
Method Article
Sequenziamento di nuova generazione di RNA e una pipeline di bioinformatica per identificare i LINE-1S espressi a livello specifico del locus
In questo articolo
Riepilogo
Qui presentiamo un approccio bioinformatico e analisi per identificare l'espressione LINE-1 al livello specifico del locus.
Abstract
Gli elementi INterspersed lunghi-1 (LINEs/L1s) sono elementi ripetitivi che possono copiare e inserire casualmente nel genoma con conseguente instabilità genomica e mutagenesi. Comprendere i modelli di espressione di L1 loci a livello individuale presterà alla comprensione della biologia di questo elemento mutageno. Questo elemento autonomo costituisce una porzione significativa del genoma umano con oltre 500.000 copie, anche se il 99% è troncato e difettoso. Tuttavia, la loro abbondanza e il numero dominante di copie difettose rendono difficile identificare L1s espressi in modo autentico da sequenze correlate a L1 espresse come parte di altri geni. È anche difficile identificare quale specifico locus L1 è espresso a causa della natura ripetitiva degli elementi. Superando queste sfide, presentiamo un approccio bioinformatico RNA-seq per identificare l'espressione L1 a livello specifico del locus. In sintesi, raccogliamo RNA citoplasmatico, selezioniamo per le trascrizioni poliadenilate e utilizziamo analisi di RNA-seq specifiche per la mappatura univoca delle letture ai loci L1 nel genoma umano di riferimento. Curiamo visivamente ogni locus L1 con letture mappate univocamente per confermare la trascrizione dal proprio promotore e regolare la trascrizione mappata letture per tenere conto della mappabilità di ogni singolo locus L1. Questo approccio è stato applicato a una linea cellulare del tumore della prostata, DU145, per dimostrare la capacità di questo protocollo di rilevare l'espressione da un piccolo numero di elementi L1 a lunghezza intera.
Introduzione
I retrotrasposti sono elementi di DNA ripetitivi che possono "saltare" nel genoma in un meccanismo di copia e incolla tramite intermedi di RNA. Un sottoinsieme dei retrotrasponi è conosciuto come Long INterspersed Elements-1 (LINEs/L1s) e costituisce un sesto del genoma umano con oltre 500, 0000 copie1. Nonostante la loro abbondanza, la maggior parte di queste copie sono difettosi e troncati con solo una stima 80-120 elementi L1 pensato per essere attivo2. Un L1 a lunghezza intera è di circa 6 KB di lunghezza con 5' e 3' regioni non tradotte, un promotore interno e un promotore anti-Sense associato, due telai di lettura aperta non sovrapposti (ORFS) e un segnale e Polya Tail3,4,5 . Nell'uomo, L1s sono costituiti da sottofamiglie distinte dall'età evolutiva con le famiglie più anziane che hanno accumulato mutazioni di sequenze più uniche nel tempo rispetto alla sottofamiglia più giovane, L1HS6,7. L1s sono gli unici retrotrasponi umani autonomi e i loro ORF codificano una trascrittasi inversa, endonuclease e RNP con attività di legame e Chaperone di RNA, necessarie per ritrasporre e inserire nel genoma in un processo indicato come bersaglio-innescato trascrizione inversa8, 9,10,11,12.
Il retrotrasposizione di L1s è stato segnalato per causare malattie della linea germinali umane da una varietà di meccanismi tra cui mutagenesi inserzionale, eliminazioni del sito bersaglio e riarrangiamenti13,14,15, 16. recentemente è stato ipotizzato che L1s possa svolgere un ruolo nell'oncogenesi e/o nella progressione tumorale poiché sono stati osservati maggiori eventi di espressione e inserimento di questo elemento mutageno in una varietà di tumori epiteliali17,18 . Si stima che vi sia un nuovo inserimento L1 in ogni 200 nascite19. Pertanto, è imperativo comprendere meglio la biologia dell'espressione attiva di L1s. La natura ripetitiva e l'abbondanza di copie difettose trovate all'interno di trascrizioni di altri geni hanno reso questo livello di analisi impegnativo.
Fortunatamente, con l'avvento delle tecnologie di sequenziamento ad alta velocità, sono stati compiuti passi avanti per analizzare e identificare autenticamente esprimendo L1s a livello di locus-specific. Ci sono diverse filosofie su come identificare al meglio espresso L1s utilizzando sequenziamento di nuova generazione di RNA. Ci sono stati solo due approcci ragionevoli suggeriti per la mappatura delle trascrizioni L1 al livello specifico del locus. Si concentra solo sulla potenziale trascrizione che legge attraverso il segnale di poliadenilazione L1 e nelle sequenze di fianchatura20. Il nostro approccio sfrutta le piccole differenze di sequenza tra gli elementi L1 e mappa solo quei RNA-seq legge che mappano in modo univoco a un locus21. Entrambi questi metodi hanno limitazioni in termini di quantificazione dei livelli di trascrizione. La quantificazione può essere migliorata potenzialmente aggiungendo una correzione per la "mappabilità univoca" di ogni locus21L1, o utilizzando algoritmi più complessi che ridistribuiscono le letture multi-mappate che non possono essere mappati in modo univoco a un locus22specifico. Qui, ci sarà dettaglio in modo graduale l'estrazione dell'RNA e il protocollo di sequenziamento e bioinformatica di nuova generazione per identificare gli elementi L1 espressi a livello specifico del locus. Il nostro approccio sfrutta al massimo la nostra conoscenza della biologia degli elementi funzionali L1. Ciò include sapere che gli elementi funzionali L1 devono essere generati dal promotore L1, iniziato all'inizio dell'elemento L1, devono essere tradotti nel citoplasma e che le loro trascrizioni devono essere co-lineari con il genoma. In breve, raccogliamo RNA citoplasmatico fresco, selezioniamo per le trascrizioni poliadenilate e utilizziamo analisi di RNA-seq specifiche per la mappatura univoca delle letture ai loci L1 nel genoma umano di riferimento. Queste letture allineate richiedono comunque una curation manuale estesa per determinare se le letture di trascrizione provengono dal promotore L1 prima di designare un locus come un L1 espresso in modo autentico. Applichiamo questo approccio sul campione di linea di cellule tumorali della prostata DU145 per dimostrare come identifica un relativamente pochi membri L1 trascritto attivamente dalla massa di copie inattive.
Protocollo
1. estrazione di RNA citoplasmatico
- Ottenere le celle tramite i seguenti metodi.
- Raccogli le cellule vive dal 2,75% – 100% confluente, T-75 fiaschi.
- Lavare il pallone 2 volte in 5 mL di PBS freddo, e nell'ultimo lavaggio rasare le cellule e trasferirla in un tubo conico da 15 mL. Centrifugare per 2 min a 1.000 x g e 4 ° c e rimuovere e scartare con cautela il supernatante (tabella dei materiali).
- Raccogli le cellule dai campioni di tessuto.
- Preparare il tessuto per l'estrazione del RNA citoplasmatico entro un'ora dall'essere dissezionato e mantenere sempre il ghiaccio. Per lo stoccaggio a lungo termine, utilizzare soluzioni di inibitori di RNA per immagazzinare i tessuti fino a 72 ore dopo la dissezione seguendo il protocollo del produttore (tabella dei materiali).
- Dadi a 10 μm3 campione e omogeneizzare il campione fresco con 5 ml di PBS freddo in un omogeneizzatore a Dounce sterile, trasferire ad un 15 ml tubo conico, centrifugare per 2 min a 1.000 x g a 4 ° c, e rimuovere con cautela e scartare supernatante (tabella dei materiali < /C8 >).
- Raccogli le cellule vive dal 2,75% – 100% confluente, T-75 fiaschi.
- Aggiungere 2 mL di tampone di lisi al pellet cellulare-mescolare e incubare su ghiaccio per 5 min.
- Preparare tampone di lisi fresca con 150 mM di NaCl, 50 mM HEPES (pH 7,4) e 25 μg/mL di digitonina (tabella dei materiali).
- Poiché la concentrazione minima di digitonina nella tampone di lisi necessaria per penetrare la membrana plasmatica può variare in base al tipo di cellula, il microscopicamente conferma che le cellule trattate con tampone di lisi perdono la membrana plasmatica e mantengono la membrana nucleare intatta.
- Appena prima dell'uso, aggiungere 1.000 inibitore della RNAsi U/mL (tabella dei materiali).
- Centrifugare per 1 min a 1.000 x g e 4 ° c e raccogliere il surnatante.
- Aggiungere surnatante a 7,5 ml pre-refrigerati di Trizol e 1,5 ml di cloroformio. Tutti i passaggi che richiedono il cloroformio devono essere eseguiti all'interno di un cappuccio chimico pulito (tabella dei materiali).
- Centrifugare per 35 min a 3.220 x g e 4 ° c.
- Trasferire la porzione acquosa (strato superiore) in un nuovo tubo pre-refrigerato da 15 mL.
- Aggiungere 4,5 mL di cloroformio e Vortex.
- Centrifugare per 10 min a 3.220 x g e 4 ° c.
- Trasferire la porzione acquosa al tubo pre-refrigerato fresco.
- Aggiungere 4,5 mL di isopropanolo, agitare bene e incubare a-80 ° c durante la notte (tabella dei materiali).
- Centrifugare a 3.220 x g e 4 ° c per 45 minuti.
- Rimuovere isopropanolo, aggiungere 15 mL di 100% etanolo (tabella dei materiali).
- Centrifugare a 3.220 x g per 10 min.
- Rimuovere l'etanolo, drenare e asciugare per circa 1 h.
- Utilizzare un batuffolo di cotone sterile per cancellare qualsiasi etanolo rimanente (tabella dei materiali).
- Risospendere il campione in 100 a 200 μL di acqua libera da RNasi a seconda della dimensione del pellet (tabella dei materiali).
- Frazionamento dei campioni utilizzando la tecnologia di elettroforesi per determinare la qualità e la concentrazione dei campioni secondo le istruzioni del produttore23 (tabella dei materiali).
- I campioni si qualificano per l'analisi RNA-seq se RIN > 824.
2. sequenziamento di nuova generazione
- Inviare campioni di RNA citoplasmatico per essere sequenziati utilizzando la piattaforma di sequenziamento di nuova generazione finalizzata alla generazione di almeno 50 milioni accoppiato-end 100 BP letture.
- Selezionare per RNA poli-adenilati e sequenziamento specifico del filamento.
3. creare annotazioni (facoltativo se si dispone di un'annotazione esistente)
- Creare un'annotazione L1 a lunghezza intera o scaricare l'annotazione L1 a lunghezza intera (file supplementare 1a-b).
- Scarica le annotazioni di Repeat Masker per gli elementi LINE-1 dal browser del genoma UCSC con lo strumento browser della tabella (https://genome.ucsc.edu/cgi-bin/hgTables). Specificare il clade mammifero, il genoma umano, l'assembly hg19 (o hg38 per un genoma più aggiornato) e filtrare per "linea 1" in nome classe. Scaricare come file. GTF ed etichettare come FL-L1-BLAST. GTF.
- Eseguire una ricerca BLAST locale del primo 300 BP dell'elemento L1 a lunghezza intera L 1.3 che comprende la regione del promotore nel genoma umano e aggiungere 6.000 BP a valle per creare una fine delle coordinate L1 al file di annotazione. Salvare in un file GTF ed etichettare come FL-L1-RM. GTF.
- Intersecare l'annotazione RepeatMasker e l'annotazione L1 basata su promotore utilizzando bedtools ed etichettare come FL-L1-BLAST_RM. txt (pacchetti software).
- Utilizzare questo comando nel terminale Linux: bedtools intersecano-un FL-L1-Blast. GTF-b FL-L1-RM. gtf > FL-L1-BLAST_RM. txt.
- Separare l'annotazione FL-L1 intersecata dal filo superiore e inferiore.
- Copia il FL-L1-BLAST_RM. txt nel software di foglio di calcolo e Ordina per il "meno" e "più" filo e quindi ordinare per posizione cromosoma.
- Creare due nuovi documenti foglio di calcolo, uno con le coordinate intersecate per l'intera lunghezza L1s sul filo meno e uno sul filo inferiore, e salvare come FL-L1-BLAST_RM_minus. xls e FL-L1-BLAST_RM_plus. xls.
- Salvare i due nuovi documenti come file. txt.
- Utilizzare il programma mac2unix per convertire i file. txt nei file di annotazione corretti (pacchetti software).
- Utilizzare questo comando nel terminale: Mac2unix.sh FL-L1-BLAST_RM_minus. GFF.
- Utilizzare questo comando nel terminale: Mac2unix.sh FL-L1-BLAST_RM_plus. GFF.
- Salvare i nuovi file con l'estensione. GFF.
- In alternativa, usare AWK per filtrare le righe associate al filo + e –.
- Utilizzare il comando seguente per ottenere il + Strand: awk '/+/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_plus. GTF.
- Utilizzare la seguente riga di comando per ottenere il-Strand: awk '/-/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_minus. GTF.
4. leggere la pipeline di allineamento per identificare L1s espressa
| Opzione | descrizione |
| – p | In questo dettaglio il numero di thread che il computer deve utilizzare l'esecuzione del tracciato. La memoria del computer più grande consentirà più thread e dovrebbe essere empiricamente d. |
| – m 1 | Questo dice al programma di accettare solo letture che hanno una corrispondenza nel genoma che è meglio di qualsiasi altra corrispondenza del genoma. |
| – y | Questo è l'interruttore rimarranno che rende la ricerca di mappatura per tutte le possibili corrispondenze e non permette di smettere dopo un numero fisso di corrispondenze viene raggiunta. |
| – v 3 | Questo consente solo al programma di utilizzare la memoria per le letture mappate con 3 o meno disallineamenti al genoma. |
| – X 600 | Questo consente solo di letture accoppiate che la mappa all'interno 600 basi di un altro. Questo assicura che le coppie di lettura siano co-lineari nel genoma e selezioni contro s che coinvolgono molecole di RNA elaborate. |
| – chunkmbs 8184 | Questo comando assegna memoria aggiuntiva per la gestione della grande quantità di allineamenti possibili per ogni lettura correlata a L1. |
Tabella 1: opzioni della riga di comando per bowtie.
- Eseguire l'allineamento dei file FASTQ di sequenziamento a fine accoppiamento con il campione di RNA-seq di interesse utilizzando bowtie.
Nota: Bowtie1 deve essere utilizzato e non Bowtie2 perché i parametri necessari per l'allineamento univoco sono specificamente presenti solo in questa versione di Bowtie (pacchetti software). Bowtie è utilizzato su allineatori consapevoli di Splice come STAR per valutare concordanti, letture contigue più rilevanti per la biologia L1 e l'espressione.- Utilizzare questa riga di comando nel terminale Linux: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_sample_1. FQ-2 hg_sample_2. FQ | samtools visualizzare-hbuS-| samtools ordinamento – hg_sample_sorted. BAM. Vedere la tabella 1 per una descrizione delle opzioni della riga di comando per bowtie.
- Strand separare il file BAM di output utilizzando samtools (pacchetti software) e i seguenti comandi Linux. Si noti che i valori di flag effettivi possono variare se non si utilizzano protocolli di sequenziamento di nuova generazione standard.
- Utilizzare questa riga di comando per selezionare il filo superiore: samtools Visualizza-h hg_sample_sorted. BAM | awk ' substr ($ 0, 1, 1) = = "@" | | $2 = = 83 | | $2 = = 163 {stampa}' | samtools Visualizza-BS-> hg_sample_sorted_topstrand. BAM.
- Utilizzare questa riga di comando per selezionare il filo inferiore: samtools Visualizza-h hg_sample_sorted. BAM | awk ' substr ($ 0, 1, 1) = = "@" | | $2 = = 99 | | $2 = = 147 {Print}' | samtools Visualizza-BS-> hg_sample_sorted_bottomstrand. BAM.
- Generare conteggi di lettura contro le annotazioni per i loci L1 utilizzando bedtools (pacchetti software).
- Utilizzare questa riga di comando per generare conteggi di lettura per L1s nella direzione di senso sul filo superiore: copertura bedtools-ABAM FL-L1-BLAST_RM_plus. GFF-b hg_sample_sorted_topstrand. bam > hg_sample_sorted_bowtie_tryhard_plus_top. txt.
- Utilizzare questa riga di comando per generare conteggi di lettura per L1s nella direzione di senso sul filo inferiore: copertura bedtools-ABAM FL-L1-BLAST_RM_minus. GFF-b hg_sample_sorted_bottomstrand. bam > hg_sample_sorted_bowtie_tryhard_minus_bottom. txt.
- File di indice BAM dal passaggio 5.1.1 per renderlo visualizzabile in Integrative Genomics Viewer (IGV)25 (pacchetti software).
- Utilizzare questa riga di comando: samtools index hg_sample_sorted. BAM
- Per utilizzare una modalità batch per aumentare il numero di campioni di RNA-seq convogliati alla volta, utilizzare uno script supercomputer per completare il passaggio 4,1 denominato human_bowtie. sh, uno script per completare i passaggi 4.2-4.3 è stato creato chiamato human_L1_pipeline. sh, e uno script per completare il passo 4,4 è stato creato chiamato bam_index. sh. Questi script possono essere trovati nel file supplementare 2 con i comandi supercomputer associati per eseguire gli script.
5. curation manuale
- Creare un foglio di calcolo per le letture mappate a ogni locus L1 annotata.
- Copia su hg_sample_sorted_bowtie_tryhard_minus_bottom. txt creato nel passaggio 4.3.2 e pagina etichetta come "meno-basso."
- Ordina tutte le colonne in base al numero di letture più alto al più basso trovato nella colonna J.
- Copia su hg_sample_sorted_bowtie_tryhard_plus_top. txt creato nel passaggio 4.3.1 ed etichetta come "Top-Plus" in un altro foglio di calcolo.
- Ordina tutte le colonne in base al numero di letture più alto al più basso trovato nella colonna J.
- Creare una terza pagina etichettata come "combinata" e aggiungere tutti i loci con dieci o più letture dalle pagine "meno-basso" e "più-alto".
- Ordina tutte le colonne in base al numero di letture più alto al più basso trovato nella colonna J.
- Caricare i seguenti file in IGV25 (pacchetti software): 1) genoma di riferimento di interesse per visualizzare i geni annotati, 2) FL-L1-BLAST_RM. GFF per visualizzare l'annotazione L1, 3) hg_sample_sorted. BAM per visualizzare le trascrizioni mappate da campione di interesse, e 4) hg_genomicDNA_sorted. BAM per valutare la mappabilità delle regioni genomiche.
- Rimuovere la copertura e le righe di giunzione associate a ciascun file BAM.
- Comprimere hg_sample_sorted. BAM e hg_genomicDNA_sorted. BAM in modo che tutte le tracce IGV siano adatte a una sola schermata.
- Copia su hg_sample_sorted_bowtie_tryhard_minus_bottom. txt creato nel passaggio 4.3.2 e pagina etichetta come "meno-basso."
- Curare manualmente.
- Utilizzando le coordinate da loci elencate nella pagina "combinata" del foglio di calcolo, visualizzare denominata loci in IGV25 (pacchetti software).
- Curare un locus per essere autenticamente espresso fuori proprio se non ci sono letture a Monte nella direzione L1 fino a 5 KB.
- Etichettare la riga di colore verde e notare perché è un L1 espresso in modo autentico.
Nota: un'eccezione a questa regola esiste se la regione a Monte del L1 non è mappabile. Se questo è il caso, etichettare la riga di colore rosso e notare che l'espressione della regione a Monte del promotore L1 non può essere valutata e quindi l'espressione L1's non è in grado di essere determinata con fiducia.
- Etichettare la riga di colore verde e notare perché è un L1 espresso in modo autentico.
- Curare un locus per non essere autenticamente espresso fuori il proprio promotore se ci sono letture upstream fino a 5 KB.
- Etichettare la riga di colore rosso e notare perché non è un L1 autenticamente espresso.
- Curare un locus come falso se viene espresso all'interno di un introne di un gene espresso nella stessa direzione con letture a Monte del L1, se è a valle di un gene espresso nella stessa direzione con letture a Monte del L1, o per modelli di espressione non annotati con re annunci a Monte del L1.
Nota: un'eccezione a questa regola si applica quando sono presenti letture minime che si sovrappongono direttamente al sito iniziale del promotore L1, ma leggermente a monte dell'L1. Se non ci sono altre letture a Monte di un caso L1 come questo, considerare questo L1 per essere autenticamente espressa. Etichettare il colore verde riga e notare perché è un L1 espresso autenticamente.
- Curare un locus L1 come probabilità di essere false se il modello di letture mappate al locus non è correlato con le specifiche aree L1's di mappabilità.
Nota: ad esempio, se un L1 è altamente mappabile ma ha solo una pila di letture in una regione condensata all'interno del L1, è meno probabile che sia correlato all'espressione L1 fuori dal proprio promotore e più probabilmente da fonti non annotate come esoni o ltr. In casi come questo, curare il loci come arancione e notare perché il locus è sospetto. Verificare le fonti di pile-up sospetti controllando la posizione L1 nel browser del genoma UCSC. - Curare un locus per non essere autenticamente espresso se si trova all'interno di un ambiente genomico di regioni non annotate sporadicamente espresse
Nota: ad esempio, le letture possono essere espresse 10 KB a Monte del L1, ma ogni 10 KB o giù di lì ci sono letture mappate e alcune di queste letture si allineano con il L1. Questi L1s hanno meno probabilità di essere espressi da un proprio promotore e più probabilmente hanno mappato le letture a causa di modelli non annotati di espressione genomica. In casi come questo, curare il loci come arancione e notare perché il locus è sospetto.
6. leggi la strategia di allineamento per valutare la mappabilità nel genoma di riferimento (facoltativo se si dispone di un DataSet di DNA genomico allineato esistente)
- Scarica interi file di sequenze di DNA del genoma e Converti in file. FQ
- Vai al sito NCBI trovato qui: https://www.ncbi.nlm.nih.gov/sra
- Digitare l' estremità accoppiata di WGS HeLa.
- Selezionare per Homo sapiens sotto risultati di Taxon.
- Selezionare un campione che è accoppiato fine e ha letture con 100 o più BP come il seguente esempio: https://www.ncbi.nlm.nih.gov/sra/ERX457838 [ACCN]
- Confermare la lunghezza di lettura selezionando Esegui e quindi i metadati come mostrato qui: https://Trace.ncbi.nlm.nih.gov/TRACES/SRA/?Run=ERR492384
- Per scaricare l'intero genoma DNA sequenza dati, inserire questo comando nel terminale Linux: sratoolkit. 2.9.2-mac64/bin/prefetch-X 100g ERR492384
Nota: la funzione di prefetch del Toolkit SRA Scarica il numero di adesione "ERR492384" che si trova nel sito NCBI (pacchetti software). Il "100G" limita la quantità di dati scaricati a 100 gigabyte. - Inserire questo comando nel terminale Linux: fastq-dump--Split-files ERR492384
Nota: questo divide il DataSet di DNA genomico scaricato in due file FASTQ.
- Eseguire l'allineamento con bowtie.
- Utilizzare questo comando in Linux per l'allineamento: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | visualizzazione samtools-hbuS-| ordinamento samtools – hg_genomicDNA_sorted. BAM.
- Fare riferimento al passaggio 4,1 per comprendere i parametri utilizzati nell'allineamento bowtie (pacchetti software).
- Scaricare il file BAM con allineamento genomico per valutare la mappabilità disponibile in base alla richiesta dell'autore.
- Utilizzare questo comando in Linux per l'allineamento: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | visualizzazione samtools-hbuS-| ordinamento samtools – hg_genomicDNA_sorted. BAM.
- File di indice BAM dal passaggio 4.2.1 utilizzando samtools per renderlo visualizzabile in IGV25 (pacchetti software) per informare ulteriormente la curation manuale.
- Utilizzare questa riga di comando in Linux: Indice samtools hg_genomicDNA_sorted. BAM
- Valutare la mappabilità di ogni loci L1
- Determinare il numero di letture mappate univocamente su L1 loci utilizzando il programma bedtools, l'annotazione FL-L1 e i dati della sequenza genomica allineata (pacchetti software).
- Utilizzare questa riga di comando in Linux: copertura bedtools-ABAM FL-L1-BLAST_RM. GTF – b hg_genomicDNA_sorted. bam ≫ L1_Mappability_hg_genomicDNA. txt.
- Designare un locus L1 per avere la mappabilità di copertura completa quando 400 letture univoche sono allineate ad esso.
- Determinare il fattore necessario per aumentare o abbassare il DNA genomico allineato legge a 400 per ogni singolo L1.
- Per avere una misura di espressione in scala in base alla mappabilità del locus L1 individuale, moltiplicare il fattore determinato nel passaggio 6.4.3 al numero di letture di trascrizione dell'RNA che si allineano a L1s espressi in modo autentico determinati nelle sezioni 4 – 5.
- Determinare il numero di letture mappate univocamente su L1 loci utilizzando il programma bedtools, l'annotazione FL-L1 e i dati della sequenza genomica allineata (pacchetti software).
Risultati
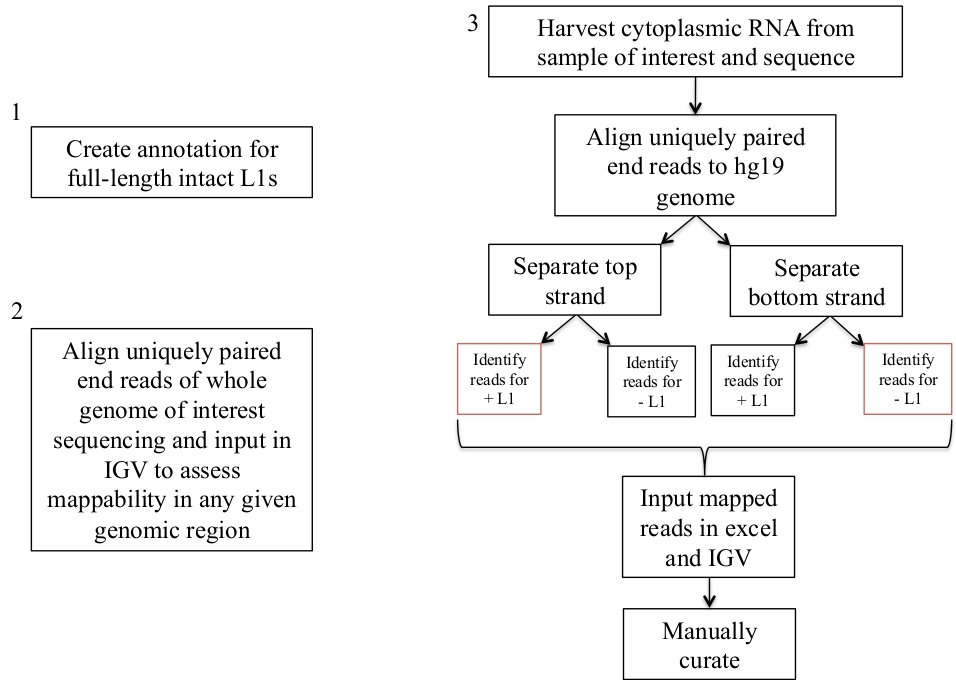
I passaggi descritti sopra e descritti graficamente in Figura 1 sono stati applicati a una linea di cellule tumorali della prostata umana DU145. Il campione di RNA è stato preparato in modo citoplasmatico ed è stato sequenziato di nuova generazione in un protocollo di estremità accoppiata, specifico di un filamento, di Poly-A. Utilizzando bowtie, i file di sequenziamento con estremità accoppiate sono stati allineati permettendo solo corrispondenze univoche in cui la lettura di estremità accoppiata corrisponde meglio a una posizione genomica rispetto a qualsiasi altra posizione genomica. I file di sequenza DU145 sono stati allineati al genoma umano di riferimento creando un file BAM, che è disponibile su richiesta dell'autore. Utilizzando bedtools, i dati sono stati estratti dai file BAM separati da Strand DU145 sul numero di letture mappate a tutta la lunghezza L1s. Tali letture sono state ordinate in un foglio di calcolo dal più grande al più piccolo e curata manualmente esaminando l'ambiente genomico intorno a ogni locus L1 in IGV per confermarne l'autenticità (tabella supplementare 1). Se un campione è stato curato per essere autenticamente espresso, è stato colorato di colore verde con una spiegazione per la sua accettazione nella colonna più a destra. Esempi di loci L1 accettati per essere autenticamente espressi seguendo le linee guida descritte nella sezione metodi sono mostrati in Figura 2a-b. Se un campione è stato rifiutato per essere autenticamente espresso, è stato codificato a colori come rosso con il motivo del rifiuto nella colonna più a destra. Esempi di L1 loci rifiutato a causa di espressione da un promotore diverso dalle proprie seguenti linee guida descritte nella sezione metodi sono dettagliati nella Figura 2c-e.
Qui, sono stati studiati solo L1s a lunghezza intera con una regione promotrice intatta. Se questa distinzione non viene fatta, viene introdotta una grande fonte di rumore trascrizionale proveniente da L1s troncato. Esempi di L1s troncati in DU145 sono illustrati nella Figura 3a-b dove sono stati identificati come hanno MAPPATO in modo univoco RNA-seq letture. In IGV, tuttavia, è evidente che tali trascrizioni non sono state avviate dall'L1 troncato, ma dall'inclusione della sequenza L1 in un gene o a valle di un gene espresso.
Nel complesso, in DU145, la percentuale di loci L1 a lunghezza intera e le letture che vengono rifiutate autenticamente espresse L1s dopo la curation manuale è di circa 50% (tabella supplementare 2) che dimostra l'elevato livello di letture di trascrizione mappata L1 che sarebbe altrimenti essere registrati come falsi positivi senza curation manuale. In particolare, in DU145 c'erano 114 totali di lunghezza totale L1 loci di avere mappate univocamente letture nella direzione del senso con un totale di 3.152 letture, ma c'erano solo 60 loci identificati per essere espressi fuori il proprio promotore dopo la curazione manuale con 1.879 letture ( Tabella supplementare 1). Questo è il caso anche quando sono state adottate misure per ridurre l'espressione irrilevante per la biologia L1 selezionando per mRNA citoplasmatico. Si noti che il locus con il più alto livello di trascrizioni mappate in DU145 è stato rifiutato perché non era un L1 espresso autenticamente (Figura 4). Complessivamente il numero di trascrizioni mappate a specifici loci L1 varia Analogamente tra i loci L1 accettati e rifiutati come autenticamente espressi dopo la curazione manuale (Figura 4).
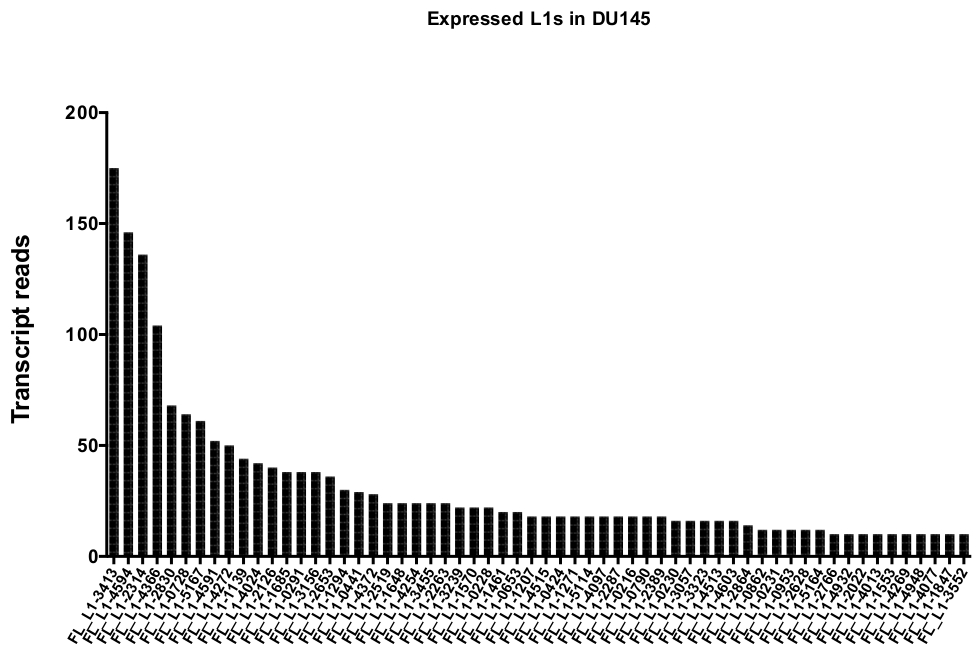
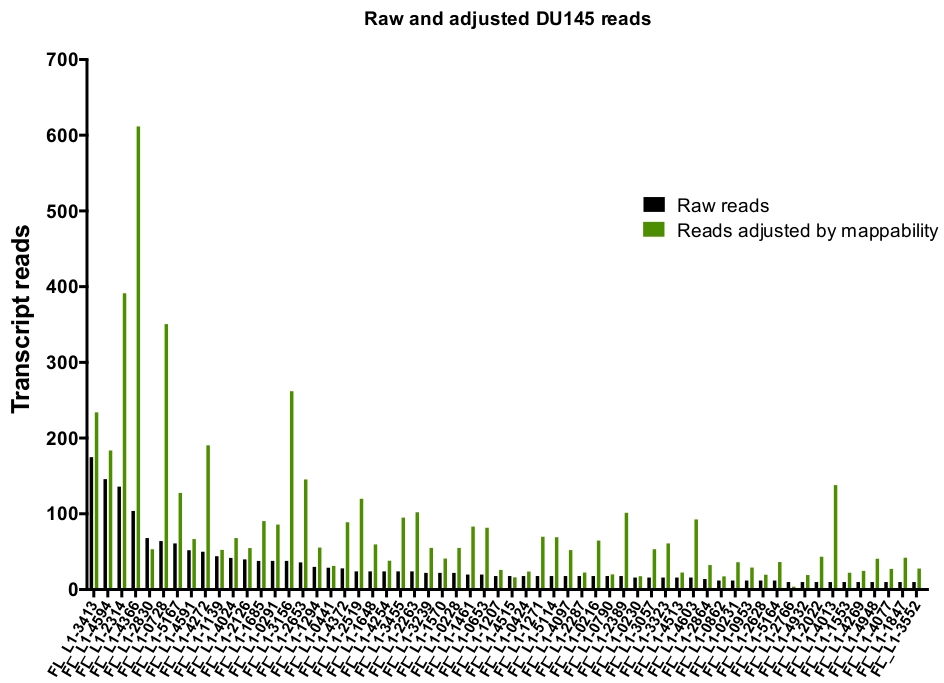
Dopo la curation manuale, il numero di letture che la mappa in modo univoco per espresso autenticamente specifico loci L1 in DU145 gamma da 175 letture a un arbitrariamente scelto taglio minimo di 10 letture (Figura 5). Questo approccio di identificazione della trascrizione mappata in modo univoco a L1s limita la capacità di quantificare accuratamente l'espressione. Per tenere conto di questo, è stato creato un fattore di correzione per ogni locus in base alla sua mappabilità. Per creare questo fattore di correzione, è stato utilizzato il primo bedtools per estrarre il numero di letture mappate univocamente dal file BAM genomico HeLa che è allineato a tutti i loci L1 a lunghezza intera e i grafici di tali loci da letture di trascrizione mappata più alte a quelle più basse (supplemento Figura 1). È stato arbitrariamente designato che L1s con 400 letture aveva piena mappability copertura. Il numero di letture in grado di mappare a un locus L1 in HeLa campione di sequenziamento genomico è stato ridimensionato rispetto a 400 letture e che il numero in scala è stato quindi moltiplicato per il numero di letture che mappato a ogni loci L1 autenticamente espressi in DU145 (tabella supplementare 2) . Come previsto, gli elementi L1 che avevano punteggi di correzione più grandi per la mappabilità provenivano da sottofamiglie più giovani come L1PA2 (tabella complementare 2). Una volta che le letture sono state regolate per i punteggi di mappabilità in ogni locus, la quantificazione per l'espressione per la maggior parte dei loci è aumentata (Figura 6). Il numero di letture mappate univocamente a loci L1 specifici espressi in modo autentico con correzioni di mappabilità in DU145 variava da 612 a 4 letture e c'era un riordino di più alto al più basso esprimendo loci (Figura 6).

Figura 1: schema del flusso di lavoro.
Graficamente descritti sono i passi per identificare L1s espressi in un campione umano. Si noti che i passaggi 1 e 2 non devono essere ripetuti se i file appropriati sono già disponibili. Questi file appropriati possono essere scaricati da supplemento file 1a-b e supplemento file 2. Le caselle in rosso indicano i passaggi in cui il programma di copertura bedtools viene utilizzato per contare il numero di letture di mappatura a L1s nella stessa direzione di senso. Questi loci con il senso orientato mappatura letture sono il L1s che dovrebbe essere curata manualmente. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 2: esempi di loci L1 curati in DU145.
Caricati in IGV sono il genoma di riferimento, il file di annotazione L1 GFF a lunghezza intera corrispondente alla versione del genoma di riferimento (supplemento file 1), il file BAM DU145, e infine il file genomico Hela BAM per valutare la mappabilità, che sono tutti disponibili su autore richiesta. Sono state aggiunte frecce per aiutare nella visualizzazione della direzione dell'L1 annotata. Le frecce e le letture in rosso sono orientate in sequenza da destra a sinistra. Le frecce e le letture in blu sono orientate in sequenza da sinistra a destra. a) in IGV, questo locus L1 sembra essere espresso dal proprio promotore in quanto non ci sono letture a Monte del L1 nell'orientamento di senso per oltre 5 KB. Questo L1 ha bassa mappabilità, non è in un gene, e ha la prova di attesa attività promotore antisenso26. b) in IGV, questo locus L1 sembra essere espresso dal proprio promotore in quanto non ci sono letture a Monte del L1 nell'orientamento del senso per oltre 5 KB. Questo L1 ha una bassa mappabilità ed è all'interno di un gene di direzione opposta. c) in IGV, questo locus L1 è stato respinto come L1 espresso in quanto vi sono letture a Monte nello stesso orientamento entro 5 KB. Questo L1 è all'interno di un gene della stessa direzione, quindi le letture di trascrizione sono molto probabilmente originarie del promotore del gene espresso. d) in IGV, questo locus L1 è stato rifiutato come L1 espresso in quanto vi sono letture a Monte nello stesso orientamento entro 5 KB. Questo L1 è a valle di un gene altamente espresso nella stessa direzione, quindi le letture di trascrizione sono probabilmente originarie del promotore di quel gene espresso e si estendono oltre il normale terminatore genico. e) in IGV, questo locus L1 è stato respinto come L1 espresso in quanto vi sono letture a Monte nello stesso orientamento entro 5 KB. Questo L1 non è all'interno o vicino a un gene annotato nel gene di riferimento in modo che l'origine di queste trascrizioni all'interno e a monte dell'elemento L1 suggerisca un promotore non annotato. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 3: il rumore di fondo proviene da L1s troncato pure.
La nostra annotazione L1 non include troncati L1s in quanto sono una fonte principale di rumore di sottofondo. Sono state aggiunte frecce per aiutare nella visualizzazione della direzione dell'L1 annotata. Le frecce e le letture in blu sono orientate in sequenza da sinistra a destra. a) dimostrato è un esempio di un tronco L1 nella SUFAMILY L1MB5 che è 2706 bps. In IGV è evidente che le letture provengono dall'estensione a valle di un gene espresso. b) mostrato è un altro esempio di un L1 troncato. Questo L1 è un L1PA11 che è 4767 BPS lungo. In IGV è evidente che la mappatura delle letture unicamente all'L1 proviene dall'Exon espresso, che è all'interno dell'L1. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 4: trascrizione legge che la mappa in modo univoco a tutti L1s intatto intero nel genoma umano espressa in DU145 linea cellulare del tumore prostatico.
In nero sono i loci specifici da identificare come autenticamente espressi dopo la curazione manuale e in rosso sono i loci specifici da respingere come letture autenticamente espresse dopo la curazione manuale. In grigio sono loci con meno di dieci letture di mappatura a ciascuno. Poiché questi loci rappresentano una piccola frazione di letture di trascrizione, non sono stati curato manualmente. I segni di graduazione dell'asse x indicano ogni 100 a lunghezza intera, intatto L1s. circa 4.500 loci non sono graficamente mostrati in quanto avevano zero letture mappate. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 5: trascrizione legge che la mappa in modo univoco per autenticamente espressa piena lunghezza intatta L1s in DU145 linea cellulare del tumore prostatico.
Mostrato sono i numeri di trascrizione legge che mappa a loci specifici in DU145 celle dopo la curazione manuale. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 6: legge mappatura per espresso autenticamente L1 quando regolato da mappabilità.
Sono mostrati i numeri delle letture di trascrizione regolate dai punteggi di mappabilità specifici dei loci che mappano i loci L1 a cura manuale nelle celle DU145. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}
File supplementare 1: annotazioni per il L1s umano intatto a lunghezza intera secondo l'orientamento. a) FL-L1-BLAST_RM_minus. GFF. b) FL-L1-BLAST_RM_plus. GFF. Per favore clicca qui per scaricare questo file.
File supplementare 2: script supercomputer utilizzati per automatizzare la pipeline bioinformatica descritta nella sezione 4. Per favore clicca qui per scaricare questo file.
Figura 1 supplementare: campione di DNA genomico utilizzato per determinare la mappabilità L1.
Mostrato sono il numero di trascrizione genomica legge dal campione di linea cellulare HeLa che mappa in modo univoco a tutti i 5.000 loci L1 a lunghezza intera nel genoma. È stato designato che un L1 ha piena mappabilità di copertura quando 400 legge mappa per il L1. Per favore clicca qui per scaricare questa cifra.
Tabella supplementare 1: curation manuale di L1s in DU145. Per favore clicca qui per scaricare questa tabella.
Tabella complementare 2: L1s curata in DU145 con regolazione della mappabilità. Per favore clicca qui per scaricare questa tabella.
Discussione
L'attività L1 ha dimostrato di causare danni genetici e instabilità contribuendo alla malattia27,28,29. Delle circa 5.000 copie L1 a lunghezza intera, solo poche dozzine di giovani evoluzionariamente L1s conto per la maggior parte dell'attività di retrotrasposizione2. Tuttavia, ci sono prove che anche alcuni vecchi, retrotraspositivamente-incompentent L1s sono ancora in grado di produrre DNA dannoso proteine30. Per apprezzare appieno il ruolo di L1s nell'instabilità genomica e nella malattia, deve essere compresa l'espressione L1 al livello specifico del locus. Tuttavia, l'elevato background delle sequenze correlate a L1 incorporate in altri RNA estranei alla retrotrasposizione L1 rappresenta una sfida significativa nell'interpretare l'autentica espressione L1. Un'altra sfida nell'identificare e quindi comprendere i modelli di espressione dei singoli loci L1 si verifica a causa della loro natura ripetitiva che non consente a molte sequenze di lettura brevi di mappare a un unico locus unico. Per superare queste sfide, abbiamo sviluppato l'approccio sopra descritto per identificare l'espressione dei singoli loci L1 utilizzando i dati di RNA-seq.
Il nostro approccio filtra l'alto livello (oltre il 99%) di rumore trascrizionale generato da sequenze L1 che non sono correlate alla retrotrasposizione L1 adottando una serie di passaggi. Il primo passo riguarda la preparazione dell'RNA citoplasmatico. Selezionando per RNA citoplasmatico, le letture correlate a L1 trovate all'interno dell'mRNA INTRONICO espresso nel nucleo sono significativamente esaurite. Nella preparazione della libreria di sequenziamento, un altro passo compiuto per ridurre il rumore trascrizionale non correlato a L1s include la selezione di trascrizioni poliadenilate. Questo rimuove il rumore di trascrizione correlato a L1 trovato in specie non-mRNA. Un altro passo include il sequenziamento specifico del filamento al fine di identificare ed eliminare le trascrizioni antisenso L1 correlate. L'uso di un'annotazione per L1s a lunghezza intera con aree promotrice funzionali quando si identifica il numero di trascrizioni RNA-seq che mappano a L1s Elimina anche il rumore di fondo che altrimenti provengono da L1s troncati. Infine, l'ultimo passo critico nell'eliminazione del rumore trascrizionale delle sequenze L1 non correlate alla retrotrasposizione L1 è la curazione manuale di L1s a lunghezza intera identificata per aver mappato le trascrizioni RNA-seq. La curazione manuale comporta la visualizzazione di ogni locus L1 bioinformaticamente identificato-to-be-espresso nel contesto del suo ambiente genomico circostante per confermare che l'espressione proviene dal promotore L1. Questo approccio è stato applicato a DU145, una linea cellulare del tumore della prostata. Anche con tutte le misure relative alla preparazione adottate per ridurre il rumore di fondo, circa il 50% dei loci L1 identificati bioinformaticamente nel DU145 sono stati rifiutati come rumore di fondo L1 proveniente da altre fonti trascrizionali (Figura 4), enfatizzando il rigore necessario per produrre risultati affidabili. Questo approccio con la curazione manuale è laborioso, ma necessario nello sviluppo di questa pipeline per valutare e comprendere l'ambiente genomico che circonda un L1 a lunghezza intera. I passi successivi includono la riduzione della necessaria curation manuale automatizzando alcune delle regole di curation, anche se a causa della natura ancora non completamente conosciuta dell'espressione genomica, delle fonti di espressione non annotate nel genoma di riferimento, delle regioni di bassa mappabilità, e anche complicare i fattori coinvolti nella costruzione di un genoma di riferimento non è possibile automatizzare completamente la curazione L1 in questo momento.
La seconda sfida nell'identificare l'espressione dei singoli loci L1 con il sequenziamento si riferisce alla mappatura delle trascrizioni L1 ripetitive. In questa strategia di allineamento, è necessario che una trascrizione deve allinearsi in modo univoco e co-linearemente al genoma di riferimento per poter essere mappata. Selezionando per le sequenze di estremità accoppiate che mappano concordantemente, la quantità di trascrizioni che si allineano in modo univoco ai loci L1 trovati nel genoma di riferimento aumenta. Questa strategia di mappatura univoca fornisce fiducia nella chiamata di lettura mappatura specificamente a un singolo locus L1, anche se potenzialmente sottovaluta la quantità di espressione di ogni identificato da essere autenticamente espresso, ripetitivo L1. Per correggere approssimativamente questa sottovalutazione, un punteggio di "mappabilità" per ogni locus L1 basato sulla sua mappabilità è stato sviluppato e applicato al numero di letture di trascrizione univocamente mappate (Figura 6). È da notare che idealmente, la mappabilità dovrebbe essere segnato a piena copertura legge attraverso l'intero L1 in base al campione WGS abbinato. Qui, usiamo WGS delle cellule HeLa per determinare i punteggi di mappabilità di ogni loci L1 al fine di gonfiare o deviare letture di mappatura a L1 loci in DU145 linee cellulari del tumore della prostata. Questo calcolo della mappabilità è un punteggio di correzione grezzo, ma la "mappabilità di copertura completa" scelta di 400 letture è stata determinata con la natura dinamica delle linee cellulari tumorali in mente. Può essere osservato nella Figura 1 supplementare, che ci sono un paio di loci L1 con Hela WGS con un numero estremamente elevato di letture mappate. Questi probabilmente provengono da sequenze cromosomi duplicate all'interno di HeLa che non sono all'interno del genoma di riferimento, motivo per cui quei loci non sono stati scelti per essere rappresentativi della copertura di mappabilità completa. Invece è stato determinato che la media di 100% di copertura di lettura si verifica intorno 400 letture in base alla Figura 1 supplementare e poi è stato ipotizzato che questa media si applica alla linea di cellule tumorali di prostata DU145 pure.
Questa strategia di allineamento con 100-200 BP legge dalla tecnologia RNA-seq seleziona anche preferenzialmente per evoluzionariamente più vecchio L1s all'interno del genoma di riferimento come più vecchio L1s hanno accumulato nel tempo mutazioni uniche che li rendono più mappabile. Questo approccio, quindi, ha una sensibilità limitata quando si tratta di identificare il più giovane di L1s così come non di riferimento, polimorfica L1s. Per identificare il più giovane di L1s, suggeriamo di utilizzare 5' RACE selezione di trascrizioni L1 e la tecnologia di sequenziamento come PacBio che fanno uso di più letture21. Ciò consente una mappatura più univoca e quindi un'identificazione sicura dei giovani L1s espressi. l'uso di RNA-seq e PacBio insieme può portare a un elenco più completo di L1s autenticamente espressi. Per identificare l'L1s polimorfico autenticamente espresso, i primi passi successivi includono la costruzione e l'inserimento di sequenze polimorfiche nel genoma di riferimento.
Le sfide biologiche e tecniche nello studio delle sequenze ripetute sono grandi, anche se con la procedura sopra rigorosa per rimuovere il rumore trascrizionale delle sequenze L1 non legati alla retrotrasposizione utilizzando la tecnologia di sequenziamento dell'RNA, iniziamo a setacciare i grandi livelli di rumore di sottofondo trascrizionale e di essere di identificare con sicurezza e rigore i modelli di espressione L1 e la quantità a livello di singolo locus.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Vorremmo ringraziare il dottor Yan Dong per le cellule del tumore della prostata DU145. Vorremmo ringraziare il dottor Nathan Ungerleider per la sua guida e consigli per la creazione di script supercomputer. Alcuni di questi lavori sono stati finanziati da NIH Grants R01 GM121812 a PD, R01 AG057597 a VPB, e 5TL1TR001418 a TK. Vorremmo anche riconoscere il sostegno dei Cancer Crusaders e del Tulane Cancer Center Bioinformatics core.
Materiali
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
Riferimenti
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860 (2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805 (1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808 (1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926 (2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- . . Agilent RNA 6000 Nano Kit Guide. , (2017).
- Mueller, O. L., Schroeder, A. . RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24 (2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati