. Più delle cellule endoteliali, epiteliali o fibroblasto-come tipi, anche piccoli come keratocytes di pesce (Vedi Dang et al. 21 e Anderson e croce22) può essere iniettati con successo. Tuttavia, ci sono eccezioni, come cellule B16-F1 seminate su laminina, che costituiscono un sistema eccellente modello di migrazione delle cellule, ma non sono compatibili con iniezione su questo tipo di substrato per motivo sconosciuto. Per le cellule dei fibroblasti NIH3T3, eseguiamo regolarmente iniezioni su substrato di fibronectina e photomanipulation ulteriori tecniche come FRAP (anche con fotoattivazione; indicato per cellule B16-F1 qui) può essere eseguita ugualmente bene in questi fibroblasti (Vedi ad esempio, Köstler et al. 3). si deve anche considerare che le proteine differenti, secondo le loro proprietà funzionali e gli obiettivi dell'esperimento, potrebbero richiedere diverse quantità di tempo per causare i cambiamenti, variando da secondi a ore. Un vantaggio della tecnica è che la dose/concentrazione di agente esogeno può essere controllata con maggiore precisione a livello di singola cellula rispetto ad esempio, quando si utilizza la transfezione di plasmide. Inoltre, etichettatura fluorescente di una proteina non è una necessità per garantire la sua presenza nella cellula, che può aumentare la flessibilità se simultanea visualizzazione multi-canale di altre proteine fluorescente-tag è necessaria. Microiniezione può essere particolarmente utile per analizzare gli effetti immediati di specifiche proteine o miscele della proteina su cambiamenti dinamici della morfologia delle cellule o del citoscheletro (es., Dang et al. 21 per un esempio di effetti istantanei sulla migrazione dall'inibitore complesso Arp2/3 Arpin). Uno svantaggio della tecnica è la sua invasività, che può causare danni alle cellule o influenzare la morfologia delle cellule. Di conseguenza, una considerazione importante quando si eseguono microiniezioni sta monitorando l'attuabilità delle cellule. Il metodo introdotto qui si basa sulla manipolazione manuale. In condizioni testate per essere compatibile con iniezioni di successo, come i fibroblasti che cresce su substrato di fibronectin, il protocollo di iniezione manuale descritto qui permette un tasso di successo del 100% nei pressi; Questo è essenziale quando si combinano questo approccio con gli esperimenti di follow-up sofisticati e richiede molto tempo tra cui video microscopia o FRAP, come pubblicato in precedenza3. Ciò non esclude che occasionalmente, singole celle potrebbero soffrire di un evento di microiniezione, che possa essere riconosciuto in modo sicuro da improvvisi cambiamenti di contrasto del nucleo e del citoplasma, seguita da retrazione del bordo di cella. Questi rari casi sperimentali sono esclusi e quindi non considerati per ulteriori analisi.
Tuttavia, un approccio semi-automatico è inoltre comunemente usato, per esempio impiegando rapida (< 300 ms) macchina controllata dell'ago abbassamento coincidente con aumento della pressione di iniezione, che l'ago deve essere posizionato sopra ogni cella prima del rispettivo iniezione. Il tasso di successo delle iniezioni semiautomatica è per definizione inferiore rispetto all'approccio manuale descritto in precedenza, semplicemente perché è ottimizzato per la velocità, seguita da analisi di più celle che con successo è sopravvissuto questo trattamento; così esso non si basa sull'iniezione di successo di una singola cella. Pertanto, al contrario di analisi unicellulare, semi-automatico iniezioni sono più adatte per analizzare gli effetti della iniezione di parecchie centinaia cellule, ad esempio, mediante video microscopia ad ingrandimento basso o su richiesta di fissazione delle cellule e la macchiatura. Qualunque sia l'approccio dettagliato impiegato, microinjection non costituisce un'analisi di punto finale, ma può essere combinato con una varietà di tecniche, tra cui FRAP o fotoattivazione3.
Quando si determina il tasso di turnover proteico da FRAP, l'intensità del laser deve essere ottimizzato, a seconda delle condizioni di installazione e la formazione immagine del microscopio (ingrandimento, obiettivi, ecc., così come il tipo di cella, la struttura e la proteina fluorescente per photobleaching). Si noti che alla potenza ottimale del laser, candeggio efficiente è combinato con il photodamage meno possibile, per evitare il restringimento o completare la retrazione della struttura sotto analisi (ad es., lamellipodi o filopodi) o persino danni a livello cellulare. Idealmente, almeno il 70 – 80% di efficienza di sbiancamento deve essere raggiunto, anche se lo sbiancamento completo può essere ostacolato da estremamente rapido turnover della proteina, in cui caso, nulla oltre il 50% potrebbe anche essere accettabile. Potenza candeggina ottimale per una determinata struttura e tintura fluorescente deve essere testata sperimentalmente, a partire da una potenza di laser a basso seguita da suo aumento graduale. Naturalmente, qualsiasi colorante fluorescente può per definizione essere sbiancato con luce vicino al suo picco di eccitazione laser (488 nm per coloranti utilizzati di frequente verde ad esempio FITC o EGFP). Tuttavia, laser con lunghezze d'onda più corta, come il vicino-UV laser, fornire potenze superiori e quindi può essere utilizzato anche per lo sbiancamento efficiente di coloranti comunemente utilizzati. Impieghiamo ordinariamente un diodo laser 405 nm (120 mW) per il candeggio di EGFP e rosse tinture fluorescenti (ad esempio mCherry), seppur con efficienza leggermente inferiore nel caso di quest'ultimi (dati non mostrati). Come il 405 nm-diodo può anche essere usato per fotoattivazione di PA-GFP (Vedi sotto), dota di questo sistema con massima flessibilità.
Per le strutture delle cellule B16-F1 e proteine fluorescenti photobleached qui, 405 potenze laser nm tra 65 – 100 mW sono state applicate. Quando si analizza una regione di photobleached, è importante considerare se la struttura specificata è conservata nella sua forma originale sopra l'analisi periodo di tempo. Per esempio, quando si analizza il turnover delle proteine alle punte di lamellipodi, prestare attenzione se la curvatura di lamellipodi è alterata significativamente nel corso del tempo, come cambiamenti nella curvatura potrebbero portare a risultati imprecisi se il regione/contorno analizzato non comprendere pienamente la totalità della struttura in ogni fotogramma misurato. Inoltre, dovrebbe essere notato che fasci incorporati in lamellipodi, come microspikes, potrebbero causare deviazioni nell'intensità di fluorescenza. Come illustrato nella Figura 2b (freccia bianca nel lasso di tempo s 9), una struttura microspike è situata vicino alla regione della photobleached misurato, ma rimane di fuori di esso per tutta la durata della misura e pertanto non causa qualsiasi inesattezza. Per l'analisi del turnover delle proteine, considerazioni importanti quando selezionando la posizione e le dimensioni di analizzato regioni sono che loro fluorescenza nel corso del tempo non dovrebbe essere significativamente influenzato dai cambiamenti nella morfologia delle cellule o fattori diversi da quelli duramente per evitare acquisizione photobleaching. Per esempio, strutture che forniscono quantitativo significativo contributo alla struttura analizzata non devono muoversi fuori della regione di misurata durante l'analisi; Inoltre, entità indipendenti, fluorescenti quali strutture vescicolari che ottenere la proteina non dovrebbe entrare nel campo di interesse durante l'analisi. Per determinare il tasso di polimerizzazione dell'actina lamellipodial, dovrebbe prestare attenzione che non retrattile o Scapigliatura (cioè, verso l'alto pieghevole) lamellipodi sono analizzati, come questo influenzerà fortemente la precisione dei risultati. Inoltre, retrazione delle regioni lamellipodial potrebbe essere visualizzato come rapido spostamento all'indietro, potrebbe condurre alla sopravvalutazione dei tassi di polimerizzazione dell'actina di lamellipodial. Un'ulteriore considerazione è la distanza delle regioni di normalizzazione intracellulare (preso come posizioni di riferimento per la correzione di acquisizione photobleaching) dalla posizione effettiva di photobleaching, che dovrebbe essere abbastanza grandi per evitare diretto influenza della zona di photobleached.
Quando si impostano le condizioni ottimali per fotoattivazione di costrutti di PA-GFP-etichettato, si dovrebbe prestare attenzione per evitare lo sbiancamento immediato durante la fotoattivazione. Nel nostro lavoro, i risultati migliori sono stati ottenuti con potenze laser 5 - 10 volte inferiore a quello normalmente impiegato per lo sbiancamento di EGFP. Per acquisizione di immagini di molecole di fotoattivazione, tempo di esposizione e intervallo di tempo tra i fotogrammi dovrebbe essere ottimizzati considerando la dimensione delle regioni e delle strutture per essere fotoattivazione e analizzati, così come la mobilità potenziale di fotoattivazione proteine ad altre posizioni sottocellulari. Per quanto riguarda tutti i tipi di formazione immagine di fluorescenza, manutenzione di attuabilità delle cellule è fondamentale per l'ottenimento di risultati fisiologicamente rilevanti.
In linea di principio, verde-rosso fotoconversione di proteine fluorescenti come mEos o Dronpa varianti12 costituisce un metodo altrettanto potente delle seguenti dinamiche e fatturato delle strutture subcellulari quali il lamellipodio (Vedi ad es., Burnette et al. 23). il vantaggio del metodo quest'ultimo al contrario di PA-GFP sarebbe la possibilità di seguire la dinamica di proteine prima e dopo la conversione con due colori distinti, senza la necessità di co-esprimono una proteina fluorescente rossa ulteriore. Tuttavia, nei nostri esperimenti preliminari, la portata del cambiamento di contrasto e l'intensità del segnale fluorescente raggiunto su fotoattivazione di PA-GFP era più grande rispetto alle sonde photoconverted, forse a causa delle caratteristiche spettrali superiore di verde contro rosso sonde fluorescenti (dati non mostrati). In ogni caso, studi dettagliati sul fatturato di filamento di actina in sporgenze di bordo di cella come lamellipodi o code di Vaccinia virus-indotta actina sono finora solo stati pubblicati utilizzando PA-GFP derivati5,6,24.
Quando si considera quale regione di cella per analizzare in seguito fotoattivazione, parecchi fattori dovrebbero essere tenuti conto, che sono discussi con lo specifico esempio mostrato qui (incorporazione di monomeri di actina al bordo delle cellule al momento dell'attivazione nel citosol), ma certamente possono essere estrapolati per vari problemi scientifici analoghi. In primo luogo, quando si misura il tasso di incorporazione di lamellipodial di cytosolically fotoattivazione di proteine, per esempio, in diverse condizioni sperimentali (come mostrato nella Dimchev et al. 6), dimensioni delle regioni citosoliche e loro distanze ai bordi di lamellipodial dovrebbero essere comparabili tra i gruppi sperimentali. È anche importante considerare che quando provveduto citosolico regioni, lo spessore delle cellule è maggiore in posizioni più vicino al nucleo. Attivazione cellulare più spessore regioni potrebbe comportare una maggiore quantità di proteine attivate, dato che la distribuzione della proteina deve essere attivato è distribuita omogeneamente nel citosol. Infine, i livelli di espressione della proteina deve essere attivato possono certamente essere altamente variabili in singole celle. A causa di tutte queste considerazioni di variabilità, è fondamentale confrontare i livelli di incorporazione di proteine cytosolically attivate altrove nella cella relativa la fluorescenza totale ottenuta al momento dell'attivazione in specifiche regioni.
Abbiamo descritto come microiniezione può essere utilizzato come strumento per indagare gli effetti delle proteine sulla morfologia delle cellule e questo hanno esemplificato dimostrando l'induzione potente delle strutture lamellipodial in NIH3T3 cellule del fibroblasto microiniettate con il piccolo GTPase Rac1. In precedenza abbiamo applicato questa tecnica per interferire con la funzione di Arp2/3 in cellule microiniettati con il dominio C-terminale WCA di Scar/WAVE3. Vari parametri in cellule iniettati possono essere analizzati da altri saggi, quali FRAP o fotoattivazione. Abbiamo descritto come FRAP e fotoattivazione può essere impiegata per investigare la dinamica subcellulare e mobilità di monomeri di actina. FRAP è stato utilizzato dal nostro gruppo in precedenza5 per indagare il fatturato delle proteine eseguendo la localizzazione lamellipodi, quali Abi, cortactin, VASP, cofilina e tappatura della proteina, o per il delucidamento il fatturato delle componenti in adesioni focali in presenza e assenza di Rac4di segnalazione. Inoltre, tassi di polimerizzazione dell'actina di misura può essere compiuta da photobleaching EGFP-Tag β-actina5, ma esistono metodi alternativi. Rilevamento delle disomogeneità fluorescente come visto dalle sonde di imaging-compatibile di cellule vive etichettatura filamenti dell'actina cellulare, ad esempio Lifeact25, può anche essere autonomo6,26. Il vantaggio è che la sovraespressione di β-actina può essere evitata, che è capace di aumentare la sporgenza del bordo di cella e migrazione e quindi potenzialmente interferisce con l'analisi specifica o la domanda sperimentale (Vedi ad es., Kage et al. 26; Peckham et al. 27). Tuttavia, un netto svantaggio della sonda Lifeact costituisce sua rapida accensione/spegnimento cinetica dell'associazione ai filamenti dell'actina, in modo che lo sbiancamento delle strutture di filamento di actina etichettati da Lifeact in cellule fornisce informazioni solo sul fatturato sonda, ma non il fatturato dei filamenti dell'actina, a cui si lega il25. Il tracciamento delle disomogeneità di fluorescenza precedentemente impiegato6,26 forniscono un compromesso pratico, molto simile al tracciamento ampiamente usato di fluorescenza macchioline incorporate in filamentose del citoscheletro strutture (Vedi ad es., salmone e Waterman28), ma non può essere come semplice da utilizzare e preciso come FRAP delle strutture di EGFP-etichetta F-actina. Fotoattivazione è stata applicata da noi per stimare i tassi di incorporazione di actina monomerica in sporgenti lamellipodi, come pure la sua mobilità in tutto il citosol, nel contesto di livelli di F-actina sperimentalmente sintonizzati citosolico6. La tecnica è utile quando esaminando la mobilità e la distribuzione delle proteine derivate da relativamente grandi aree, quali le regioni citosoliche. Tuttavia, esaminando la distribuzione delle proteine derivate da strutture di fotoattivazione relativamente piccola; ad esempio, i coni di crescita potrebbero essere impegnativi a causa del basso numero di molecole fluorescenti attivati, segnali deboli e così mancanza di sensibilità. Tecniche alternative potenziali di fotoattivazione o fotoconversione della fluorescenza (Vedi sopra) possono includere inverso FRAP, che si basa sul photobleaching l'intera cella tranne il ROI, seguito da rilevamento la mobilità delle molecole fluorescenti lontano da in questa regione. La tecnica non richiede che overexpressing fuse versioni delle proteine, ma comporterà sempre l'esposizione a una dose insolitamente elevata di potenza laser, potenzialmente causando effetti collaterali indesiderati quali photodamage.
Chiaramente, la fotoattivazione e FRAP non può distinguere se le proteine si stanno muove come monomeri, dimeri o anche piccoli oligomeri e se si muovono insieme a partner di associazione aggiuntive. Informazioni di questo tipo possono essere ottenuti invece da fluorescenza correlazione spettroscopia tecniche29 o, in alternativa, FLIM-FRET30. Ciò nonostante, FRAP e fotoattivazione costituiscono semplici approcci per valutare direttamente le dinamiche locali e globali della proteina in cellule, indipendentemente dalla proteina di interesse, posizione subcellulare o tipo di cellula ha studiato.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Siamo grati per la Fondazione di ricerca tedesca (DFG) per il sostegno finanziario (grant Nr. RO2414/5-1 a KR).
Materiali
| Name | Company | Catalog Number | Comments |
| B16-F1 mouse skin melanoma cells | American Type Culture Collection, Manassas, VA | CRL-6323 | |
| NIH-3T3 cells | American Type Culture Collection, Manassas, VA | CRL-1658 | |
| DMEM 4.5g/L glucose | Life Technologies, Thermno Fisher Scientific, Germany | 41965-039 | |
| Ham’s F-12 medium | Sigma-Aldrich | N8641 | |
| Fetal calf serum (FCS) | PAA Laboratories, Linz, Austria | A15-102 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich, Germany | F7524 | Lot054M3396 |
| MEM Non essential amino acids | Gibco, ThermoFisher Scientific, Germany | 11140035 | |
| L-Glumatine 200mM (100x) | Life Technolgies | 25030-024 | |
| Pen-Strep 5000 U/mL | Life technologies | 15070063 | |
| Sodium Pyruvate (100 mM) | Gibco, ThermoFisher Scientific, Germany | 11360-039 | |
| Laminin | Sigma-Aldrich | L-2020 | |
| Laminin coating buffer | Self-made: 50mM Tris ph7.4, 150mM NaCl | ||
| Fibronectin from human plasma | Roche Diagnostics, Mannheim, Germany | 11 051 407 001 | |
| Jetpei | Polyplus Transfection, Illkirch, France | 101-10N | |
| JetPei buffer | Polyplus Transfection, Illkirch, France | 702-50 | 150mM NaCl |
| PA-GFP-actin plasmid DNA | described in Koestler et al.2008 | ||
| pEGFP-actin plasmid DNA | Clontech, Mountain View, CA, USA | ||
| Rac1 protein for microinjection | Purified as GST-tagged version, and cleaved from GST prior to injection | ||
| Microinjection buffer | Self-made: 100mM NaCl, 50mM Tris-HCl ph7.5, 5mM MgCl2, 1mM DTT | ||
| Dextran, Texas Red, 70,000 MW, Lysine Fixable | Molecular Probes, Thermno Fisher Scientific, Germany | D1864 | |
| Microscope circular cover glasses 15mm, No.1 | Karl Hecht, Aisstent, Sondheim, Germany | 1001/15 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf, Hamburg, Germany | 5242 956 003 | |
| Eppendorf Femtotip Microinjection Capillary Tips | Eppendorf, Hamburg, Germany | 930000035 | |
| Silicone Grease | ACC Silicones, Bridgewater, England | SGM494 | |
| Aluminium Open Diamond Bath Imaging Chamber | Warner instruments | RC-26 | |
| Automatic temperature controller | Warner Instruments | TC-324B | |
| Microscope: Axio Observer | Carl Zeiss, Jena, Germany | ||
| CoolSnap-HQ2 camera | Photometrics, Tucson, AZ | ||
| Lambda DG4 light source | Sutter Instrucment, Novato, CA | ||
| Laser source | Visitron Systems | ||
| Eppendorf FemtoJet microinjector | Eppendorf, Hamburg, Germany | With built-in compressor for pressure supply | |
| Nikon Narishige Micromanipulator system | Nikon Instruments, Japan | ||
| Visiview software v2.1.4 | Visitron Systems, Puchheim, Germany | ||
| Metamorph software v7.8.10 | Molecular Devices, Sunnyvale, CA | ||
| Sigma Plot v.12 | Systat Software Inc. |
Riferimenti
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 38 (10), 2887-2921 (2009).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Koestler, S. A., et al. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol Biol Cell. 24 (18), 2861-2875 (2013).
- Steffen, A., et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 126, Pt 20 4572-4588 (2013).
- Lai, F. P., et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27 (7), 982-992 (2008).
- Dimchev, G., et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol Biol Cell. 28 (10), 1311-1325 (2017).
- Koppel, D. E., Axelrod, D., Schlessinger, J., Elson, E. L., Webb, W. W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys J. 16 (11), 1315-1329 (1976).
- Patterson, G. H., Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297 (5588), 1873-1877 (2002).
- McKinney, S. A., Murphy, C. S., Hazelwood, K. L., Davidson, M. W., Looger, L. L. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 6 (2), 131-133 (2009).
- Gurskaya, N. G., et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 24 (4), 461-465 (2006).
- Lippincott-Schwartz, J., Patterson, G. H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 19 (11), 555-565 (2009).
- Kremers, G. J., Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J Vis Exp. (40), (2010).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 4988(2008).
- Small, J. V., Rottner, K. Actin-based Motility. Carlier, M. F. , Springer. Dordrecht. (2010).
- Kaverina, I., et al. Enforced polarisation and locomotion of fibroblasts lacking microtubules. Curr Biol. 10 (12), 739-742 (2000).
- Small, J., Rottner, K., Hahne, P., Anderson, K. I. Visualising the actin cytoskeleton. Microsc Res Tech. 47 (1), 3-17 (1999).
- Mikhailov, A. V., Gundersen, G. G. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskeleton. 32 (3), 173-186 (1995).
- Rottner, K., Behrendt, B., Small, J. V., Wehland, J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1 (5), 321-322 (1999).
- Svitkina, T. M., et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 160 (3), 409-421 (2003).
- Small, J. V., Isenberg, G., Celis, J. E. Polarity of actin at the leading edge of cultured cells. Nature. 272 (5654), 638-639 (1978).
- Dang, I., et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 503 (7475), 281-284 (2013).
- Anderson, K. I., Cross, R. Contact dynamics during keratocyte motility. Curr Biol. 10 (5), 253-260 (2000).
- Burnette, D. T., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 13 (4), 371-381 (2011).
- Humphries, A. C., et al. Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell Host Microbe. 12 (3), 346-359 (2012).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 5 (7), 605-607 (2008).
- Kage, F., et al. FMNL formins boost lamellipodial force generation. Nat Commun. 8, 14832(2017).
- Peckham, M., Miller, G., Wells, C., Zicha, D., Dunn, G. A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 114, Pt 7 1367-1377 (2001).
- Salmon, E. D., Waterman, C. M. How we discovered fluorescent speckle microscopy. Mol Biol Cell. 22 (21), 3940-3942 (2011).
- Machan, R., Wohland, T. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 588 (19), 3571-3584 (2014).
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J Microsc. 247 (2), 119-136 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati