
Method Article
Gocciolina basati su codici a barre singola cella trascrittomica dei tessuti dei mammiferi adulti
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Questo protocollo descrive le procedure generali e controlli di qualità necessari per la preparazione di cellule singole mammifera adulte sane per le preparazioni di RNA-Seq unicellulare gocciolina-based, alta velocità di trasmissione. Parametri di sequenziazione, leggi l'allineamento e l'analisi bioinformatica di cella singola a valle sono inoltre forniti.
Abstract
L'analisi dell'espressione genica di singola cellula attraverso migliaia di singole celle all'interno di un tessuto o un microambiente è un prezioso strumento per l'identificazione delle cellule composizione, discriminazione degli stati funzionali e vie molecolari che sono alla base del tessuto osservato funzioni e comportamenti degli animali. Tuttavia, l'isolamento di cellule singole intatte, sane da tessuti di mammiferi adulti per l'analisi molecolare successiva cella singola a valle può essere impegnativo. Questo protocollo descrive le procedure generali e controlli di qualità necessario per ottenere preparazioni di cellule singole adulto di alta qualità dal sistema nervoso o della pelle che è stato attivato analisi e sequenziamento di successive imparziale singola cella RNA. A vostra disposizione anche linee guida per l'analisi bioinformatica e a valle.
Introduzione
Con lo sviluppo di elevato throughput unicellulare tecnologia1,2 e gli avanzamenti in strumenti di bioinformatica user-friendly nel corso degli ultimi dieci anni3, è emerso un nuovo campo di analisi di espressione genica ad alta risoluzione – sequenziamento di RNA di singola cellula (scRNA-Seq). Lo studio dell'espressione genica singola cella è stato sviluppato per identificare eterogeneità all'interno di popolazioni di cellule definito, come in cellule staminali o cellule tumorali, o per identificare rare popolazioni di cellule4,5, che erano irraggiungibili utilizzando tecniche di sequenziamento di RNA tradizionale alla rinfusa. Strumenti di Bioinformatic hanno permesso l'identificazione di sub-popolazioni romanzo (Seurat)2, visualizzazione dell'ordine di cellule lungo un psuedotime spazio (monocolo)6, definizione delle reti di segnalazione attive all'interno o tra popolazioni ( SCENIC)7, la previsione dell'Assemblea del singolo-cellule in uno spazio 3D artificiale (Seurat e più)8. Con queste nuove e interessanti analisi disponibili alla comunità scientifica, scRNA-Seq è rapidamente diventando il nuovo approccio standard per l'analisi dell'espressione genica.
Nonostante il vasto potenziale della scRNA-Seq, la skillsets tecnica necessaria per produrre un set di dati puliti e interpretare con precisione i risultati può essere difficile per i nuovi arrivati. Qui, un protocollo semplice, ma completo, a partire dall'isolamento di singole cellule da tessuti intero primari per visualizzazione e presentazione dei dati per la pubblicazione è presentato (Figura 1). In primo luogo, l'isolamento di singole cellule sane possa essere considerato impegnativo, come tessuti differenti variano nel loro grado di sensibilità alla digestione enzimatica e successiva dissociazione meccanica. Questo protocollo fornisce una guida nella procedura di isolamento e identifica i checkpoint importante controllo di qualità durante tutto il processo. In secondo luogo, comprendere la compatibilità e requisiti tra tecnologia single cell e sequenziamento di nuova generazione può essere confusa. Questo protocollo fornisce linee guida per implementare una piattaforma facile da usare, basato su goccia unicellulare codici a barre ed eseguire l'ordinamento. Infine, programmazione di computer è un presupposto importante per l'analisi unicellulare Transcrittomica DataSet. Questo protocollo fornisce risorse per iniziare a utilizzare il linguaggio di programmazione R e fornisce indicazioni sull'implementazione di due popolari pacchetti di R scRNA-Seq-specifici. Insieme, questo protocollo può guidare i nuovi arrivati in esecuzione di analisi scRNA-Seq per l'ottenimento di risultati chiari e interpretabili. Questo protocollo può essere registrato alla maggior parte dei tessuti nel topo e cosa importante potrebbe essere modificato per l'utilizzo con altri organismi, compresi tessuti umani. Regolazioni a seconda del tessuto e l'utente sarà richieste.
Esistono alcune considerazioni da tenere a mente mentre a seguito di questo protocollo; compreso, 1) a seguito di tutte le linee guida di controllo di qualità nei passaggi 1 e 2 del presente protocollo è consigliato per garantire una sospensione di cellule singole vitali di tutte le cellule all'interno del campione di interesse assicurando numero conta accurata e totale delle cellule (riassunti in Figura 2 ). Una volta che questo è realizzato, e se vengono rispettate tutte le condizioni ottimizzate, la procedura di controllo di qualità può essere eliminata (per risparmiare tempo - preservando la qualità di RNA e riducendo cella perdita). Confermando il successo isolamento di singole cellule di alta redditività dal tessuto di interesse è altamente consigliato a valle prima di qualsiasi elaborazione. 2) dato che alcuni tipi di cellule sono più sensibili di altre allo stress, tecniche di dissociazione eccessivo possono inavvertitamente pregiudizi la popolazione, quindi confusione dell'analisi a valle. Dissociazione dolce senza inutili tosatura cellulare e digestione è fondamentale per il raggiungimento di alti rendimenti cellulari e una rappresentazione accurata della composizione del tessuto. Le forze di taglio si verificano durante le operazioni di triturazione, FACS e risospensione. 3) come con qualsiasi altra attività di RNA, è meglio introdurre come little RNasi aggiuntiva nel campione come possibile durante la preparazione. Questo aiuterà a mantenere alta qualità RNA. Utilizzare soluzioni di inibitore della ribonucleasi con risciacquo per attrezzi puliti e qualsiasi apparecchiatura che non sia RNAsi-libera ma evitare prodotti trattati con DEPC. 4) eseguire preparazioni più rapidamente possibile. Questo vi aiuterà a mantenere alta qualità RNA e ridurre la morte delle cellule. A seconda della lunghezza di dissezione dei tessuti e numero degli animali, è consigliabile iniziare dissezioni/preparazioni multiple allo stesso tempo. 5) preparare cellule su ghiaccio quando possibile per mantenere alta qualità RNA, ridurre la morte delle cellule e lento attività trascrizionale e segnalazione delle cellule. Seppur, elaborazione ghiacciata è ideale per la maggior parte delle cellule, alcuni tipi di cellule (per esempio, neutrofili) siano migliori quando trattati a temperatura ambiente. 6) evitare di calcio, magnesio, EDTA e prodotti trattati con DEPC durante la preparazione delle cellule.
Protocollo
Tutti i protocolli descritti qui sono conformi e approvato dal comitato di cura degli animali dell'Università di Calgary.
1. dissociazione dei tessuti (giorno 1)
- Eutanasia di topi con una dose eccessiva di sodio pentobarbital (i.p., 50 mg/kg) o come appropriato secondo il protocollo di etica animale. Quindi rimuovere i peli superflui dalla schiena e le gambe del mouse ed etanolo-sterilizzare la regione della dissezione.
- Sezionare il tessuto o il microambiente di interesse. Per questo protocollo, usiamo tessuti della pelle e del nervo per dimostrare il generalizability di gocciolina basati su codici a barre singola cella trascrittomica dopo dissociazione tessuto adulto.
- Per il nervo sciatico, utilizzare il protocollo dettagliato trovato al Stratton et al. 9. brevemente, tagliare la pelle dalla regione posteriore della schiena/gambe del mouse. Fare un'incisione lungo la lunghezza della coscia con un bisturi sterile. Utilizzare una pinzetta e forbici per esporre e rimuovere il nervo sciatico.
- Per la pelle indietro, utilizzare il protocollo dettagliato trovato al Biernaskie et al. 10. brevemente, sezionare la pelle dorsale posteriore facendo incisioni da spalla a spalla, lungo il groppone e lungo la schiena utilizzando una pinzetta e forbici. Tagliare la pelle in fette sottili (spessore 0,5 cm) usando un bisturi sterile.
- Lavare il tessuto 2 volte con HBSS ghiacciata e rimuovere indesiderati del tessuto connettivo, i depositi di grasso o detriti sotto un microscopio per dissezione.
- Per il derma della pelle solo, float le fette in dispase (5 mg/mL, 5 U/mL) in HBSS per 30-40 min a 37 ° C. Chirurgicamente è possibile separare l'epidermide dal derma. Scartare l'epidermide o dissociare ulteriormente usando tripsina se di interesse.
- Tritare il campione in pezzi di 1-2 mm usando un bisturi sterile e mettere in enzima di freddo collagenosi-IV appena scongelati 2 mg/mL (2 mg/mL, 125 CDU/mg, media F12).
- Per il nervo, utilizzare ~ 500 µ l 2 x nervo sciatico. Per la pelle, uso ~ 8 mL per topo 1x indietro della pelle.
Nota: I tessuti devono essere completamente immersi nella soluzione della collagenosi-IV. È fondamentale che qualsiasi enzimi di digestione sono gestiti, archiviati e preparati in modo corretto. Se gli enzimi sono lasciati a temperatura ambiente per lunghi periodi di tempo, isolamento di singole cellule richiederà triturazione meccanica eccessiva e ridurre la vitalità cellulare. Collagenasi-IV può essere altresì composto in terreni di coltura delle cellule dove la vitalità cellulare è più ottima. Tuttavia, questo potrebbe alterare l'attività dell'enzima o firma trascrizionale quindi dovrebbe essere ottimizzato dall'utente.
- Per il nervo, utilizzare ~ 500 µ l 2 x nervo sciatico. Per la pelle, uso ~ 8 mL per topo 1x indietro della pelle.
- Incubare il campione nell'enzima in un bagno di 37 ° C per 30 minuti con agitazione delicata ogni 10 min. Un agitatore posizionato a 37 ° C è anche un'alternativa appropriata.
- Triturare con un pipettatore P1000 20 - 30 volte all'aggiunta degli post-enzimi 30 min.
- Ripetere triturazione ogni 30 min fino a quando la soluzione appare torbida e pezzi di tessuto in gran parte sono dissociati.
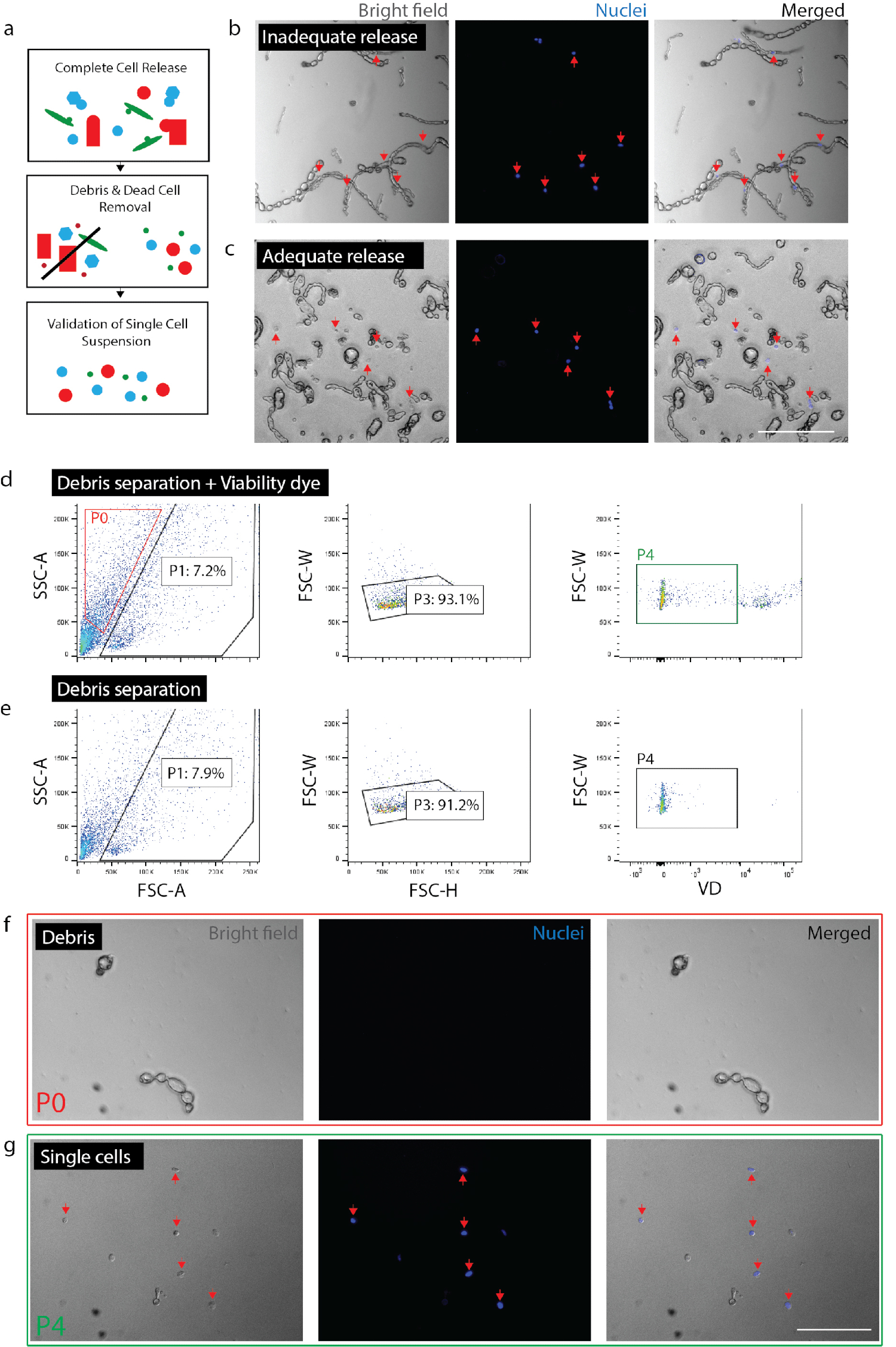
Nota: Assicurarsi di rilascio completo delle cellule (Figura 2b, 2C). Per confermare il rilascio completo, piatto di cellule con Nuc blu (2 gocce per 1 mL) e dopo 20 minuti, controllare sotto il microscopio per garantire a tutti i nuclei sono associati a singole cellule piuttosto che detriti. È fondamentale per verificare il grado di rilascio delle cellule all'interno di un determinato esperimento per ogni tipo di tessuto o condizione. In tessuto fibrotico (cioè, lesione cronica) o tessuto adulto illeso, il rilascio di cellule variano notevolmente da lesione acuta o tessuto embrionale. Questo è particolarmente importante perché alcuni tipi di cellule hanno meno probabili di rilascio dal tessuto rispetto ad altri, preferenzialmente escludendo quindi quelle cellule da analisi a valle.- Per il nervo, dissociare tessuto per 0,5-1,5 ore totali. Per la pelle, dissociare il tessuto per un totale di 2 ore (nell'ultima ora di incubazione, aggiungere dnasi (1 mg/mL) al campione della pelle).
- Filtro due volte con un filtro di 40 µm. Risciacquare il filtro con gelida 1% BSA/HBSS.
- Centrifugare a 260 x g per 8 min. Quindi rimuovere il surnatante.
- Risospendere il pellet cellulare in HBSS contenente BSA 1% utilizzando una punta di wide-bore e metterli su ghiaccio. Il volume di risospensione è basato sul volume di tessuto (il peso bagnato 800mg per pelle = 800 µ l volume; 10 mg peso per nervi bagnato = 100 µ l).
- Facoltativamente, iniziare con un volume basso risospensione e quindi regolare come necessario sulla base del tasso di flusso (eventi al secondo) il sorter FACS. La densità di ordinamento più efficiente (massimizzare il numero di cellule raccolte mentre limitandone tempo) per le collezioni è 3.000-7.000 eventi al secondo.
- Se usando la tintura di attuabilità, togliere un subaliquot per un controllo non colorato. Quindi aggiungere colorante vitalità 1:15,000 (stock: 20.000 nM / µ l) di campione (concentrazione finale di 1,3 nM / µ l) utilizzando una punta di wide-bore per ridurre la tosatura.
Nota: È fondamentale per verificare il grado di morte cellulare all'interno di un determinato esperimento per ogni tipo di tessuto o condizione. Alcuni tipi di cellule all'interno di un campione sono più probabili di morire rispetto ad altri, quindi preferenzialmente essere stati esclusi dall'analisi a valle.- Incubare il campione con la tintura di attuabilità per 5-10 min sul ghiaccio al buio. Quindi aggiungere 4 mL di gelida 1% BSA/HBSS al campione. Centrifugare a 260 x g per 8 min rimuovere tinture per eccesso di vitalità. Trattare il subaliquot con nessuna vitalità tingere non macchia controllo nello stesso modo.
2. isolamento di cellule vitali e sane (giorno 1)
- Assicurarsi che l'impianto di FACS segue delle cellule attivate la fluorescenza appropriati (FACS) parametri di ordinamento.
- Preparare la macchina di FACS in anticipo per assicurare che sia pronta una volta completata la centrifuga finale nel passaggio 1 e garantire che il vano di raccolta è mantenuto freddo utilizzando blocchi di ghiaccio.
- Utilizzare i seguenti parametri: velocità di flusso: 1.0 (corrispondente all'incirca a 10 µ l/min); Filtro: 1.5 ND; Formato dell'ugello: 100 µm; Forward scatter: 80-180 V (cambia come necessario al fine di distinguere la dimensione degli eventi); A dispersione laterale: 150-220 V (cambia come necessario al fine di distinguere la granularità/forma degli eventi); Laser: 100-400 V (cambia come necessario al fine di distinguere gli eventi negativi di attuabilità tintura positivo vs & controllare questo contro alcun controllo di tintura di redditività); Gates: Modificare come necessario per garantire che tutte le celle sono raccolti. Vedere figura 2d-2 g.
Nota: I parametri FACS sono altamente dipendente dai tipi delle cellule e il sorter impiegato e pertanto deve essere ottimizzato dall'utente.
- Preparare 15 mL provette stretto sotto con 8 mL di gelida 1% BSA/HBSS per campionari. Statica all'interno del tubo e la tensione superficiale può influenzare l'efficienza di raccolta. Invertire i tubi prima di collezioni per assicurare l'interfaccia tra la superficie del liquido e l'interno del tubo è umido.
Nota: Se si lavora con numeri molto bassi delle cellule, aggiustare per nave piccola collezione come appropriato. - Una volta che tutte le celle vengono raccolti, centrifugare il campione a 260 x g per 8 min.
Nota: Prima di centrifugazione, aggiungere 1% BSA/HBSS a lavare/push cellule giù dalla superficie laterale e Inverti/mix il tubo subito dopo FACS. - Risospendere le cellule in 1% BSA/HBSS e tenere sul ghiaccio. È il volume massimo per esempio che è compatibile con l'elaborazione di passaggio 3 µ l 33,8, pertanto assicurarsi che volume di diluizione/risospensione delle cellule finale è opportuno ottenere il numero di cellulare ideale a 33,8 µ l. Altre opzioni di supporto di diluizione per questo passaggio (e tutte le diluizioni di precedente in 1% BSA/HBSS) includono DMEM e fino a 40% di siero, ma evitare di calcio, magnesio o EDTA contenente i reagenti.
- Lasciare le cellule sul ghiaccio per un importo minimo di tempo. Co-worker idealmente dovrebbe preparare tutte le attrezzature e reagenti per il passo successivo (passaggio 3) durante le fasi finali della fase 2.
- Cella preparazione controlli critici
- Confermare le stime del numero di cellule ottenute da FACS. A seconda del tipo di tessuto e di dissociazione lunghezze, detriti e cellule possono essere molto simili in forma e dimensione. Così, a meno che non viene utilizzato un reporter fluorescente, FACS non può escludere tutti i detriti. È consigliabile che un conteggio di cella finale dopo raccolta FACS è effettuata per capire qual è la percentuale di eventi (secondo FACS) sono in realtà le cellule per una determinata preparazione (Figura 2 g). Eseguire il conteggio delle cellule usando un emocitometro o contatore di cellule automatizzato (Ripeti due volte) e calcolare la percentuale di cellule vitali che è rappresentata dagli eventi totali raccolti secondo macchina FACS.
- Convalidare la preparazione delle cellule. Verificare che nessun particelle di grandi dimensioni (> 100 µm) sono presenti come essi possono intasare attrezzature nei passaggi a valle. Rimozione inadeguata dei detriti può rischiare di intasamento il chip microfluidici unicellulare. Piastra di celle rimanenti con Nuc Blue (come sopra) affinché che non vi siano frammenti grandi detriti sono presenti. Questo permetterà anche la conferma che le cellule sono singolare (cioè, non attacchino) dando fiducia che l'analisi genetica a valle singola cella rappresenta singole cellule, piuttosto che più celle.
- Decidere sui numeri di cellulare per sequenza: c'è una vasta gamma di numeri di adulto delle cellule tessuto-derivato per campione che possa essere caricato nel sistema con fino a 8 campioni che possono essere eseguiti in una sola volta. Gli autori hanno caricato ovunque da 500 – 50.000 cellule per campione e ottenuto set di dati di buona qualità scRNA-Seq. Più discussione per quanto riguarda i numeri di cellulare più appropriati per caricare può essere trovato nella sezione discussione. L'output finale di numeri in sequenza delle cellule dipende in larga misura la qualità del singolo-cellule isolate. Caricamento di 10.000 cellule adulte tessuto-derivato può restituire ovunque da 1.000 a 4.000 sequenziate cellule (10-40% di ritorno). Se siete interessati a sequenziare numeri alti delle cellule (~ 10.000 cellule, il numero massimo consigliato per questo sistema), quindi 25.000-100.000 celle di carico sarà richiesta.
3. GEM generazione (Gel tallone in emulsione) e codici a barre (giorno 1)
Nota: I passaggi da 3 a 6 del presente protocollo sono progettati per essere utilizzato in combinazione con la piattaforma più comune basato su microdroplet unicellulare, Prodottoda da 10 X Genomics. Linee guida dettagliate per i passaggi 3 e 4 sono definiti del produttore protocollo (Vedi il protocollo di cromo singola cella 3')11,12 e devono essere seguite in combinazione con questo protocollo. Per risultati ottimali, passaggio 3 deve essere completata immediatamente dopo dissociazione (passaggio 1) e cella isolamento (passaggio 2) passi il giorno 1 del presente protocollo.
- Preparare il chip secondo protocollo 11,12 del produttore. Questa piattaforma di cella singola basata su microdroplet utilizza la tecnologia che i codici a barre campioni ~ 750.000 per indicizzare separatamente del trascrittoma di ogni cella. Ciò si ottiene suddividendo le cellule in Gel tallone in emulsioni (gemme) cui cDNA generato condividono un comune codice a barre. Durante la generazione del GEM, le cellule vengono consegnate in modo che la maggior parte (90-99%) di gemme generati non contiene alcuna cella, mentre il resto, per la maggior parte, contengono una singola cella.
- Posizionare il chip nel supporto chip.
- Preparare il mix master cella sul ghiaccio.
- Aggiungere glicerolo al 50% a pozzi inutilizzati e aggiungere 90 µ l di mix master cella a ben 1, 90 µ l di gel perline a ben 2 e 270 µ l di olio per ben 3 di partizionamento.
- Coprire il chip con la guarnizione.
- Caricare il chip ed eseguire in un controller di cella singola.
- Il vassoio di uscita, posizionare il chip nel cassetto, chiudere il cassetto e premere il tasto Play. Un singola cellula 3' gel tallone in un gioiello include primer contenente una sequenza parziale di Illumina R1 (leggere 1 primer di sequenziamento), un 16 nucleotidi (nt) 10 x Barcode, un 10 nt identificatore univoco molecolare (UMI) e una sequenza di poli-dT primer. Durante l'esecuzione, perline di gel nel controller vengono rilasciati e mescolati con mix di lisato e maestro di celle.
- Raccogliere 100 µ l di campione e posto in un tubo PCR.
- Provette per PCR posto nella pre-impostati PCR macchina ed eseguire la PCR secondo il kit. A seguito di incubazione, gemme includerà integrale, con codice a barre del cDNA da mRNA poli-adenylated.
- Dopo l'esecuzione, posto a-20 ° C durante la notte per fino a 1 settimana prima precedente al passaggio successivo.
4. clean-Up, amplificazione, la costruzione della libreria e Biblioteca quantificazione (2 ° giorno in poi)
Nota: Linee guida dettagliate per i passaggi da 4 sono descritte in protocollo 11,12 del produttore e devono essere seguite in combinazione con questo protocollo.
- Utilizzare biglie magnetiche silano per rimuovere residui Reagenti biochimici/primer dalla miscela di reazione di GEM.
- Amplificare il cDNA integrale, con codice a barre per generare una massa sufficiente per la costruzione della libreria.
- Valutare il rendimento del DNA. Prima della costruzione della libreria, valutare il rendimento del DNA del campione. Questo determinerà quanti cicli da utilizzare nella fase PCR a valle (esempio indice PCR durante la costruzione della libreria). A seconda del contenuto di RNA di un dato campione, che può variare a seconda degli Stati di attivazione (ad es., controllo vs feriti, ecc.), tipo di cella e il rendimento delle celle, il numero di ciclo consigliato potrebbe cambiare.
- Per sequenziamento ~ 3.000 tessuto-derivato le cellule (irrilevante agli Stati di attivazione), gli autori hanno trovato che 14 cicli (esempi: ~ 10-100 ng DNA) è standard.
- Utilizzare un Bioanalyzer per l'analisi del DNA. Fare riferimento all'utente Guida13.
- Frammento di esempio e selezionare il formato del DNA. Prima della costruzione della libreria, utilizzare frammentazione enzimatica e protocolli di selezione del formato per ottenere cDNA appropriato amplicone dimensione.
- Preparare il campione per la costruzione della libreria. Mentre R1 (leggi 1 sequenza primer) viene aggiunto alle molecole durante l'incubazione GEM; P5, P7 (un indice di esempio), e R2 (leggere 2 sequenza primer) vengono aggiunti durante la costruzione della libreria.
- Valutare il rendimento del DNA. La maggior parte delle strutture di sequenziamento necessitano presentazione di finale librerie che includono informazioni di resa e la qualità del DNA. Pertanto, eseguire il bioanalyzer a seguito del completamento del protocollo intero e prima del trasporto al centro di sequenziamento.
- Conservare i campioni a-80 ° C fino a 2 mesi.
- Prima di sequenziamento, quantificare i campioni utilizzando un kit di quantificazione del DNA. Questo può essere fatto presso l'impianto di sequenziamento.
5. Biblioteca sequenziamento (giorno 3 in poi)
Nota: La piattaforma di codici a barre del trascrittoma cella singola utilizzata nel presente protocollo genera librerie di accoppiato-fine Illumina-compatibile inizia e termina con sequenze P5 e P7. Anche se la profondità minima necessaria per risolvere il tipo di cella identità può essere da un minimo di 10.000-50.000 letture/cella15,16, ~ 100.000 letture/cella è raccomandato come un compromesso ottimo di copertura dei costi per adulte cellule in vivo (tenendo a mente alcune delle cellule tipi o stati minimamente attivata delle cellule raggiungerà saturazione a 30.000-50.000 letture/cella).
- Trasporto di cDNA su ghiaccio secco ad un impianto di sequenziamento dotato di un sequencer Illumina appropriato.
- Fornire le seguenti informazioni al centro di sequenziamento:
- Fornire i dettagli del campione: campione indice ID corrispondenti a ogni libreria; specie; database di genomica per assembly primario (cioè, GRCm38 per mouse); elettroferogramma mostrando dimensioni del frammento dal bioanalyzer (tra 200 e 9.000 bp); cDNA concentrazione (ng / µ l) e concentrazione totale biblioteca (rendimenti totali variano da 200-1400 ng); volume (µ l) di campione.
- Fornire le richieste di sequenziamento: quantificare i campioni utilizzando un kit di quantificazione del DNA; tipo di adattatore/indice (TruSeq DNA); tipo di piastra (Eppendorf Twin. TEC, completo gonna - consigliato per DNA); sequenziamento tecnologia/libreria tipo (10 x, sequenziamento completo istruzioni e consigli di ciclo)17.
- Eseguire sequenziamento superficiale (opzionale): studi di analisi di più campioni biologici potranno beneficiare di pool di campioni (aggregazione) per generare una matrice di singolo gene-codice a barre contenente i dati da tutti i campioni. Per ridurre al minimo gli effetti di batch tra campioni quando il pool, la profondità di lettura tra diverse biblioteche dovrebbe essere standardizzata. A tal fine è necessaria un'approssimazione dei numeri di singola cellula. Il sequencer MiSeq permetterà sequenziamento superficiale ed è un modo pratico e conveniente per ottenere stime accurate delle cellule.
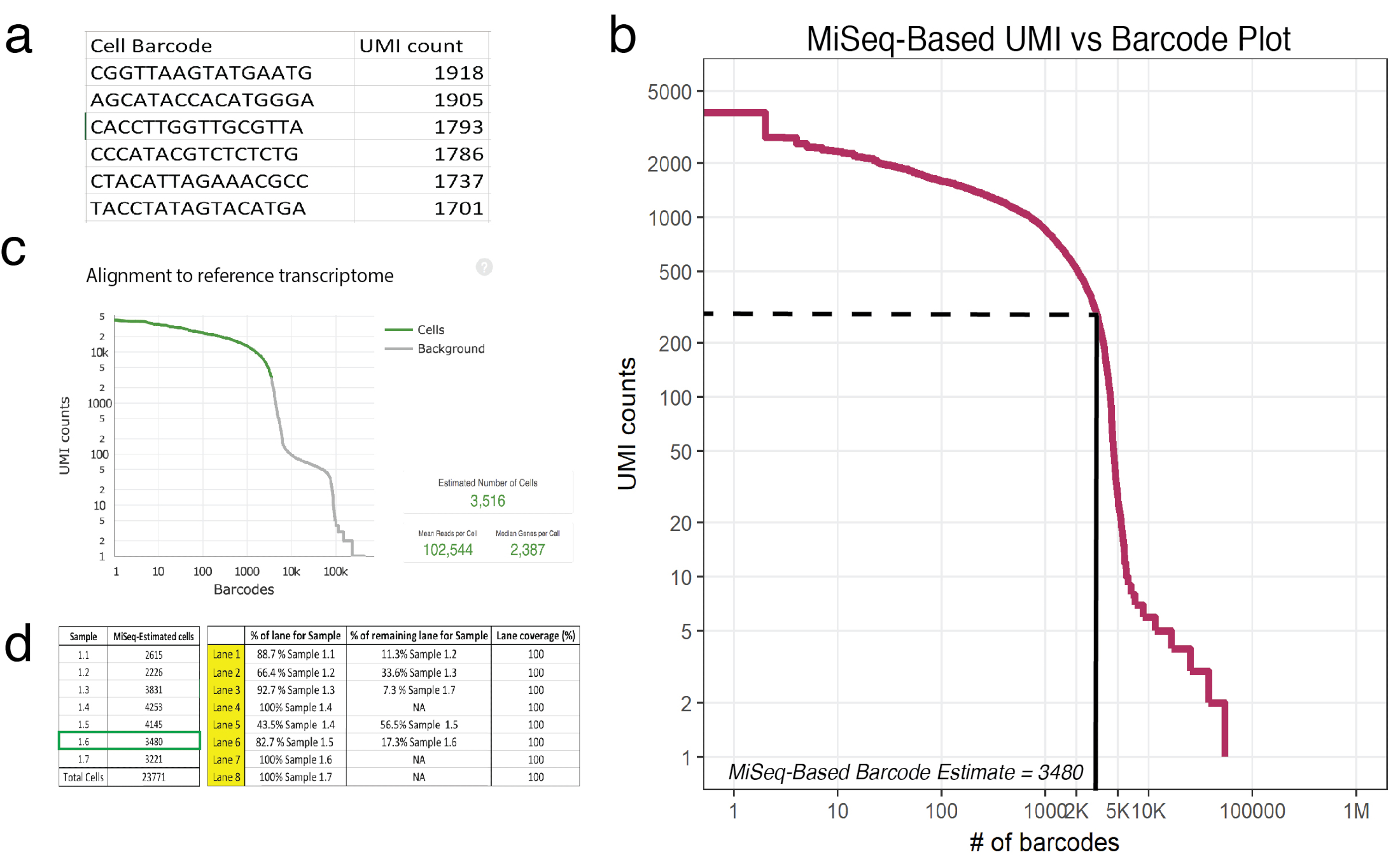
Nota: Una corsa con un sequencer MiSeq SR50 fornisce una copertura sufficiente per stimare con precisione circa 20.000 cellule. Questa corsa si avvicinerà il numero di UMI recuperati per ogni codice a barre univoco. In Figura 3a, viene visualizzato l'intestazione di un output di esempio (esempio 1.6) (. csv), inserzione di codici a barre e suo corrispondente UMI conta come determinato da letture con fiducia mappate.- Consultarsi con un bioinformatico per acquisire familiarità con il linguaggio di programmazione R. Fare riferimento al tutorial di DataCamp per più informazioni18.
- Valutare dati grezzi ottenuti dal sequencer utilizzando lo script fornito di R come un modello19. Dati grezzi si riferiscono al numero di UNMIS mappato a ogni codice a barre di cella univoci. Lo script legge un file con estensione CSV, dove la prima colonna è un elenco di codici a barre e la seconda colonna sono suoi corrispondenti UMI conta. Questo script vi fornirà una trama (Figura 3b) così come il numero stimato di cellule con codice a barre in ciascun campione. Regolare script per garantire che il numero immesso di UMI conteggi per un dato campione è presso il punto di un terzo della prima tendina ripida. In Figura 3b, questo gomito cade circa 225 UNMIS corrispondente a 3.480 cellule con codice a barre.
- Paragonabile a piena profondità sequenziamento utilizzando HiSeq (dove 3.516 cellule erano con successo sequenziato, Figura 3C), stime di sequenziamento superficiale predetto 3.480 cellule.
- Uso cellulare recupero approssimazioni (dal punto 5.3) o utilizzare il grafico di recupero trovati a protocollo20 per pianificare la distribuzione di lane per il sequenziamento di più profondo del produttore. Ogni campione deve ricevere copertura paragonabile, quindi se sequenziamento superficiale rivela che ci sono numeri differenti delle cellule in ogni campione (che è spesso il caso) quindi distribuzione lane dovrebbe essere calcolato di conseguenza. Una cella di flusso HiSeq (che comprende 8 corsie) possibile sequenza fino a 2,4 miliardi personalizzato fine accoppiato letture. Set-up delle cellule di flusso esempio è presentato in figura 3d.
6. elaborazione leggere i file
Nota: Ordinando una singola cella 3' libreria utilizzando questo protocollo genera dati grezzi in formato binario chiamata base (BCL). La cella Ranger pacchetto viene utilizzato per generare il file FASTQ basati su testo da file BCL, eseguire genomica e trascrittomica allineamenti, gene conteggi, demultiplazione e aggregazione dei campioni. In questa sezione, i passaggi chiave che consentono agli utenti di scaricare i dati grezzi di BCL da un impianto di sequenziamento e generare codice a barre-gene filtrata matrici pronte per la bioinformatica a valle è presentato.
- Utilizzare un server centralizzato per l'esecuzione del programma. BCL file, i file FASTQ e la maggior parte della bioinformatica a valle l'elaborazione richiede potenza di elaborazione significativi.
- Scarica tutti i file raw di lettura sul server (o file FASTQ se sono disponibili).
- Rivolgersi all'amministratore del server per impostare un account su un server centralizzato o un cluster e per acquisire familiarità con Unix21.
- Utilizzare un comando fetch appropriato per il sistema operativo del server per scaricare tutti i file dal server della struttura di sequenziamento.
- La maggior parte delle strutture di sequenziamento forniscono un comando per scaricare file da un percorso sicuro che può essere eseguito dalla riga di comando (Vedi esempio sotto).
- Sostituire la "< nomeutente >" e "< password >" segnaposto nella riga di comando con le credenziali fornite.
wget - O - "https://your_sequencing_facilitys_server.com/path_to_raw_read_files/ - no-cookies-no-controllo-certificato - post-dati ' j_username = username & j_password = password' | wget-- no-cookies-no-controllo-certificato - post-dati ' j_username = username & j_password = password' - ci -
- Se un percorso assoluto al file viene fornito solo (vale a dire https://your_sequencing_facilitys_server.com/path_to_raw_read_files/), inserire un comando fetch in questo percorso.
- Decomprimere i file: se scaricato fine di file con estensione ". gz", è stato compresso utilizzando il comando "gzip". Per decomprimere il file, Esegui decomprimere il comando nella riga di comando (Vedi esempio sotto).
gunzip raw_read_files.gz - Scarica l'ultima versione di cella Ranger sul server come un self-contained. tar22.
-
Critico: Prima di scaricare, verificare che il sistema Linux soddisfi requisiti minimi23. Garantire un minimo di 8-core Intel con 64 GB di RAM e 1 TB di spazio libero su disco.
Nota: Cella Ranger fornisce trascrittomi precostruite riferimento umano e del roditore. Questi può essere modificati utilizzando il comando cellranger mkref per rilevare geni come GFP24.
-
Critico: Prima di scaricare, verificare che il sistema Linux soddisfi requisiti minimi23. Garantire un minimo di 8-core Intel con 64 GB di RAM e 1 TB di spazio libero su disco.
- Generare file FASTQ da file di base chiamata del sequencer (BCL) utilizzando il comando di mkfastq cellranger.
Nota: Il programma verrà allineare letture crude (da file FASTQ) di un genoma di riferimento e generare matrici di cellula del gene per l'analisi a valle. Esso utilizza Allineatore di stelle che viene eseguito l'allineamento splicing-consapevoli di letture di un genoma di riferimento. Sola con fiducia mappate letture (cioè, letture compatibili con un'annotazione di singolo gene) sono utilizzate per il conteggio di UMI.- Ad esempio, utilizzare il comando di mkfastq cellranger:
mkfastq cellranger - id = sample_name \
-eseguire = / percorso/per/sample \
..--csv=csv_file_containing_lane_sample_index.csv
- Ad esempio, utilizzare il comando di mkfastq cellranger:
- Eseguire cellranger conteggio FASTQ file generati utilizzando mkfastq per generare unicellulare gene conteggi.
- Ad esempio, utilizzare il comando del conte di cellranger:
cellranger totali -- id = sample_name \
-trascrittoma = refdata-cellranger-mm10-1.2.0 \
-fastqs = / assoluto/percorso/per/fastq/files \
-campione = same_sample_name_supplied_to_cellranger_mkfastq \
-localcores = 30
- Ad esempio, utilizzare il comando del conte di cellranger:
- Multi-libreria aggregazione (opzionale): per combinare i campioni, piscina cellranger totali uscite usando cellranger aggr. In questo modo una matrice di singolo gene-codice a barre contenente dati riuniti da più librerie. Comando di esempio cellranger aggr:
aggr CellRanger - id = sample_name \
-csv = csv_with_libraryID_ & _path_to_molecule_h5.csv \
-normalizzare = mappato
Nota: Le librerie possono essere aggregate utilizzando tre modalità di normalizzazione (mappata, crudo, nessuno). Mappato è raccomandato in quanto esso Sottocampiona librerie di profondità superiore fino a quando tutte le librerie hanno uguale sequenziamento profondità25. - Per l'immediata visualizzazione/analisi dei dati, importare il file di output di .cloupe (generato utilizzando cellranger conteggio o cellranger aggr) 10x lente di ingrandimento cellulare Browser26.
7. avanzate analisi dei DataSet scRNA-Seq
Nota: Un database di strumenti completa scRNA-Seq può essere trovato alla scRNA-strumenti3,27. Sotto è un framework per cella senza supervisione pseudotemporal e clustering utilizzando Seurat2 ordinamento utilizzando Monocle6. Anche se molto di questo lavoro può essere fatto su un computer locale, la procedura seguente presuppone che calcolo sarà completato utilizzando un server istituzionale.
- Scarica la versione più recente di Miniconda sul account server utilizzando la piattaforma Linux28.
- Installare l'ultima versione di R utilizzando conda29.
- Tracciare i dati utilizzando lo script fornito di Seurat R come un modello30.
Nota: Seurat è un toolkit R-based che consente a controlli di qualità, clustering, differenziale analisi dell'espressione genica, identificazione di geni marcatori, riduzione della dimensionalità e visualizzazione dei dati di scRNA-Seq. Il Satija Lab sito Web31troverete una descrizione completa di Seurat codifica e tutorial. - Tracciare i dati utilizzando lo script fornito di Monocle R come un modello32.
Nota: Monocle è un altro toolkit R-basato che permette la visualizzazione dei cambiamenti di espressione sopra pseudotime e identifica i geni alla base di decisioni di destino delle cellule. Sul sito Web di Monocle33troverete una descrizione completa di Monocle codifica e tutorial. - R-pacchetti come kBET possono essere impiegati per testare e correggere gli effetti di batch come conseguenza di pool di DataSet34.
8. NCBI GEO e SRA Submissions
Nota: Poiché permette di raggiungere facilmente i file raw sequenziamento garantire riproducibilità e rianalisi, impacchettati iscrizioni alla repository pubblicamente disponibili online sono raccomandati o richiesti prima della presentazione del manoscritto. National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) e sequenza di lettura archivio (SRA) sono archivi di dati pubblicamente accessibili per sequenziamento high throughput dati35,36.
- Registratevi per GEO Submitter conto37 di NCBI.
- Presentazione di GEO completo che include tre componenti compilati in una directory/cartella (intitolato come nome utente del chi si sottomette GEO): 1) record di metadati (un foglio di calcolo per la presentazione del progetto); 2) file di dati raw; 3) trattati in file di dati.
- Scaricare e compilare il foglio di calcolo di metadati38. La seguente presentazione pubblica di GEO può essere utilizzata come una guida (GSE100320)39. Posizionare il foglio di calcolo nella directory.
- File di dati Raw posto generati da uno script di cellranger totali per tutte le librerie nella directory.
- Posto elaborati dati file (barcodes.tsv, genes.tsv e matrix.mtx i file filtrati) generati da uno script di cellranger totali per tutte le librerie nella directory.
- Utilizzare le credenziali di submitter GEO FTP server per trasferire la directory contenente tutti i tre componenti. Per gli utenti Linux/Unix: ncftp, lftp, ftp, sftp e ncftpput può essere utilizzati.
- Notificare GEO per tutti i trasferimenti38.
Risultati
Il repertorio di pacchetti open source progettato per analizzare DataSet scRNA-Seq è aumentato drammaticamente40 con la maggior parte di questi uso di pacchetti di linguaggi basati su R3. Qui, vengono presentati risultati rappresentativi utilizzando due di questi pacchetti: valutando raggruppamento senza supervisione di basato sull'espressione genica di cellule singole e singole cellule lungo una traiettoria di ordinazione al fine di risolvere delle cellule di eterogeneità e decostruire biologico processi.
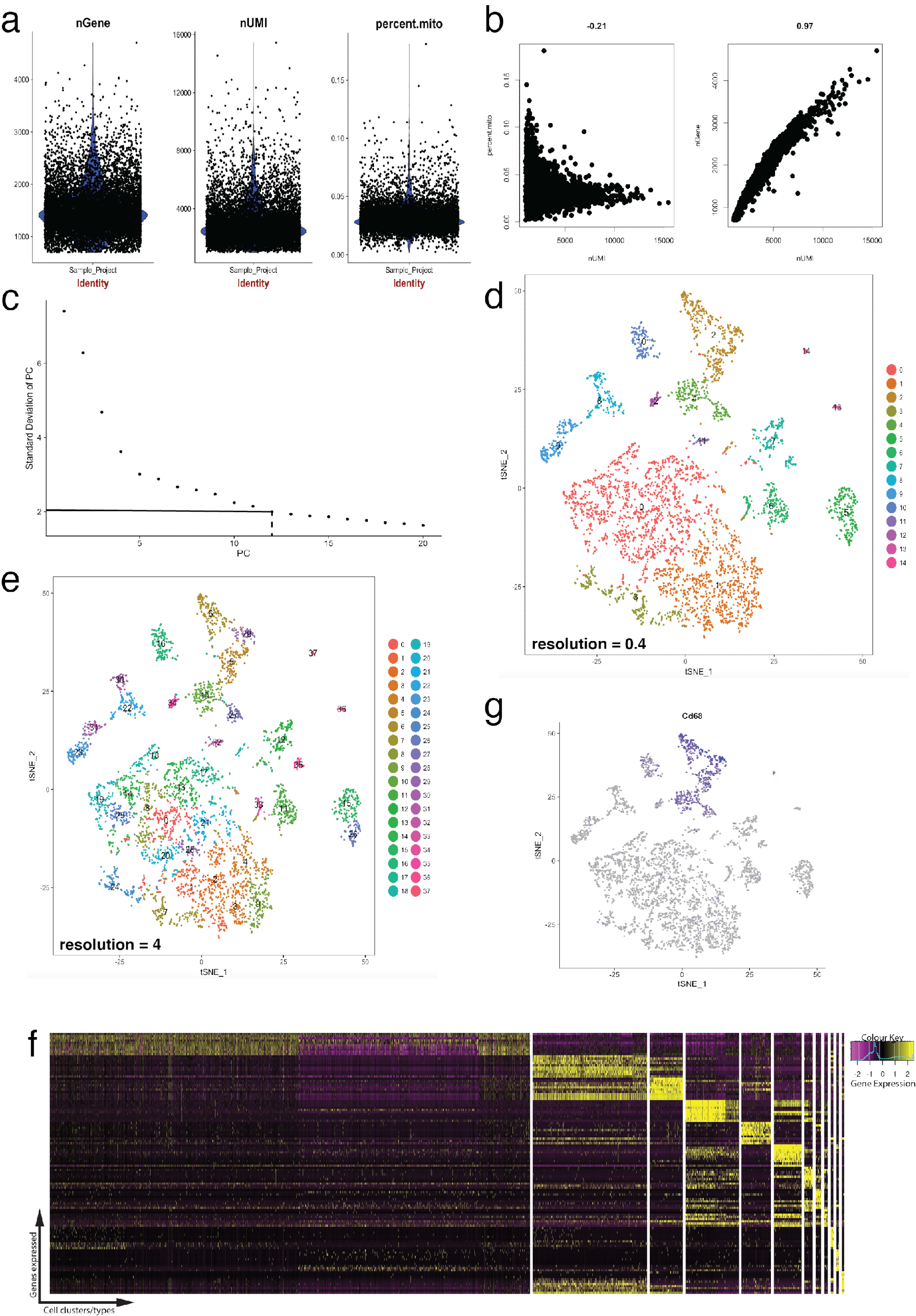
La figura 4 illustra l'uso di Seurat per controlli di qualità e l'analisi bioinformatica a valle di pre-elaborazione. In primo luogo, filtrazione e rimozione delle cellule devianti dall'analisi è essenziale per il controllo qualità. Questo è stato fatto utilizzando violino (Figura 4a) e dispersione trame (Figura 4b) per visualizzare la percentuale di geni mitocondriali, numero di geni (cuore) e numero di UMI (nUMI) per identificare la cella doppietti e outlier. Ogni cella con un numero di chiaro valore erratico dei geni, UMI o percentuale di geni mitocondriali è stato rimosso utilizzando la funzione FilterCells di Seurat. Dal momento che Seurat utilizza componenti principali (PC) gol di analisi alle cellule di cluster, determinare statisticamente significativa pz da includere è un passo fondamentale. Trame di gomito (Figura 4c) sono stati utilizzati per la selezione di PC, in quali PC oltre l'altopiano della 'deviazione standard del PC' asse sono stati esclusi. La risoluzione di clustering è stata maneggiata anche dimostrando che è possibile modificare il numero di cluster, che vanno da 0.4 (bassa risoluzione che conduce a meno mazzi delle cellule, Figura 4D) a 4 (alta risoluzione che porta maggiore mazzi delle cellule, Figura 4e ). A bassa risoluzione, è probabile che ogni cluster rappresenta un tipo di cella definita, mentre ad alta risoluzione questo può rappresentare anche sottotipi o stati transitori di una popolazione delle cellule. In questo caso, le impostazioni del cluster a bassa risoluzione sono state utilizzate per analizzare ulteriormente espressione heatmaps (utilizzando la funzione DoHeatmap di Seurat) per identificare i geni più altamente espressi in un determinato cluster (Figura 4f). In questo caso, sono stati identificati i geni più altamente espressi valutando l'espressione differenziale in un determinato cluster contro tutti gli altri gruppi combinati, dimostrando che ogni cluster è stato rappresentato in modo univoco dai geni definiti. Inoltre, i geni candidati individuali possono essere fruiti su tSNE trame utilizzando la funzione di FeaturePlot di Seurat (Figura 4 g). Questo ha permesso per decifrare se c'erano i cluster che rappresentato i macrofagi. Usando FeaturePlot, abbiamo trovato che entrambi cluster 2 e 4 sono state esprimendo Cd68 - un marcatore pan-macrofago.
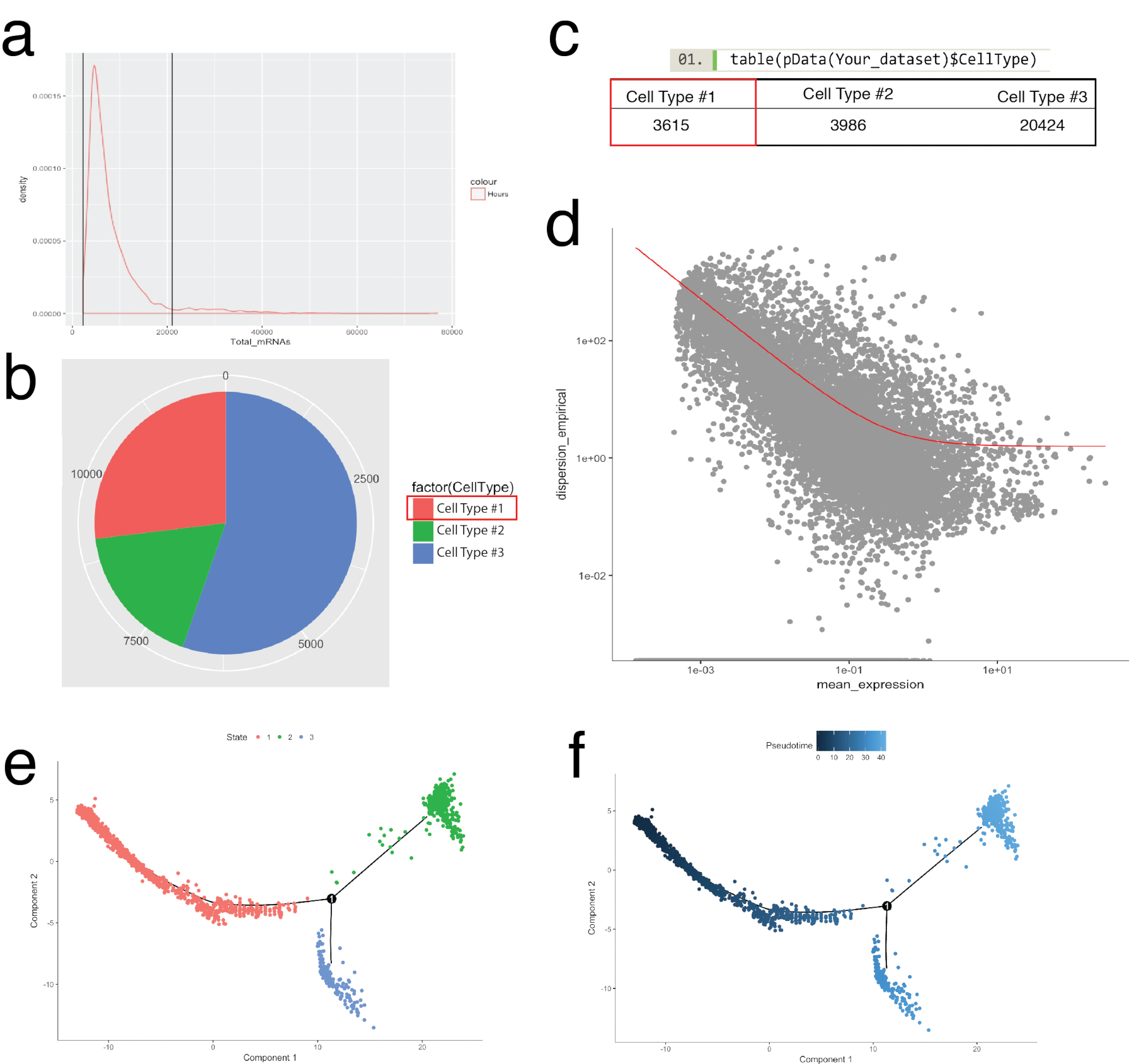
Il pacchetto di Monocle è stato utilizzato per corroborare la mazzi delle cellule identificate in Seurat e per la costruzione di traiettorie delle cellule, o ordinare pseudotemporal, ricapitolare i processi biologici (Figura 5). Ordinazione pseudotemporal può essere utilizzato per gli esempi dove i profili di espressione di singole cellule sono tenuti a seguire un corso di tempo biologico. Le cellule possono essere ordinate lungo un continuum pseudotemporal per risolvere stati intermedi, punti di biforcazione di due destini cellulari alternativi e identificano firme di gene alla base di acquisizione di ogni destino. In primo luogo, simile a filtrazione di Seurat, scarsa qualità celle sono state rimosse tali che la distribuzione di mRNA attraverso tutte le cellule è stato registro normale ed è caduto tra i limiti superiore e inferiore come indicato in Figura 5a. Quindi, utilizzando la funzione newCellTypeHierarchy di Monocle, singole cellule sono state classificate e contati usando geni marcatori noti lignaggio (Figura 5b, 5C). Ad esempio, le cellule che esprimono PDGF recettore alfa o del fibroblasto specifico della proteina 1 sono state assegnate a Cell tipo #1 per creare un criterio per la definizione di fibroblasti. Successivamente, questa popolazione (Cell tipo #1) è stata valutata per decifrare le traiettorie del fibroblasto. Per effettuare questa operazione, è stata utilizzata la funzione GeneTest differenziale di Monocle, che rispetto le cellule che rappresentano gli stati estremi all'interno della popolazione e trovati geni differenziale per ordinare le celle rimanenti nella popolazione (Figura 5 d). Applicando i metodi di apprendimento collettore (un tipo di riduzione della dimensionalità non lineare) su tutte le celle, è stata assegnata una coordinata lungo un percorso di pseudotemporal. Questa traiettoria quindi è stata visualizzata da stato della cellula (Figura 5e) e pseudotime (Figura 5f).

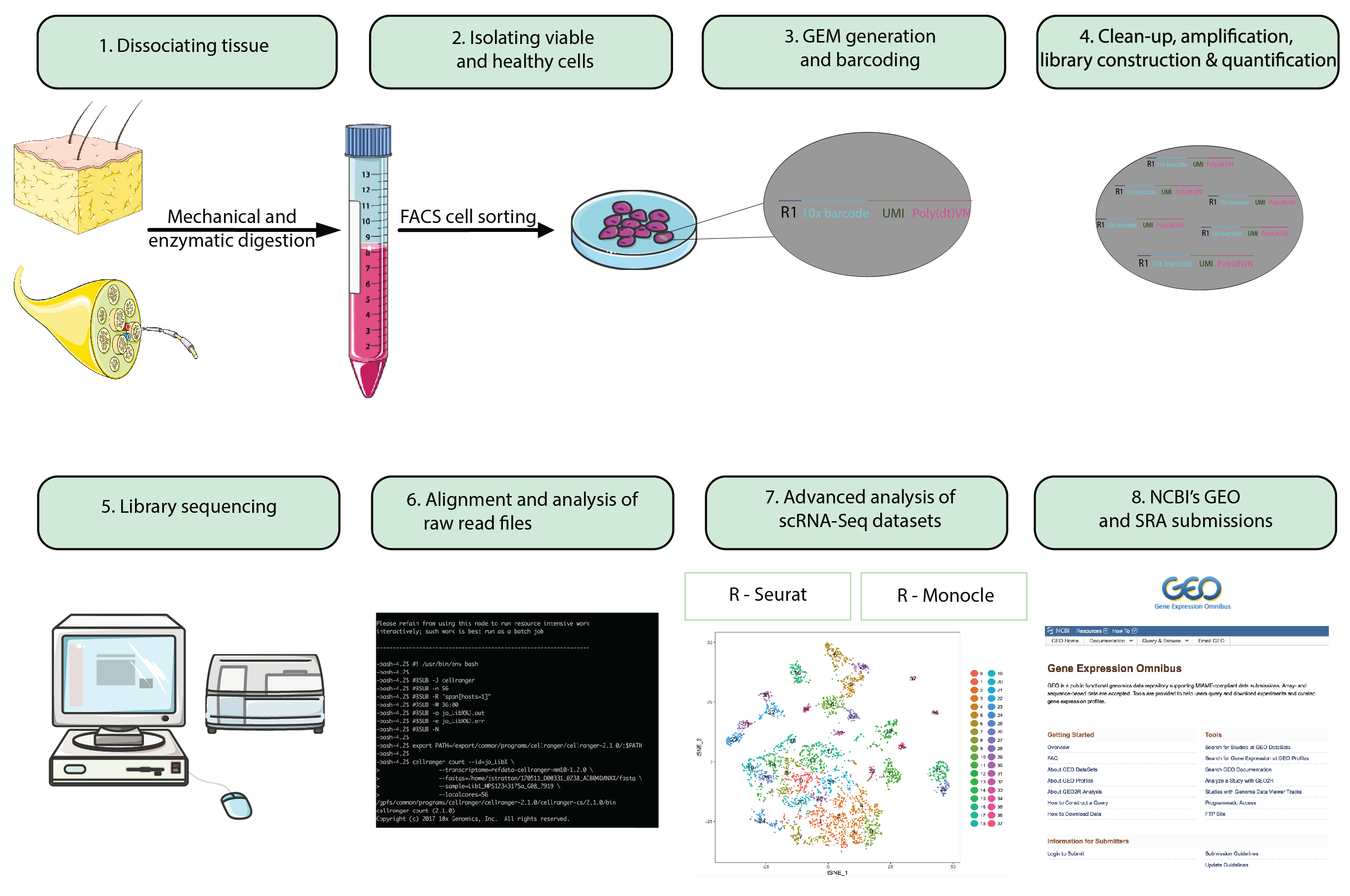
Figura 1: diagramma di flusso. Passi dall'animale intero preparazione all'analisi di singole cellule RNA-Seq DataSet alla presentazione finale di set di dati in un repository pubblicamente disponibile. Perline di gel in emulsione (gemme) riferiscono a perline con oligonucleotidi con codice a barre che racchiudono migliaia di singole cellule. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: creazione di sospensione unicellulare praticabile da tessuto nervoso. (a) del fumetto Panoramica dei controlli di qualità. (b) cellule e detriti con cellule ancora incorporato in detriti (frecce rosse). (c) cellule rilasciate da detriti (frecce rosse). (d) isolamento delle cellule di FACS. P0: frazione detriti; P1: cellula-come frazione; P3: esclusione di Duine; P4: frazione negativo colorante (Sytox Orange) di attuabilità. (e) nessun controllo di tintura di attuabilità. (f) immagine di P0 frazione rappresentare isolato detriti. (g) immagine di P4 frazione rappresentare isolato cellule vitali (frecce rosse). (b) (c) (f) e (g) aveva nucleare colorante aggiunto 20 minuti prima di formazione immagine. Barre della scala: 80 µm. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: sequenziamento superficiale predice il numero delle cellule recuperate in campioni trattati: 10x. (a) un esempio (esempio 1.6) di csv generati MiSeq Inserzione cellulare i codici a barre e suo corrispondente UMI conta come determinato da letture con fiducia mappate. (b) codice a barre trama rango per esempio 1.6 Mostra un calo significativo nel conteggio UMI in funzione del cellulare i codici a barre. Le linee tratteggiate e solide rappresentano il cut-off tra cellule e sfondo come determinato mediante ispezione visiva. (c) codici a barre delle cellule osservate utilizzando la cella Ranger pipeline post-HiSeq rivela superficiale sequenziamento approssimata con precisione il numero di cellule per esempio 1.6. (d) un esempio di un set-up di cella di flusso basato sul sequenziamento poco profonda derivate stime delle cellule. Per esempio 1.6, poiché poco profonda sequenziamento predetto 3480 cellule, 1,17 corsie sono state assegnate per garantire > 100.000 letture per copertura di sequenziamento di cella in HiSeq. Nota: Tutte le corsie devono aggiungere al 100%. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: controllo di qualità e bioinformatica di singola cellula RNA-Seq dataset utilizzando pacchetto Seurat R. (un) trame di metriche di controllo di qualità che includono il numero di geni, numero di identificatori univoci molecolari (UNMIS) e la percentuale di trascrizioni mappatura al genoma mitocondriale. (b) gene campione tracciate individuare celle con livelli deviante di trascrizioni mitocondriali e UNMIS. (c) trama di gomito campione utilizzato per la determinazione ad hoc di PC statisticamente significativa. Le linee tratteggiate e dot-tratteggiata rappresentano il taglio dove un chiaro "gomito" diventa evidente nel grafico. Dimensioni PC prima questo gomito sono inclusi nell'analisi a valle. (d, e) Aggregati di cellule basato su grafico visualizzati in due diverse risoluzioni in uno spazio basso-dimensionale utilizzando una trama tSNE. (f) top geni marcatori (gialli) per ogni cluster visualizzati su un'espressione heatmap utilizzando la funzione DoHeatmap di Seurat. (g) visualizzazione dell'espressione dei marker di, ad esempio, Cd68 gene che rappresenta i macrofagi (viola) utilizzando la funzione FeaturePlot di Seurat. Ciò suggerisce che quel cluster 2 e 4 (in pannello d) di questo dataset rappresenta i macrofagi. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: cellulare categorizzazione e ordinazione lungo la traiettoria di peudotemporal utilizzando il toolkit di Monocle. (a) controllare la distribuzione di mRNA (derivata dal conteggi UMI) su tutte le celle in un campione. Solo le celle con mRNA tra 0 - ~ 20.000 sono stati usati per l'analisi a valle. (b, c) Assegnazione e contando tipi cellulari basati su indicatori delle cellule lignaggio noto. Ad esempio, le cellule che esprimono PDGF recettore alfa o del fibroblasto specifico della proteina 1 erano assegnate a Cell tipo #1 che rappresentano pan-fibroblasti utilizzando la funzione newCellTypeHierarchy di Monocle. Numero di diversi tipi di cellule possa essere visualizzato come un grafico a torta (b) e come una tabella (c). (d) mediante cella tipo n. 1 (fibroblasti) ad esempio, i geni per ordinare le celle possono essere visualizzate utilizzando un grafico a dispersione che illustra la dispersione del gene vs espressione media. La curva rossa mostra il taglio per geni utilizzati per l'ordinamento calcolato dal modello media-varianza mediante la funzione estimateDispersions di Monocle. Geni che soddisfano questa frequenza di taglio sono stati utilizzati per l'ordinazione pseudotime a valle. (e, f) Visualizzazione delle traiettorie di cella in un ridotto spazio bidimensionale colorata della cella "stato" (e) e di Monocle-assegnato "Pseudotime" (f). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Questo protocollo dimostra come la corretta preparazione delle singole cellule possa scoprire l'eterogeneità trascrizionale di migliaia di singole cellule e discriminare gli stati funzionali o identità univoche cellulare all'interno di un tessuto. Il protocollo non richiede proteine reporter fluorescenti o strumenti transgenici e può essere applicato per l'isolamento di singole cellule da vari tessuti di interesse compresi quelli dagli esseri umani; tenendo in considerazione ogni tessuto è unico e questo protocollo richiederà un certo grado di regolazione/modifica.
I programmi trascrizionali vari e altamente dinamici all'interno delle cellule hanno sottolineato il valore della cella singola genomica. Oltre a isolamento di RNA di alta qualità, una fase di preparazione del campione critici necessaria per i set di dati di alta qualità è garantire che le cellule sono completamente liberate dal tessuto e che le cellule sono sane e intatte. Questo è relativamente semplice per raccolta di cellule che sono facilmente rilasciato, come cellule circolanti o nei tessuti dove le cellule senza bloccare vengono mantenute, come nei tessuti linfoidi. Ma questo può essere difficile per altri tessuti dell'adulto, a causa dell'architettura cellulare altamente sviluppata che coprono grandi distanze, matrice extracellulare e le proteine citoscheletriche rigida spesso coinvolti nel mantenimento della struttura delle cellule circostanti. Anche con tecniche di dissociazione appropriato per la versione completa di cellule, esiste la possibilità che l'elaborazione rigorosa e spesso lunga richiesta avrebbe alterato l'integrità delle cellule e di qualità di mRNA. Inoltre, le temperature elevate utilizzate per dissociazione enzima-assistita anche sui transcriptional firme29,30. L'intento del protocollo è di presentare il controllo di qualità controlla, usando tessuti quali il nervo myelinated adulto e pelle adulta ricchi di matrice extracellulare, per dimostrare come ottimizzazione può aiutare a superare questi ostacoli.
Una considerazione importante quando si progetta qualsiasi esperimento scRNA-Seq è la scelta della profondità di sequenziamento. L'ordinamento può essere altamente multiplexato e leggere profondità può variare da essere molto basso facendo uso di goccia-Seq2 a fino a 5 milioni di letture/cella14 utilizzando un metodo di RNA-Seq full-length come Smart-Seq. Maggior parte degli esperimenti di scRNA-Seq può rilevare trascrizioni moderata-alta espressione con sequenziamento à partir 10.000 letture/cella, che è solitamente sufficiente per cella tipo classificazione41,42. Profondità poco profonda sequenziamento è di valore per risparmiare sui costi di sequenziamento in quando si cerca di rilevare popolazioni di cellule rare attraverso tessuti complessi dove migliaia di cellule può essere necessario attribuire con fiducia popolazioni rare. Ma sequenziamento di profondità non è adeguata quando informazioni dettagliate sull'espressione genica e processi associati con sottile transcriptional firme sono necessari. Attualmente, si stima che la grande maggioranza dei geni in una cella vengono rilevata con 500.000 letture/cella, ma questo può variare a seconda del protocollo e tessuto tipo43,44. Mentre trascrizione integrale sequenziamento aggira la necessità per l'assemblaggio e può, pertanto, rilevare romanzo o varianti di splicing rara, costi di sequenziamento spesso limitano tali approcci per esaminare migliaia di cellule che compongono un sistema complesso tessuto di ridimensionamento. Al contrario, 3' etichetta singola cellula librerie come quelli descritti nel presente protocollo in genere hanno una minore complessità e richiedono meno profonda sequenziamento. È importante notare che le librerie generate utilizzando il protocollo descritto possono essere sequenziate su uno dei cinque sequencer supportati: 1) NovaSeq, 2) HiSeq 3000/4000, 3) HiSeq 2500 esecuzione rapida e ad alto rendimento, 4) NextSeq 500/550 e 5) MiSeq.
Un approccio alternativo a singola cella RNA-Seq, che riduce la necessità di tessuti delicati e manipolazione ancora celle mantiene alcuni dei vantaggi della singola cella RNA-Seq, è l'analisi del RNA da singoli nuclei45. Questo approccio permette l'elaborazione più rapida, riducendo la degradazione di RNA e altre misure estreme per garantire adeguata rilascio dei nuclei e così probabilmente consente un'acquisizione più sicura dei profili trascrizionali che rappresenta tutte le celle all'interno di un dato tessuto. Questo sarebbe, naturalmente, fornire solo una parte dell'attività trascrizionale presente all'interno di una cella specificata, quindi a seconda di quali sono gli obiettivi sperimentali di interesse questo approccio potrebbe o potrebbe non essere appropriato.
Oltre alla caratterizzazione completa delle identità cellulare all'interno di un dato tessuto, una delle analisi più preziosi per i DataSet scRNA-Seq è la valutazione di stati intermedi trascrizionale attraverso le popolazioni 'definito' delle cellule. Questi stati intermedi possono impartire le relazioni di discendenza tra le celle all'interno di popolazioni identificati, che non era possibile con tradizionale alla rinfusa che si avvicina a RNA-Seq: intuizioni. Diversi strumenti di bioinformatic scRNA-Seq ora sono stati sviluppati per delucidare questo. Tali strumenti possono valutare i processi coinvolti in, ad esempio, le cellule tumorali transizione a uno stato di oncogeni e/o metastatico, cellule staminali, maturando in diversi destini terminali o cellule immunitarie spola tra Stati attivi e quiescenti. Transcriptome sottili differenze nelle cellule possono anche essere indicative delle polarizzazioni di lignaggio che, strumenti bioinformatici sviluppati di recente come FateID, in grado di dedurre47. Poiché le distinzioni tra cellule di transizione possono essere difficile accertare dato le differenze trascrizionale possono essere sottili, sequenziamento più profonda può essere necessario46. Fortunatamente, copertura di una libreria che digrada dolcemente in sequenza può essere aumentata se siete interessati a sondare il dataset ulteriormente rieseguendo la libreria su un'altra cella di flusso.
Presi insieme, questo protocollo fornisce un flusso di lavoro facile adattare che consente agli utenti di profilo trascrizionalmente centinaia o migliaia di single-celle all'interno di un esperimento. La qualità finale di un dataset scRNA-Seq si basa sull'isolamento delle cellule ottimizzato, citometria a flusso, generazione della libreria di cDNA e interpretazione del codice a barre-gene crudo matrici. A tal fine, questo protocollo fornisce una panoramica completa di tutti i passaggi chiave che possono essere facilmente modificati per attivare studi di tipi di tessuti diversi.
Divulgazioni
Nessun informazioni integrative
Riconoscimenti
Riconosciamo che il personale di supporto presso l'impianto di servizi UCDNA, così come il personale della struttura Animal Care presso l'Università di Calgary. Grazie a Matt Workentine per il suo sostegno di bioinformatica e Jens Durruthy per il suo supporto tecnico. Quest'opera è stata finanziata da una sovvenzione CIHR (R.M. e J.B.), un CIHR nuovo Investigator Award a J.B. e Health Research Institute Fellowship (J.S. bambini all'Alberta).
Materiali
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
Riferimenti
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati