
Method Article
Applicazioni di ibridazione in situ di RNA/DNA virale di nuova generazione nella ricerca sul virus dell'immunodeficienza umana/virus dell'immunodeficienza delle scimmie
In questo articolo
Riepilogo
Qui presentiamo un saggio di ibridazione in situ di nuova generazione per identificare specifiche sequenze di RNA virale o DNA in tessuti inclusi in paraffina fissata in formalina (FFPE). Questo approccio consente la visualizzazione di basse copie di RNA e DNA in meno di 24 ore con sensibilità e specificità molto elevate.
Abstract
L'ibridazione in situ è una tecnica potente per identificare specifiche sequenze di RNA o DNA all'interno di singole cellule in sezioni di tessuto, fornendo importanti informazioni sui processi fisiologici e sulla patogenesi della malattia. L'ibridazione in situ (ISH) è stata utilizzata per molti anni per valutare la posizione delle cellule infettate da virus, ma recentemente è stato sviluppato un approccio ISH di nuova generazione con una strategia di progettazione della sonda unica che consente l'amplificazione simultanea del segnale e la soppressione del fondo per ottenere la visualizzazione di una singola molecola preservando la morfologia dei tessuti. Questa ISH di nuova generazione si basa su un approccio simile alla PCR ramificata, ma eseguita in situ ed è più facile, sensibile e riproducibile rispetto ai metodi ISH classici o agli approcci di PCR in situ nel rilevare di routine l'RNA o il DNA nei tessuti inclusi in paraffina fissata in formalina (FFPE). Negli ultimi anni, il nostro laboratorio ha applicato questa piattaforma ISH per la rilevazione di immunodeficienza umana (HIV) e immunodeficienza delle scimmie (SIV), RNA virale (vRNA) e/o DNA virale (vDNA) positive all'interno di una moltitudine di tessuti FFPE. Con questo manoscritto tecnico dettagliato, vorremmo condividere le nostre conoscenze e i nostri consigli con tutte le persone interessate a utilizzare l'ISH di nuova generazione nella loro ricerca.
Introduzione
L'ISH è l'approccio sperimentale utilizzato per indirizzare e visualizzare filamenti complementari di DNA, RNA o acido nucleico modificato (cioè sonde) a specifiche sequenze di DNA o RNA all'interno di una cellula o di una sezione di tessuto. L'ISH consente la localizzazione e la visualizzazione specifiche di specifiche sequenze nucleiche nei tessuti, importanti per comprendere il livello di espressione, l'organizzazione, la distribuzione e le interazioni tra il bersaglio e il suo ambiente cellulare, che sono informazioni preziose non ottenibili con l'uso di altre tecniche popolari, come la qPCR. Fino a poco tempo fa, l'ISH veniva comunemente eseguita con un DNA complementare marcato o un RNA complementare (riboprobe). Queste sonde sono state coniugate direttamente con basi marcate con radio, fluorescenza o antigene (ad esempio, 35S, FITC e digossigenina), quindi localizzate e quantificate nel tessuto utilizzando rispettivamente approcci di rilevamento autoradiografico, microscopia a fluorescenza o immunoistochimica. Sebbene queste tecnologie in situ continuino a essere approcci validi, c'è ampio margine di miglioramento per sviluppare approcci meno laboriosi, più semplici, più veloci, sensibili e specifici.
Un approccio ISH commerciale alternativo di nuova generazione (ad esempio, il saggio RNAscope), descritto per la prima volta nel 2012, per la rilevazione dell'RNA messaggero dell'ospite (mRNA) si basa sulla PCR ramificata. La rilevazione dell'mRNA viene eseguita in cellule e tessuti FFPE, con una sensibilità che si avvicina alla visualizzazione di una singola molecola di RNA nelle singole cellule1. La specificità di questo approccio si ottiene nella condizione unica che due sonde bersaglio a doppia Z si leghino contiguamente alle rispettive sequenze complementari di RNA (o DNA) per un preamplificatore di segnale per legare sequenzialmente1. Ciò consente l'avvio di una cascata di amplificazione del segnale attraverso successive fasi di ibridazione simili al DNA ramificato (bDNA)1,2. Inoltre, questo approccio è notevolmente rapido e facile, con risultati ottenuti in appena 1 giorno (<8 h), un vantaggio significativo rispetto a un massimo di 4 settimane con tecniche alternative, tra cui Radio-ISH 1,2. Questa ISH di nuova generazione ha aperto nuove prospettive e opportunità per la ricerca sull'HIV/SIV. I principali ostacoli a una cura dell'HIV sono i serbatoi cellulari e tissutali che si stabiliscono durante le prime fasi della malattia 3,4. L'obiettivo generale di questa tecnica è identificare, localizzare e, infine, comprendere i principali compartimenti tissutali che fungono da serbatoio virale e sono persistenti all'interno di un ospite infetto. Ciò contribuirà a sua volta allo sviluppo di strategie di cura efficaci contro l'HIV.
In questo manoscritto, spieghiamo in dettaglio il nostro protocollo ISH multiplex RNA/DNA duplex di nuova generazione (ad esempio, RNAscope/DNAscope) e spieghiamo come abbiamo modificato il protocollo RNA ISH esistente per ottimizzare l'ISH di nuova generazione per i nostri campioni e bersagli specifici. Questo protocollo consente la visualizzazione, la localizzazione e la quantificazione dell'RNA virale e del DNA virale dell'HIV/SIV all'interno di sezioni di tessuto di 5 μm. La visualizzazione simultanea di vRNA e vDNA viene eseguita combinando due set di sonde personalizzate: uno senso, mirato al filamento codificante il vDNA (sonda C1 SIVmac239 Gag-Pol-Sense [416141-C1]), e uno antisenso, mirato ai trascritti di vRNA (sonda C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Sense [416131-C2]) che copre diverse regioni del genoma virale (Tabella 1), utilizzando due diversi canali di visualizzazione, C1 e C2. In questo protocollo, i canali C1 e C2 ci permettono di visualizzare i segnali in diversi colori (ad esempio, AP in rosso e HRP in marrone) e di rilevare le sonde con approcci diversi. Escludendo il processo di fissazione tissutale e il taglio, questo test richiede 2 giorni. Qui viene presentato il protocollo di ibridazione in situ duplex vRNA e vDNA che può essere eseguito su pellet cellulari o sezioni di tessuto.
Protocollo
1. Preparazione di sezioni e vetrini
- Tagliare i blocchi di paraffina e utilizzare un microtomo per tagliare sezioni da 5 +/-1 μm. Montare le sezioni o i pellet cellulari su vetrini da microscopio carichi in un bagno d'acqua privo di RNasi a 40-45 °C. Scivoli asciutti all'aria durante la notte a 37 °C o RT.
NOTA: I vetrini possono essere conservati per un massimo di 3 mesi a temperatura ambiente (RT) e 6 mesi a 4 °C. - Deparaffinizzare i vetrini FFPE.
- Cuocere i vetrini in forno asciutto per 1 ora a 60 °C.
- In una cappa aspirante, riempire due piastre di colorazione dei vetrini con ~200 ml di xilene fresco e due piastre di colorazione aggiuntive con ~200 ml di etanolo fresco al 100%. Coprite i contenitori con i coperchi.
- Posizionare i vetrini in una griglia e immergerli nel primo piatto contenente xilene. Incubare per 5-10 minuti a RT con agitazione.
- Posizionare i vetrini nella seconda piastra contenente xilene e incubare per 5-10 minuti a RT con agitazione.
- Posizionare immediatamente i vetrini nella capsula contenente etanolo al 100%. Incubare i vetrini per 5-10 minuti a RT con agitazione.
- Posizionare immediatamente i vetrini nella seconda piastra contenente etanolo al 100% e incubare per 5-10 minuti a RT con agitazione.
- Rimuovere la griglia dall'etanolo, picchiettare delicatamente il lato della griglia per rimuovere l'etanolo in eccesso e sciacquare con acqua priva di RNasi per 5-15 minuti.
2. Preparazione del forno
- Accendere il forno di ibridazione e impostare la temperatura a 40 °C.
- Metti un panno o un tovagliolo di carta assorbente robusto in un vassoio e bagnalo completamente con acqua a doppia distillazione per consentire il controllo dell'umidità.
- Inserire la teglia coperta nel forno e chiudere la porta del forno. Riscaldare la teglia per almeno 30 minuti a 40 °C prima dell'uso. Conservare la teglia in forno quando non viene utilizzata.
3. Recupero dell'epitopo indotto dal calore
- Preparare un tampone di recupero del target di ibridazione ISH a base di citrato 0,5x (10 nmol/L, pH = 6, vedere la Tabella dei materiali). Portare a ebollizione in un bicchiere sulla piastra riscaldante.
- Eseguire il recupero dell'epitopo indotto dal calore posizionando i vetrini nel tampone di recupero del bersaglio bollente per 30 minuti.
- Rimuovere i vetrini dal tampone di recupero del bersaglio e lavarli immediatamente in acqua bidistillata. Disidratare in etanolo al 100% per 5 minuti prima dell'essiccazione all'aria.
- Una volta che i vetrini si sono asciugati all'aria, applicare una penna barriera idrofobica per circondare la sezione di tessuto sul vetrino. Assicurati di lasciare asciugare completamente la barriera idrofobica all'aria.
4. Pretrattamento con proteasi
- Posizionare i vetrini essiccati su un rack di bloccaggio per vetrini, quindi preparare i reagenti di pretrattamento della proteasi (soluzione di digestione della proteasi, 2,5 μg/mL) diluendo con PBS sterile, freddo, in rapporto 1:5. Mescola bene.
NOTA: Nel kit disponibile in commercio sono forniti tre diversi reagenti per proteasi con diverse concentrazioni. Proteasi III (standard), Proteasi IV (forte) e Proteasi Plus (lieve). Testare empiricamente il tempo di digestione e la diluizione della proteasi prima dell'implementazione in uno studio perché le condizioni ottimali variano in base al tipo di tessuto, alla fissazione e allo spessore (vedi Discussione). - Erogare la soluzione di proteasi diluita sui vetrini per coprire completamente le sezioni di tessuto. Incubare immediatamente i vetrini per 20 minuti a 40 °C in un forno (preparato al punto 1.4), assicurandosi che i vetrini siano sigillati nel vassoio di ibridazione umido. Non lasciare che le sezioni di tessuto si asciughino per il resto del protocollo.
- Sciacquare immediatamente 3 volte immergendo il cestello di bloccaggio in una vaschetta di lavaggio riempita con acqua bidistillata.
- Eseguire il blocco endogeno della perossidasi facendo cadere la soluzione di perossidasi su ciascuna sezione di tessuto per coprirla completamente. Incubare i vetrini per 10 minuti a RT. Una volta pronte, sciacquare le sezioni 3 volte in acqua bidistillata.
5. Ibridazione della sonda e amplificazione del segnale
NOTA: Per evitare l'evaporazione, assicurarsi che il vassoio a umidità controllata si chiuda correttamente in modo che i tessuti non si secchino durante le fasi di incubazione. Rimettere la camera di umidità nel forno durante la fase di lavaggio per assicurarsi che rimanga a 40 °C.
- Miscelare la sonda C2 e la sonda C1 in un rapporto 1:50 pipettando 1 volume di sonda C2 in 50 volumi di sonda C1 in una provetta, come suggerito dal produttore. Capovolgere il tubo più volte. Preriscaldare la miscela della sonda target in forno a 40 °C per ~10 minuti per sciogliere eventuali precipitazioni prima dell'uso.
NOTA: Le sonde target misti possono essere conservate a 4 °C per un massimo di 6 mesi. - Togliete gli scivoli dall'acqua. Risciacquare e picchiettare o scorrere i vetrini per rimuovere l'acqua in eccesso dalle sezioni di tessuto. Erogare immediatamente la sonda sui vetrini, assicurandosi che ogni sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare la miscela di sonde nella camera di umidità per una notte a 40 °C.
- Il giorno successivo, togliere i vetrini dal forno e metterli nella vaschetta di lavaggio contenente 0,5x tampone di lavaggio per 5 minuti a RT. Ripetere la fase di lavaggio ancora una volta.
- Rimuovere i vetrini dal tampone di lavaggio. Risciacquare e picchiettare o scorrere i vetrini per rimuovere il tampone di lavaggio in eccesso dalle sezioni di tessuto.
- Erogare commercialmente il reagente AMP 1 pronto all'uso (2 nmol/L) nel tampone di ibridazione B (20% formammide, 5x SSC, 0,3% litiododecil solfato, 10% destrano solfato, reagenti bloccanti) sui vetrini, garantendo una copertura completa della sezione di tessuto senza bolle d'aria. Incubare per 30 minuti a 40 °C nella camera di umidità. Ripetere i passaggi 1.6.3-1.6.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 2 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 15 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Erogare AMP 3 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 30 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Erogare AMP 4 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 15 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 5 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 30 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 6 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 15 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Prima del rilevamento, sciacquare i vetrini una volta in 1x TBS-Tween 20 (0,05% v/v). Rimuovere i vetrini dal tampone di lavaggio, risciacquare e picchiettare o scorrere i vetrini per rimuovere il tampone di lavaggio in eccesso dalle sezioni di tessuto. Posizionare immediatamente nella vaschetta di lavaggio riempita con 1x tampone TBS-Tween.
6. Rilevamento del segnale target del canale 1 (C1)
NOTA: Questa operazione viene eseguita utilizzando la fosfatasi alcalina rossa e l'amplificazione rapida del cromogeno rosso 6 dai kit di rilevamento a 2 plessi (vedere la tabella dei materiali) contenenti etichette di fosfatasi alcalina e rilevamento cromogenico. Utilizza il rosso veloce come substrato per generare un segnale rosso.
- Preparare la soluzione di lavoro Fast Red (FR) utilizzando una diluizione 1:60 di Fast RED-B in Fast RED-A. Mescola bene. Per ridurre il precipitato e ottenere un segnale più pulito, filtrare la soluzione cromogenica attraverso una membrana MCE da 0,45 μm utilizzando una siringa.
NOTA: Utilizzare la soluzione Fast RED-B entro 5 minuti. Non esporre alla luce solare diretta o ai raggi UV. - Rimuovere i vetrini dal TBS-Tween, risciacquare e picchiettare o scorrere i vetrini per rimuovere il tampone in eccesso dalle sezioni di tessuto.
- Erogare la soluzione FR miscelata e filtrata su ciascuna sezione di tessuto, assicurandosi che ogni sezione sia completamente coperta. Incubare a RT per 6-8 min. Osservare al microscopio.
- Sciacquare i vetrini in 0,5x tampone di lavaggio 2x. Rimuovere i vetrini dal tampone di lavaggio, risciacquare e picchiettare o scorrere i vetrini per rimuovere il tampone di lavaggio in eccesso dalle sezioni di tessuto.
- Eroga AMP 7 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 10 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 8 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 15 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 9 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 30 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
- Eroga AMP 10 disponibile in commercio. Assicurarsi che la sezione di tessuto sia completamente coperta senza bolle d'aria. Incubare i vetrini nella camera di umidità per 30 minuti a 40 °C. Ripetere i passaggi 5.3-5.4, eseguendo il lavaggio per 2 minuti ciascuno.
7. Rilevamento del segnale target del canale 2 (C2).
NOTA: Questa operazione viene eseguita utilizzando i kit di cromogeni Brown HRP e DAB disponibili in commercio (vedere la Tabella dei materiali). L'amplificazione 10 della rilevazione a 2 plessi contiene etichette di perossidasi di rafano e la rilevazione cromogenica viene eseguita utilizzando DAB per generare un segnale marrone.
- Per un rilevamento ottimale del segnale DAB, utilizzare i kit disponibili in commercio e seguire le istruzioni del produttore (vedere la Tabella dei materiali). Osservare al microscopio.
ATTENZIONE: Il DAB è tossico. Seguire le precauzioni appropriate e le linee guida di sicurezza durante la manipolazione e lo smaltimento di questa sostanza chimica.
8. Controcolorazione e montaggio
- Controcolorare i vetrini con ematossilina.
- Controcolorare i vetrini per 30 s con ematossilina filtrata fresca al 50% mentre si agitano i vetrini. Le diapositive appariranno viola. Sciacquare immediatamente con acqua corrente agitando gli scivoli su e giù fino a quando l'acqua non è limpida. Le sezioni di tessuto rimarranno violacee.
- Per ottenere un migliore contrasto, posizionare i vetrini controcolorati in acqua distillata satura di carbonato di litio per 1 minuto. Sciacquare abbondantemente in acqua corrente almeno 3 volte mentre si agitano i vetrini. Utilizzare acqua bidistillata per il risciacquo finale.
- Poiché l'ignifugo è sensibile ai solventi organici, i vetrini colorati con ignifugo dovranno essere coperti con un mezzo di montaggio a base d'acqua e asciugati durante la notte a RT.
- Montare le diapositive.
- Assicurarsi che le sezioni di tessuto ricoperte con un mezzo di montaggio a base d'acqua siano asciutte.
- Immergere i vetrini nello xilene prima di scivolare il coperchio utilizzando il reagente di montaggio. Assicurarsi di prevenire o rimuovere eventuali bolle d'aria tra il vetrino coprioggetti e la sezione di tessuto e lasciare asciugare per 16 ore a RT.
9. Protocollo di analisi quantitativa delle immagini per RNAscope utilizzando CellProfiler5
- In breve, assicurarsi che il software separi le macchie di ematossilina e FR in immagini separate. Identificare e misurare gli oggetti di interesse: nuclei, virioni, cellule positive e colorazione aggregata FR positiva. Memorizza le misurazioni in un file CSV e salva l'immagine analizzata.
- Seleziona l'opzione "Mescola colori", che separa le macchie, suddividendo la regione di interesse originale (ROI) in immagini separate di ematossilina e FR.
- Se l'emosiderina, il tatuaggio o caratteristiche simili interferiscono con l'analisi, aggiungere un secondo passaggio "Unmix" con ematossilina, FR e DAB. Usa la seconda immagine FR per trovare i pixel FR più intensi. Questa seconda immagine verrà utilizzata come maschera per la vera colorazione FR.
- Facoltativamente, levigare le immagini macchiate prima di limitarle. Si tratta di una decisione empirica e non sempre necessaria. Soglia le singole immagini colorate utilizzando "IdentifyPrimaryObjects" per selezionare i pixel positivi.
- Identificare i tre diversi tipi di oggetti (virione, aggregati di virioni, cellula produttiva).
- Assicurarsi che i nuclei siano oggetti di diametro compreso tra 4 e 100 pixel (px) colorati con ematossilina. Raggruppare e riempire i fori dopo la soglia. Gli oggetti "IntenseFastRed" hanno un diametro di 4-100 px e includono virioni, cellule positive e colorazioni positive aggregate, come quelle osservate sulle cellule dendritiche follicolari (FDC) nei follicoli delle cellule B (BCF). Questa immagine viene utilizzata per filtrare i falsi positivi (ad esempio, l'emosiderina).
- Assicurati che i piccoli positivi FR siano oggetti di diametro compreso tra 2 e 12 px. Questa misurazione include virioni e cellule vDNA+. Scarta tutti gli oggetti al di fuori di questo intervallo. Raggruppare e riempire i fori dopo la soglia.
- Assicurati che FR grandi positivi: oggetti di diametro 9-100 px. Questa misurazione include le cellule positive per il vRNA+ e la colorazione positiva aggregata. Raggruppare dopo la soglia. Le dimensioni degli oggetti FR positivi piccoli e grandi possono sovrapporsi. Saranno separati in una fase successiva.
NOTA: Il software (ad esempio, Cellprofiler) utilizza i pixel per le dimensioni degli oggetti e quelli utilizzati qui derivano da diapositive scansionate a 0,2510 μm/pixel (40x).
- Definisci ed estrai i risultati.
- Dopo aver identificato gli oggetti, definisci ed estrai i risultati per il numero di virioni, cellule infette produttive e nuclei.
- Identificare i virioni mascherando FastRedSmallPositives con l'oggetto IntenseFastRed impostato per rimuovere i falsi positivi (ad esempio, emosiderina).
- Successivamente, identificare le cellule positive e aggregare la colorazione FR positiva. Rimuovere i falsi positivi e i virioni da FastRedLargePositives mascherandolo con IntenseFastRed e con l'oggetto virion impostato.
- Estrarre le cellule positive dai raffinati FastRedLargePositives mascherando nuovamente con i nuclei. Dividi gli oggetti che non si toccano più e filtra i risultati in base all'area dell'oggetto, rimuovendo gli oggetti piccoli (≤6 px). Questo rimuove i granelli creati mascherando la sovrapposizione dei nuclei. Il risultato sono le celle positive.
- Infine, definisci i positivi FR aggregati. In questo passaggio viene utilizzato IdentifyTertiaryObjects, per trovare gli oggetti contenuti in un oggetto padre più grande. In questo caso, il set di oggetti FastRedLargePositives perfezionato è l'elemento padre e le celle positive vengono sottratte.
- Conta il numero di virioni e cellule positive.
- Misurare l'area positiva aggregata e convertirla da pixel a mm2. Facoltativamente, registrare l'area occupata dai virioni e dalle celle positive in mm2 se l'analisi richiede l'utilizzo di celle standard e dimensioni di virioni invece dei conteggi diretti.
- Sovrapponi gli oggetti positivi all'immagine originale e salva il risultato.
NOTA: I dati ISH sono riportati dal numero di virioni per 106 nuclei (cellule) e dal numero di cellule vRNA+ infettate in modo produttivo per 106 nuclei (cellule) per una migliore comprensione e per facilitare il confronto con i dati qPCR, ma i risultati potrebbero anche essere riportati per area di tessuto in mm2.
Risultati
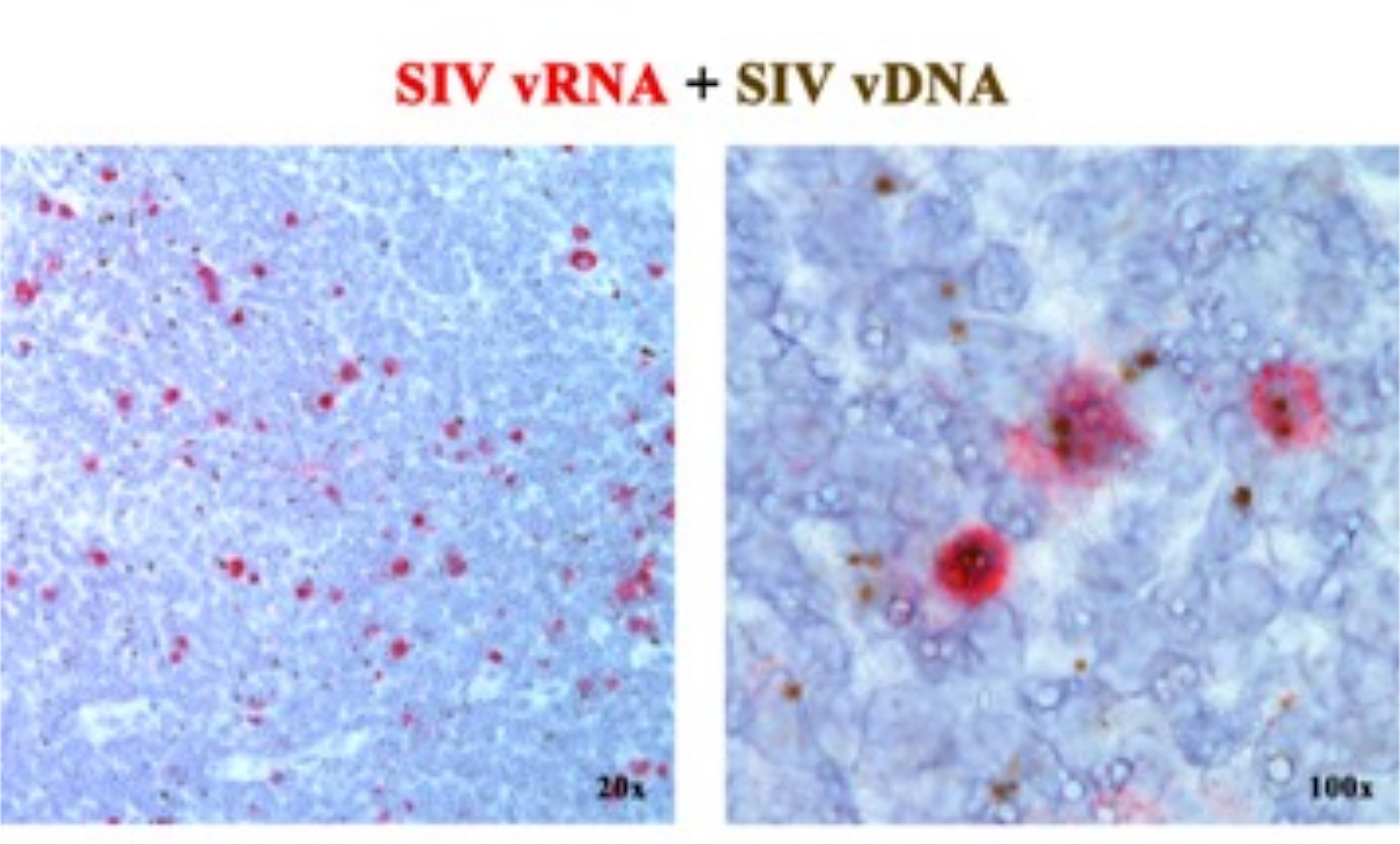
In un precedente manoscritto 2,6,7,8,9,10,11, abbiamo riportato che le piattaforme ISH di nuova generazione che rilevano sia il vRNA che il vDNA possono essere combinate, utilizzando sonde di senso (vDNA) che hanno come bersaglio la porzione 5' gag-pol del genoma SIV/HIV e sonde antisenso (vRNA) che hanno come bersaglio i geni nella metà 3' del genoma (vif, vpx, vpr, tat, env e nef) così come l'elemento TAR nel genoma 5' (Tabella 1). Questo approccio distingue le cellule trascrizionalmente attive (vRNA+, vDNA+) dalle cellule infette trascrizionalmente inattive (putativamente latenti) o dalle cellule che ospitano provirus trascrizionalmente incompetenti (vRNA-, vDNA+) nella stessa sezione tissutale2 (Figura 1).

Figura 1: Rilevamento di RNA virale e vDNA nella stessa sezione di tessuto. Combinazione del saggio di ibridazione dell'RNA (rosso) e del DNA (marrone) in un linfonodo RM acutamente infetto da SIV, che dimostra la capacità di rilevare vRNA e vDNA nella stessa sezione di tessuto e fornisce un potente approccio per identificare in situ le cellule vDNA+ vRNA trascrizionalmente silenti. Questa cifra è stata modificata da Deleage C. et al.2. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Set di sonde per RNA/DNAscope a plesso singolo | |||
| Nome | Catalogo ACD # | Numero di ZZ | Descrizione |
| SIVmac239 (Anti-senso) | 312811 | 83 | Puntamento della sonda anti-senso entro 1251-9420bp di D01065.1 (gag, pol, vif, vpx, vpr, tat, env e nef) |
| SIVmac239 (senso) | 314071 | 83 | Sonda di rilevamento che mira al filamento inverso entro 1251-9420bp di D01065.1 (gag, pol, vif, vpx, vpr, tat, env e nef) |
| V-HIV1-clade A (anti-senso) | 416101 | 80 | Targeting della sonda antisenso entro 879-7629 bp dal consenso del Clade A dell'HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-clade A (senso) | 426341 | 80 | Sonda di rilevamento mirata al filamento inverso entro 879-7629bp dal consenso del clade A dell'HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-clade B (anti-senso) | 416111 | 78 | Targeting della sonda antisenso entro 854-8291bp di AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-Clade B (Senso) | 425531 | 78 | Sonda di rilevamento mirata al filamento inverso entro 854-8291bp di AF324493.2, HIV-1 Clade B NL4-3 (gag, pol, vif, vpr, tat, rev, vpu, env e nef) |
| V-HIV1-clade D (anti-senso) | 416121 | 76 | Targeting della sonda antisenso entro 894-7697bp dal consenso del Clade D HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| V-HIV1-Clade D (Senso) | 426351 | 76 | Sonda di rilevamento mirata al filamento inverso entro 894-7697bp dal consenso del Clade D HIV-1 (gag, pol, vif, vpr, tat, rev, vpu, env, nef) |
| Set di sonde per RNA/DNAscope multi-plex | |||
| Nome | Catalogo ACD # | Numero di ZZ | Descrizione |
| V-SIVmac239-gag-pol-Sense-C1 | Codice 416141-C1 | 40 | Sonda di rilevamento che mira al filamento inverso entro 1251-4093bp di D01065.1 (gag e pol) |
| V-SIVmac239-vif-env-nef-tar-C2 (Antisenso) | Codice 416131-C2 | 47 | Puntamento della sonda anti-senso entro 5381-10257bp di D01065.1 (vif, vpx, vpr, tat, env, nef e l'elemento TAR) |
| V-HIV1-Clade_B-gag-pol-sense-C1 | Codice 444051-C1 | 40 | Sonda di rilevamento che mira al filamento inverso entro 854-3940bp di AF324493,2, HIV-1 Clade B NL4-3 (gag e pol) |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (Anti-senso) | Codice 444061-C2 | 40 | Targeting della sonda antisenso entro 5042-9673bp di AF324493,2,, HIV-1 Clade B NL4-3 (vif, vpr, tat, env, nef e l'elemento TAR) |
| V-HIV1-Clade_C-gag-pol-sense-C1 | Codice 444021-C1 | 48 | Sonda di rilevamento che mira al filamento inverso entro 888-5032 bp dalla sequenza di consenso del Clade C dell'HIV-1 (gag e pol) |
| V-HIV1-Clade_C-vif-vpr- rev-vpu-env-nef-tar-C2 (Anti-senso) | Codice 444041-C2 | 49 | Targeting della sonda antisenso entro 5078-9698bp della sequenza di consenso del clade C dell'HIV-1 (vif, vpr, tat, env, nef e l'elemento TAR) |
| V-HIV1-Clade_AE-gag-pol-sense-C1 | Codice 444011-C1 | 55 | Sonda di rilevamento mirata al filamento inverso entro 890-4812 bp di AF259954,1, HIV-1 Clade AE (gag e pol) |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (antisenso) | Codice 444031-C2 | 57 | Targeting della sonda anti-senso entro 5052-9694bp di AF259954.1, HIV-1 Clade AE (vif, vpr, tat, env, nef e l'elemento TAR) |
Tabella 1: Elenco delle sonde per il targeting del vRNA e del vDNA di HIV-1 e SIV.
Discussione
L'ibridazione in situ è un saggio meticoloso che richiede rigore e conoscenze di base della chimica degli acidi nucleici, della biologia cellulare e dell'istologia per essere in grado di adattare ogni passaggio critico per localizzare un bersaglio in un ambiente ben conservato. In questa discussione vorremmo evidenziare i passaggi critici in cui la risoluzione dei problemi è fondamentale per ottenere risultati accurati e interpretabili.
La fissazione e l'elaborazione dei tessuti sono fondamentali e devono essere affrontate in anticipo per assicurarsi che il test possa produrre i migliori risultati. Il fissativo PFA tamponato neutro (4% appena preparato) è ottimale per il test duplex. Tuttavia, il test può essere eseguito anche su tessuti congelati (OCT) con le condizioni di fissazione post-criosezione appropriate.
Il pretrattamento delle sezioni di tessuto è una fase cruciale. In questo test sono previste due fasi di pretrattamento: la prima è il recupero dell'epitopo indotto dal calore (HIER). Questo passaggio è importante per l'inversione dei legami incrociati dei ponti di metilene e il ripristino delle strutture proteiche, che è necessario nei tessuti fissati. L'efficienza di questo trattamento dipende dal tempo, dalla temperatura, dal tipo di tampone di recupero e dal pH. Il secondo pretrattamento è il recupero dell'epitopo indotto dalla proteasi (PIER). Questa fase scinde i peptidi, esponendo l'antigene o i nucleotidi, e utilizza enzimi tra cui la proteinasi K, la tripsina e la pepsina. Si tratta di un passaggio estremamente delicato che potrebbe potenzialmente danneggiare sia la morfologia tissutale che il bersaglio di interesse. La concentrazione dell'enzima, così come il tempo e la temperatura di incubazione sono fondamentali in questo processo. L'iperdigestione porta a una scarsa demarcazione dei nuclei e a difficoltà nelle fasi di quantificazione. È fondamentale trovare un equilibrio tra l'accesso ottimale al bersaglio di RNA/DNA e le condizioni di pretrattamento che non danneggino il tessuto o il bersaglio di interesse. Ogni tipo di tessuto ha un diverso livello di sensibilità a ciascuno di questi pretrattamenti e ogni parametro (concentrazione enzimatica, tempo, temperatura) deve essere testato empiricamente.
Il rigore del tampone di lavaggio si basa su tre parametri principali: temperatura, concentrazione di sali e detersivo e tempo. Il tampone di lavaggio è un tampone salino di citrato di sodio (SSC) e la concentrazione di sale all'interno del tampone controlla il rigore durante le fasi di lavaggio. Nel loro protocollo, ACD consiglia di utilizzare il tampone di lavaggio a una concentrazione finale di 0,1x SSC, 0,03% di dodecil solfato di litio. Mentre lavoravamo sull'ottimizzazione del DNAscope e del multiplex, abbiamo determinato che l'utilizzo del tampone di lavaggio a una concentrazione finale di 0,05x SSC ci ha dato risultati migliori per visualizzare il segnale del DNA e ha contribuito notevolmente a ridurre l'ibridazione non specifica fuori bersaglio derivante dall'incubazione notturna della sonda sensoriale.
La scelta dell'approccio di rilevamento, cromogeno (rosso o marrone) rispetto alla fluorescenza, deve essere ponderata in base al tipo di tessuto e all'obiettivo prima di iniziare il saggio. L'approccio cromogenico rosso darà un bel contrasto, perché il rosso non si trova naturalmente nei tessuti. Il cromogeno marrone darà risultati simili al cromogeno rosso. Tuttavia, è importante tenere presente che alcuni prodotti di degradazione del sangue presenti nel tessuto hanno un colore simile e l'inchiostro del tatuaggio sarà difficile da separare dal segnale marrone durante la quantificazione. Un approccio di rilevazione a fluorescenza consentirà una chiara distinzione dei diversi marcatori cellulari e il multiplexing offrirà un saggio perfetto per fenotipizzare le cellule che ospitano vRNA e/o vDNA.
Sono necessari controlli multipli per garantire la specificità delle sonde e la qualità del test. Ogni sonda di nuova concezione deve essere testata su tessuti di controllo positivi e negativi noti o su pellet cellulari. Spesso generiamo plasmidi contenenti la nostra sequenza mirata ed eseguiamo la trasfezione in linee cellulari per generare controlli positivi. Per ogni corsa aggiungiamo un tessuto negativo noto (HIV o SIV negativo), un controllo senza sonda contenente solo il diluente della sonda e un controllo trattato con RNasi per garantire la qualità e la specificità del test.
La quantificazione è un passaggio estremamente importante e deve essere eseguita utilizzando gli strumenti e gli algoritmi appropriati in base alla domanda posta. In questo manoscritto abbiamo presentato un software di analisi delle immagini (ad esempio, Cellprofiler), che abbiamo scelto dopo aver valutato diverse opzioni. Abbiamo stimato che questo software fosse il migliore per le nostre esigenze, ma ci sono numerosi programmi software di analisi delle immagini che potrebbero essere utilizzati.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo progetto è stato interamente finanziato con fondi federali del National Cancer Institute, National Institutes of Health, nell'ambito del contratto n. HHSN261200800001E e dall'Oregon National Primate Research Center NIH grant award P51OD011092 (J.D.E). Il contenuto di questa pubblicazione non riflette necessariamente le opinioni o le politiche del Dipartimento della Salute e dei Servizi Umani, né la menzione di nomi commerciali, prodotti commerciali o organizzazioni implica l'approvazione da parte del governo degli Stati Uniti. Il duplex è stato sviluppato con l'aiuto di Advanced Cell Diagnostics.
Materiali
| Name | Company | Catalog Number | Comments |
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
Riferimenti
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis. 14 (1), 22-29 (2012).
- Deleage, C., et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity. 1 (1), 68-106 (2016).
- Sengupta, S., Siliciano, R. F. Targeting the Latent Reservoir for HIV-1. Immunity. 48 (5), 872-895 (2018).
- Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F., Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology. 14 (1), 55-60 (2016).
- McQuin, C., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology. 16 (7), e2005970 (2018).
- Deleage, C., Chan, C. N., Busman-Sahay, K., Estes, J. D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology. 15 (1), 4 (2018).
- Deleage, C., Turkbey, B., Estes, J. D. Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology. 19, 77-84 (2016).
- Estes, J. D., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine. 23 (11), 1271-1276 (2017).
- Mavigner, M., et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology. 92 (18), (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen. 14 (4), e1006956 (2018).
- Deleage, C., et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight. 1 (10), e87065 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati