
Method Article
Mappatura delle relazioni struttura-funzione dei fattori di trascrizione oncogenica disordinati utilizzando l'analisi trascrittomica
In questo articolo
Riepilogo
I domini intrinsecamente disordinati sono importanti per la funzione del fattore di trascrizione di fusione oncogenica. Per indirizzare terapeuticamente queste proteine, è necessaria una comprensione più dettagliata dei meccanismi di regolazione impiegati da questi domini. Qui, usiamo la trascrittomica per mappare importanti caratteristiche strutturali del dominio EWS intrinsecamente disordinato nel sarcoma di Ewing.
Abstract
Molti tumori sono caratterizzati da traslocazioni cromosomiche che provocano l'espressione di fattori di trascrizione di fusione oncogenici. Tipicamente, queste proteine contengono un dominio intrinsecamente disordinato (IDD) fuso con il dominio di legame al DNA (DBD) di un'altra proteina e orchestrano cambiamenti trascrizionali diffusi per promuovere la malignità. Queste fusioni sono spesso l'unica aberrazione genomica ricorrente nei tumori che causano, rendendoli bersagli terapeutici attraenti. Tuttavia, il targeting dei fattori di trascrizione oncogenici richiede una migliore comprensione del ruolo meccanicistico che gli IDD a bassa complessità svolgono nella loro funzione. Il dominio N-terminale di EWSR1 è un IDD coinvolto in una varietà di fattori di trascrizione di fusione oncogenica, tra cui EWS / FLI, EWS / ATF e EWS / WT1. Qui, usiamo il sequenziamento dell'RNA per studiare le caratteristiche strutturali del dominio EWS importanti per la funzione trascrizionale di EWS / FLI nel sarcoma di Ewing. Viene eseguita la prima deplezione mediata da shRNA della fusione endogena da cellule di sarcoma di Ewing accoppiata con l'espressione ectopica di una varietà di costrutti mutanti EWS. Quindi il sequenziamento dell'RNA viene utilizzato per analizzare i trascrittomi delle cellule che esprimono questi costrutti per caratterizzare i deficit funzionali associati alle mutazioni nel dominio EWS. Integrando le analisi trascrittomiche con le informazioni precedentemente pubblicate sui motivi di legame del DNA EWS / FLI e la localizzazione genomica, nonché i saggi funzionali per la capacità di trasformazione, siamo stati in grado di identificare le caratteristiche strutturali di EWS / FLI importanti per l'oncogenesi e definire un nuovo set di geni bersaglio EWS / FLI critici per il sarcoma di Ewing. Questo articolo dimostra l'uso del sequenziamento dell'RNA come metodo per mappare la relazione struttura-funzione del dominio intrinsecamente disordinato dei fattori di trascrizione oncogenici.
Introduzione
Un sottogruppo di tumori, tra cui molte neoplasie maligne dell'infanzia e dell'adolescenza, sono caratterizzate da traslocazioni cromosomiche che generano nuovi oncogeni di fusione1,2,3,4,5,6. Le proteine di fusione risultanti funzionano frequentemente come fattori di trascrizione oncogenici, orchestrando cambiamenti diffusi nella regolazione trascrizionale per promuovere la tumorigenesi7,8. I tumori con queste traslocazioni possiedono comunemente un paesaggio mutazionale altrimenti tranquillo, con poche aberrazioni genomiche ricorrenti a parte la fusione patognomonica4,9. Pertanto, il targeting diretto della proteina di fusione è una strategia terapeutica interessante in queste malattie. Tuttavia, questi fattori di trascrizione oncogenici consistono comunemente in un dominio a bassa complessità, intrinsecamente disordinato, che attiva trascrizionalmente fuso con un dominio di legame al DNA (DBD)10,11, 12,13,14. Sia i domini intrinsecamente disordinati (IDD) che i DBD di queste proteine si sono dimostrati difficili da colpire con approcci farmacologici convenzionali. Lo sviluppo di nuovi approcci terapeutici, quindi, richiede una comprensione molecolare più dettagliata dei meccanismi impiegati da queste fusioni per regolare aberrantemente l'espressione genica.
La porzione IDD N-terminale di EWSR1 è comunemente fusa con una DBD nel cancro, tra cui EWS / FLI nel sarcoma di Ewing, EWS / WT1 nel tumore diffuso a piccole cellule rotonde e EWS / ATF1 nel sarcoma a cellule chiare delle parti molli10. Il ruolo meccanicistico dell'IDD EWS in ciascuna di queste fusioni non è completamente compreso. La famiglia di fusioni EWS/ETS, in particolare EWS/FLI, è la più funzionalmente caratterizzata fino ad oggi. EWS/FLI coordina i cambiamenti epigenetici e trascrizionali a livello di genoma che portano all'attivazione e alla repressione di migliaia di geni7,11,15,16. Gli studi hanno dimostrato che l'IDD è importante per il reclutamento sia di co-attivatori trascrizionali (come p300, WDR5 e il complesso BAF), sia di co-repressori (come il complesso NuRD)11,15,17. La fusione dell'IDD EWS con la porzione C-terminale di FLI1 conferisce una nuova specificità di legame del DNA alla DBD ETS di FLI1, tale che l'oncoproteina di fusione (EWS/FLI) si lega a regioni ripetitive del genoma GGAA-microsatellite oltre al motivo ETS di consenso18,19,20. In combinazione con la funzione di reclutamento del co-attivatore, questa attività emergente di legame al DNA di EWS / FLI promuove la formazione di potenziatori de novo nei siti di inizio della trascrizione distale a GGAA -microsatelliti distali (TSS) (microsatelliti "enhancer-like") e recluta RNA polimerasi II per promuovere la trascrizione a GGAA-microsatelliti prossimali a TSS (microsatelliti "promoter-like")11,15,16,21.
Presi insieme, questi dati ci hanno portato a ipotizzare che elementi discreti all'interno del dominio EWS contribuiscano al reclutamento di co-regolatori distinti in diversi tipi di siti di legame EWS / FLI. Tuttavia, discernere questi elementi all'interno della porzione EWS di EWS / FLI e come funzionano, è stato ostacolato dalla natura altamente ripetitiva e disordinata del dominio. Qui utilizziamo un sistema di knockdown-rescue precedentemente pubblicato nelle cellule del sarcoma di Ewing per mappare funzionalmente questi elementi nell'IDD EWS. In questo sistema EWS/FLI viene impoverito utilizzando uno shRNA mirato al 3'UTR del gene FLI1, e l'espressione viene salvata con vari costrutti cDNA mutanti EWS/FLI privi del 3'UTR7,17,22. Questi esperimenti si sono concentrati su costrutti con varie delezioni per mappare la relazione struttura-funzione tra l'IDD EWS e importanti fenotipi oncogeni, tra cui l'attivazione di un costrutto reporter GGAA-microsatellite, saggi di formazione di colonie e convalida mirata di geni attivati e repressi da EWS / FLI7,17,22 . Tuttavia, questi studi non sono riusciti a trovare sottodomini discreti all'interno dell'IDD EWS in EWS / FLI che sono di importanza univoca per l'attivazione o la repressione. Tutti i costrutti testati erano in grado di attivare e reprimere specifici geni bersaglio, portando a un'efficiente formazione di colonie, o incapaci di regolare nessuno dei geni bersaglio EWS / FLI, portando alla perdita della formazione della colonia7,17,22.
Le analisi trascrittomiche rese possibili dall'adozione diffusa del sequenziamento di nuova generazione sono comunemente utilizzate per confrontare le firme di espressione genica in due condizioni, spesso nel contesto di screening o studi descrittivi. Volevamo invece sfruttare la capacità di acquisire dati di espressione a livello di genoma utilizzando il sequenziamento dell'RNA (RNA-seq) per caratterizzare i contributi degli IDD alla funzione del fattore di trascrizione. In questo caso RNA-seq è accoppiato con il sistema knockdown-rescue per esplorare la relazione struttura-funzione del dominio EWS. Questo approccio è applicabile ad altri fattori di trascrizione di fusione, comprese altre fusioni EWS o fattori di trascrizione wildtype con funzione poco compresa, e presenta molteplici vantaggi rispetto agli altri saggi utilizzati per studi di mappatura funzionale, come saggi reporter o qRT-PCR mirata. Questi includono il test dei determinanti strutturali della funzione nel contesto della cromatina pertinente, la capacità di testare più tipi di elementi di risposta in un unico test (cioè attivato e represso, GGAA-microsatellite e non microsatellite, ecc.) e la conseguente capacità di rilevare meglio la funzione parziale.
Il successo dell'implementazione di questo approccio dipende da un sistema basato su cellule che cattura i fenotipi di interesse (in questo caso cellule A673 con deplezione EWS/FLI mediata da shRNA) e da un pannello di costrutti mutanti in un vettore di espressione appropriato per il sistema basato su cellule (in questo caso, pMSCV-hygro con vari mutanti EWS/FLI 3x-FLAG-tagged da erogare per trasduzione retrovirale). La trasduzione virale di costrutti di deplezione basati su CRISPR, costrutti di deplezione basati su shRNA e costrutti di espressione del cDNA con una selezione appropriata per generare linee cellulari stabili è raccomandata rispetto alla trasfezione transitoria. L'interpretazione a valle dei risultati è rafforzata quando i dati trascrittomici possono essere abbinati ad altri dati relativi alla localizzazione del fattore di trascrizione e ad altre letture fenotipiche ove disponibili.
In questo articolo, applichiamo questo approccio per caratterizzare l'attività del mutante DAF di EWS / FLI14. Il mutante DAF ha 17 mutazioni da tirosina ad alanina nelle regioni ripetitive dell'EWS IDD di EWS/FLI14. Questo particolare mutante EWS era stato precedentemente riportato e non è in grado di attivare l'espressione genica reporter quando fuso con l'ATF1 DBD14. Tuttavia, i dati preliminari della qRT-PCR hanno suggerito che questo mutante è stato in grado di attivare la trascrizione del bersaglio EWS/FLI NR0B123. L'approccio trascrittomico qui descritto ha permesso di rilevare con successo la funzione parziale del mutante DAF. Accoppiando questi dati trascrittomici con informazioni sui motivi di legame e riconoscimento EWS/FLI dimostriamo inoltre che il mutante DAF mantiene la funzione alle ripetizioni GGAA-microsatellite. Questi risultati identificano DAF come il primo mutante EWS/FLI parzialmente funzionale ed evidenziano la funzione nei geni non microsatelliti come importante per l'oncogenesi (come riportato23). Ciò dimostra la potenza di questo approccio di mappatura struttura-funzione trascrittomica per fornire informazioni sulla funzione dei fattori di trascrizione oncogenici.
Protocollo
1. Impostare un pannello di costrutti in vitro
NOTA: Questo passaggio varierà a seconda della proteina specifica da analizzare.
- Preparare aliquote del virus per l'esaurimento e i costrutti di espressione, se necessario.
- Seminare un piatto di coltura tissutale di 10 cm con 3-5 x10 6 cellule HEK293-EBNA o HEK293T per ogni costrutto necessario per la trasduzione virale. Lascia che le cellule aderiscano durante la notte nel Modified Eagle Media (DMEM) di Dulbecco integrato con il 10% di siero bovino fetale (FBS), penicillina / streptomicina / glutammina (P / S / Q) e 0,3 mg / mL G418.
NOTA: le cellule HEK293-EBNA e HEK293T sono raccomandate per la produzione virale perché sono facili da coltivare, hanno un'elevata efficienza di trasfezione ed esprimono in modo efficiente le proteine ricombinanti dai plasmidi episomiali. Le cellule dovrebbero essere tra il 50-70% confluenti il giorno della trasfezione. - Preparare una miscela di trasfezione per ogni costrutto di trasduzione virale. Combinare 2 mL di mezzi sierici ridotti con 90 μL di reagente di trasfezione.
NOTA: si consiglia di preriscaldare i supporti sierici ridotti. - Aggiungere 10 μg ciascuno di un plasmide di imballaggio virale (ad esempio, gag-pol), plasmide di involucro virale (ad esempio, VSV-G) e uno di esaurimento basato su CRISPR, esaurimento basato su shRNA o costrutto di espressione di cDNA (ad esempio, pMKO o pMSCV) alla miscela di trasfezione. Mescolare bene con un leggero pipettaggio.
- Lasciare riposare la miscela di trasfezione per 20 minuti a temperatura ambiente. Rimuovere i mezzi di crescita HEK293-EBNA dai piatti di coltura tissutale e aggiungere 3 ml di DMEM integrati con il 10% di FBS, P / S / Q e 10 mM di piruvato di sodio. Ad ogni piatto, aggiungere 2 ml di mix di trasfezione a goccia. Lasciare che le cellule siedano in mezzi di trasfezione durante la notte in un incubatore a 37 °C e 5% co2.
- La mattina seguente aggiungere 20 ml di dmem media con il 10% di FBS, supplementazione P / S / Q e 10 mM di piruvato di sodio. Incubare le cellule in esso a 37 ° C e 5% CO2 per la notte.
- La mattina successiva, sostituire i supporti con 5 mL di media di raccolta virale (VCM) (DMEM integrato con FBS inattivato al 10% di calore, P / S / Q e 20 mM HEPES).
- Dopo 4 ore, raccogliere VCM dalle piastre e conservare in un tubo conico da 50 ml su ghiaccio a 4 °C. Sostituire con 5 ml di VCM fresco.
- Dopo 4 ore, raccogliere VCM dalle piastre nello stesso tubo conico da 50 ml e conservare su ghiaccio a 4 °C. Sostituire con 8 ml di VCM fresco per la raccolta notturna.
- Al mattino raccogliere VCM dalle piastre e conservare nel tubo conico da 50 ml su ghiaccio a 4 °C. Sostituire con 5 ml di VCM fresco.
- Dopo 4 ore, raccogliere VCM dalle piastre e conservare nel tubo conico da 50 ml su ghiaccio a 4°C. Sostituire con 5 ml di VCM fresco. Dopo 4 ore, raccogliere VCM dalle piastre e aggiungere al tubo conico da 50 ml.
- Aliquot raccolte da un tubo da 50 mL in criotubi (2 mL per aliquota) dopo filtrazione attraverso un filtro da 0,45 μm. Conservare le aliquote virali a -80 °C fino all'uso.
NOTA: il protocollo può essere messo in pausa qui e le aliquote virali possono essere conservate fino a quando non sono pronte per l'uso.
- Seminare un piatto di coltura tissutale di 10 cm con 3-5 x10 6 cellule HEK293-EBNA o HEK293T per ogni costrutto necessario per la trasduzione virale. Lascia che le cellule aderiscano durante la notte nel Modified Eagle Media (DMEM) di Dulbecco integrato con il 10% di siero bovino fetale (FBS), penicillina / streptomicina / glutammina (P / S / Q) e 0,3 mg / mL G418.
- Cellule di semi alla densità appropriata in un piatto di coltura tissutale di 10 cm. Obiettivo 50% di confluenza. Lasciare che le cellule aderiscano durante la notte posizionandole nell'incubatore a 37 °C contenente il 5% di CO2.
NOTA: Per le cellule A673 si tratta di 5 x 106 celle in 10 mL di supporti DMEM con il 10% di FBS, supplementazione P / S / Q e 10 mM di piruvato di sodio. Queste condizioni possono variare a seconda del tasso di crescita delle cellule utilizzate. - Esaurire il fattore endogeno di interesse. Se le cellule non hanno bisogno di avere la proteina endogena di interesse esaurita, saltare avanti al passaggio 1.4.
- Aliquota virale di scongelamento per la trasduzione di shRNA o costrutto CRISPR mirato alla proteina di interesse. Scongelare rapidamente le aliquote congelate a bagnomaria a 37 °C.
- Aggiungere 2,5 μL di 8 mg/mL di polibrene a ciascuna aliquota virale e mescolare mediante un delicato pipettaggio. Rimuovere i mezzi dalle piastre di cellule e aggiungere delicatamente l'aliquota virale alla piastra di 10 cm pipettando lungo il lato della piastra. Scuotere la piastra per diffondere i 2 ml di aliquota virale.
- Incubare a 37 °C nell'incubatore di colture tissutali per 2 ore. Scuotere la piastra ogni 30 minuti per evitare che eventuali aree della piastra si secchino.
- Aggiungere 5 mL di supporti DMEM con il 10% di FBS, supplementazione P/S/Q e 10 mM di piruvato di sodio, con 5 μL di 8 mg/mL di polibrene. Lascia che le cellule incubano durante la notte.
- Al mattino rimuovere i mezzi dalle cellule e le cellule di passaggio nei mezzi integrati con un reagente di selezione. Quando si passano le cellule, seminarle in modo da consentire loro di crescere per 48-72 ore e raggiungere il 50% di confluenza.
NOTA: Per le cellule A673 con pSRP-iEF-2, le cellule vengono seminate in una divisione 1:5 e selezionate per 72 ore con 2 μg/mL di puromicina.
- Trasdurre i costrutti di espressione cDNA.
- Controllare le celle per confermare la confluenza del 50-70%.
- Aliquota virale di scongelamento per la trasduzione di costrutti di cDNA di interesse. Scongelare rapidamente le aliquote congelate a bagnomaria a 37 °C. Aggiungere 2,5 μL di 8 mg/mL di polibrene a ciascuna aliquota virale e mescolare delicatamente pipettando.
- Rimuovere il fluido dalle cellule placcate e aggiungere delicatamente l'aliquota virale alla piastra di 10 cm pipettando lungo il lato della piastra. Scuotere la piastra per diffondere i 2 ml di aliquota virale.
- Incubare a 37 °C nell'incubatore di colture tissutali per 2 ore. Scuotere la piastra ogni 30 minuti per evitare che eventuali aree della piastra si secchino.
- Aggiungere 5 mL di supporti DMEM con il 10% di FBS, supplementazione P/S/Q e 10 mM di piruvato di sodio, con 5 μL di 8 mg/mL di polibrene. Lascia che le cellule incubano durante la notte.
- Al mattino rimuovere i supporti dalle celle e passare le celle in supporti a doppia selezione. Crescere e passare le cellule secondo necessità per 7-10 giorni per consentire la doppia selezione ed espressione del costrutto cDNA.
NOTA: questa suddivisione di questo passaggio potrebbe richiedere l'ottimizzazione per diverse linee di cella. Per le cellule A673 con pSRP-iEF-2 e un costrutto pMSCV-igro, le cellule vengono passate senza dividersi in 2 μg/mL puromicina e 100 μg/mL di igromicina.
2. Raccogliere cellule, convalidare l'espressione di costrutti e impostare saggi fenotipici correlativi
- Dopo 7-10 giorni di doppia selezione raccogliere le cellule in un tubo conico da 15 ml. Conta le cellule raccolte con un emocitometro. Aliquot ha raccolto cellule per il sequenziamento dell'RNA e per convalidare l'espressione di costrutti di cDNA.
NOTA: Impostare eventuali saggi fenotipici correlativi richiesti dalla domanda di ricerca in esame. I saggi di formazione di colonie sono un esempio di un saggio fenotipico correlativo che viene utilizzato qui.- Raccogliere tra 5 x 105 e 1 x 106 cellule per il sequenziamento dell'RNA e 2 x 106 cellule per l'estrazione delle proteine. Celle a pellet mediante centrifugazione a 1.000 x g a 4 °C per 5 minuti e rimuovere il surnatante.
- Lavare il pellet con 1 mL di PBS freddo. Pellet mediante centrifugazione a 1.000 x g a 4 °C per 5 min e rimozione del surnatante. Flash congelare i pellet in azoto liquido e conservare a -80 °C.
- Impostare eventuali saggi correlativi con le cellule rimanenti.
NOTA: Il protocollo può essere messo in pausa qui con i campioni raccolti conservati nel congelatore a -80 °C.
- Convalidare il knockdown della proteina di interesse (se utilizzata) e l'espressione del pannello di costrutti.
- Pellet di cellule di scongelamento per l'estrazione di proteine su ghiaccio. Cellule risospese in tampone di estrazione nucleare ghiacciato da 500 μL (20 mM HEPES pH 7,9, 140 mM NaCl, 10% glicerolo, 1,5 mM MgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) con inibitore della proteasi. Lasciare riposare per 5 minuti sul ghiaccio.
- Nuclei di pellet mediante centrifugazione a 1.000 x g a 4 °C per 5 min e rimozione del surnatante. Lavare i nuclei in tampone di estrazione nucleare ghiacciato da 500 μL (20 mM HEPES pH 7,9, 140 mM NaCl, 10% glicerolo, 1,5 mM MgCl2,1 mM EDTA, 1 mM DTT, 1% IGEPAL) con inibitore della proteasi.
- Nuclei di pellet per centrifugazione a 1.000 x g a 4 °C per 5 min e rimuovere il surnatante. Nuclei risospesi in tampone RIPA freddo da 200 μL con inibitore della proteasi (regolare il volume del tampone RIPA in base alle dimensioni del pellet). Lasciare riposare sul ghiaccio per 45-60 minuti con un vigoroso vortice ogni 15 minuti.
- Detriti di celle a pellet per centrifugazione a 16.000 x g a 4 °C per 45-60 min. Conservare il surnatante e trasferirlo in un tubo freddo fresco
- Preparare i campioni per l'elettroforesi SDS-PAGE facendo bollire 5-10 μg di proteine con 1x tampone di carico per 5 min. Eseguire un gel SDS-PAGE come richiesto per la proteina di interesse.
- Trasferire su una membrana di nitrocellulosa o PVDF secondo necessità per la proteina di interesse. Bloccare e tamponare con gli anticorpi primari e secondari appropriati per confermare l'abbattimento della proteina endogena (se utilizzata) e l'espressione ectopica del costrutto cDNA.
NOTA: il protocollo può essere messo in pausa qui.
- Estratto di RNA. Valutare la qualità e la quantità dell'RNA.
- Scongelare i pellet di cellule sul ghiaccio. Estrarre l'RNA totale utilizzando un kit di estrazione basato su colonna di spin di silice secondo le istruzioni del produttore.
- In breve, lisare le cellule utilizzando il tampone di lisi dal kit. Applicare il lisato a una colonna di spin di silice con una breve rotazione a >13000 rpm per 30-60 secondi o rimuovere il gDNA applicando il lisato a una colonna di rimozione gDNA con una breve rotazione a >13000 rpm per 30-60 secondi.
- Eseguire una digestione del DNA su colonna se il lisato è stato applicato direttamente a una colonna di spin di silice. Se si utilizza una colonna di rimozione gDNA, applicare l'eluato a una colonna di rotazione della silice con una breve rotazione a >13000 rpm per 30-60 s.
- Lavare l'RNA sulla colonna secondo le istruzioni del produttore. RNA eluto in 30 μL di tampone di eluizione.
- Valutare la qualità e la quantità dell'RNA utilizzando un fluorometro o qualsiasi altro strumento comparabile. Assicurarsi che il rapporto 260/280 sia vicino a 2 e che ci siano almeno 2,5 μg di RNA da sottoporre al sequenziamento.
NOTA: man mano che le repliche vengono raccolte, ogni replica deve essere elaborata con lo stesso protocollo di estrazione dell'RNA. - Utilizzare una piccola aliquota di RNA per confermare l'abbattimento stabile della proteina di interesse, se richiesto, mediante qRT-PCR. Conservare il campione di RNA rimanente a -80 °C.
- Raccogli le repliche biologiche ripetendo i passaggi 1-2 fino a quando non sono stati raccolti 3-4 set completi di RNA. Assicurarsi che ogni replica mostri un'espressione adeguata dei costrutti del cDNA e un knockdown stabile della proteina endogena (se utilizzata).
3. Sequenziamento di nuova generazione
- Invia l'RNA estratto per essere sequenziato utilizzando una piattaforma di sequenziamento di nuova generazione con un obiettivo di 50 milioni di letture finali accoppiate a 150 coppie di basi (bp). Seguire le istruzioni della struttura che elabora i campioni. Selezionare per RNA poli-adenilati e sequenziamento specifico del filamento.
4. Pipeline di allineamento e conteggio delle trascrizioni
Nota : questo protocollo presuppone che dopo l'invio e l'elaborazione di esempio, viene restituito un set di file FASTQ accoppiati per ogni campione. Questi file sono spesso compressi con un suffisso di "fastq.gz". Un'ulteriore analisi di questi file FASTQ richiederà l'accesso a una struttura di calcolo ad alte prestazioni (HPC) che esegue un sistema operativo Linux.
- Trasferire file
- Aprire un terminale all'ambiente HPC con PuTTY. Crea una directory per l'analisi chiamata "progetto".
- Passare alla directory "path_to/project" e creare una nuova directory per i file compressi fastq.gz raw chiamata "fastq". Crea anche una directory chiamata "trimmed". Questo è mostrato nella Figura S1A-C.
- Trasferisci i file compressi fastq.gz raw dalla memoria locale alla directory "path_to/project/fastq/" utilizzando WinSCP o un programma simile. Verificare che siano presenti un file "R1" e un file "R2" per ogni campione, come mostrato nella Figura S1B.
- Facoltativo: se necessario, installare TrimGalore. Impostare la directory contenente il file eseguibile trim_galore nella variabile di ambiente PATH in Linux.
NOTA: le letture e gli adattatori di bassa qualità sono rifiniti con TrimGalore. TrimGalore è disponibile presso https://github.com/FelixKrueger/TrimGalore. - Facoltativo: passare alla directory dei pacchetti software scaricati (ad esempio "path_to/software"). Scaricare l'ultimo pacchetto TrimGalore utilizzando il comando "curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[version].tar.gz -o trim_galore-[version].tar.gz".
- Facoltativo: decomprimere il file tar.gz. Usa il comando "tar -xvzf trim_galore-[version_number].tar.gz".
- Opzionale: rendi eseguibile TrimGalore. Utilizzare il comando "chmod a+x path_to/software/TrimGalore-[version]/trim_galore". Assicurarsi che questa nuova directory sia nel PATH. Utilizzare il comando "export PATH=path_to/software/TrimGalore-[version]:$PATH".
- Passare a path_to/project/fastq/. Utilizzare TrimGalore per tagliare le letture di bassa qualità dai file fastq.gz utilizzando il comando mostrato nella Figura S1C.
NOTA: ulteriori flag per questo comando possono essere rilevanti e possono essere trovati qui: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Verificare la presenza dei file fastq.gz tagliati nella directory path_to/project/trimmed. Assicurarsi che siano denominati sample1_R1_val_1.fq.gz e sample1_R2_val_2.fq.gz
- Allinea i file FASTQ tagliati con STAR e genera conteggi di trascrizione.
NOTA: STAR è disponibile presso https://github.com/alexdobin/STAR)- Facoltativo: installare STAR versione 2.6 o successiva. Impostate l'eseguibile STAR nel percorso.
- Facoltativo: passare alla directory dei pacchetti software scaricati (ad esempio "path_to/software").
- Facoltativo: Scaricare il pacchetto STAR utilizzando il comando "curl -SLO https://github.com/alexdobin/STAR/archive/[version].tar.gz". Decomprimere il file tar.gz.
- Facoltativo: utilizzare il comando "tar -xzf [version].tar.gz". Rendi STAR eseguibile. Utilizzare il comando "chmod a+x path_to/software/STAR-[version]/bin".
- Facoltativo: assicurarsi che questa nuova directory sia nel percorso. Utilizzare il comando "export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH".
NOTA: Il manuale STAR è disponibile all'indirizzo: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Assicurarsi che ci sia un indice del genoma da utilizzare con STAR. Inseriscilo in una directory separata dalla directory path_to/project/. Se in precedenza è stato generato un indice per esperimenti precedenti, utilizzalo. In alternativa, utilizzare un indice pregenerato appropriato, se disponibile qui: http://refgenomes.databio.org/. In caso contrario, costruire un nuovo indice utilizzando il comando "STAR--runMode genomeGenerate" utilizzando le istruzioni del manuale STAR.
NOTA: Per il resto di questo protocollo il percorso dell'indice STAR sarà indicato come "path_to/STAR_index". - Passare alla directory path_to/project/. Creare una nuova directory denominata "STAR_output" come mostrato nella Figura S1D.
- Passare alla directory path_to/project/trimmed/. Utilizzare il comando mostrato nella Figura S1D per eseguire STAR per allineare i file fastq.gz tagliati.
NOTA: questo passaggio è il più impegnativo dal punto di vista computazionale e si consiglia di eseguirlo su un cluster HPC con più thread (ad esempio >16) designato per l'attività di allineamento. A seconda del numero di campioni e delle risorse computazionali disponibili, questo passaggio potrebbe richiedere molte ore o giorni. - Trova l'output richiesto per i passaggi successivi che contengono i conteggi per trascrizione nella seguente posizione: path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
Nota : nella colonna 1 del file ReadsPerGene.out.tab contiene informazioni sulla funzionalità da contare. La colonna 2 contiene i conteggi di lettura non instuntati, la colonna 3 contiene i conteggi di lettura a trefoli in avanti e la colonna 4 contiene i conteggi di lettura a trefoli inversi. Le prime quattro righe di questo file avranno informazioni sulle letture allineate che non si allineano a un singolo gene. Questo protocollo richiede i conteggi di lettura senza insida. - Utilizzare RStudio (preferibile) o R nell'ambiente HPC per compilare i dati dalla riga 5 e inferiore per le colonne 1 e 2 per ogni campione. Impostare la directory di lavoro su "project" in R.
- Leggere in ogni file ReadsPerGene.out.tab utilizzando il comando nella Figura S2A. Per la prima colonna, prendi solo i caratteri prima di "." nella colonna "Ensembl gene ID" per facilitare l'elaborazione a valle.
- Compilare i conteggi da tutti gli esempi in un dataframe denominato "totcts" utilizzando i comandi nella Figura S2B. Salvare questa nuova tabella di dati di conteggio grezzi come file .txt delimitato da tabulazioni, ovvero sample_counts.txt, se lo si desidera, utilizzando il comando "write.table".
NOTA: l'ordine dell'ID del gene Ensembl è lo stesso per ogni file ReadsPerGene.out.tab tra i campioni.
5. Espressione differenziale e analisi a valle
- Normalizza per gli effetti batch tra i campioni con ComBat.
NOTA: Ci sono due possibili variabili che spiegano i cambiamenti nell'espressione genica, la prima è il costrutto utilizzato (cioè il campione) e la seconda sono fattori esterni associati al passaggio delle cellule nel tempo (cioè il lotto). Si consiglia di normalizzare i campioni per la variazione da lotto a lotto con il comBat del pacchetto R.- Installa se necessario e carica le librerie per sva, DESeq2, AnnotationDBI, org. Hs.eg.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl, e reshape2 come mostrato nella Figura S2C. Per l'installazione, utilizzare il comando "install.packages" o Bioconductor per la documentazione di ciascun pacchetto.
- Per prima cosa filtra i dati solo per quei geni che hanno almeno un conteggio per lettura. Salvare questa nuova tabella per indicare il filtro come mostrato nella Figura S2D.
NOTA: Spesso, molti geni avranno conteggi di lettura molto bassi o assenti. - Preparare una seconda tabella per la normalizzazione dei lotti chiamata "vars" come mostrato nella Figura S2E. Impostare i nomi delle righe sui nomi univoci di ogni esempio. Impostare i nomi delle colonne su "sample", "batch" e "construct".
- Assegna a tutti i campioni un numero univoco nella colonna "campione" da 1 a n, con n che è il numero di campioni. Assegnare numeri di lotto a tutti i campioni nella colonna "batch" in modo che a_1 condizione e b_1 condizione siano entrambi assegnati 1 e a_2 condizione e b_2 condizione siano entrambi assegnati 2. Assegnare tutte le designazioni di condizione a tutti i campioni nella colonna "costrutto" in modo tale che i campioni di condizione-a siano tutti campioni "A" e i campioni di condizione-b siano tutti "B".
- Definite anche la variabile batch e una matrice di modello nullo specifica per ComBat, come illustrato nella Figura S2F. Eseguire ComBat con il comando definito nella Figura S2F.
- Curare ulteriormente i dati arrotondando al numero intero più vicino. Rimuovere anche i geni con un valore negativo. Utilizzare i comandi riportati nella Figura S3A.
NOTA: l'output della normalizzazione dei batch avrà conteggi di lettura non interi e alcuni geni con valori negativi. Questo passaggio è necessario perché l'analisi dell'espressione differenziale a valle non supporta i conteggi di lettura negativi. - Definire il profilo di espressione differenziale per ogni costrutto utilizzando DESeq2.
- Immettere il progetto dell'esperimento per DESeq2 come mostrato nella Figura S3B. Costruire un DESeqDataSet (dds) utilizzando la funzione DESeqDataSetFromMatrix, stimare i fattori di dimensione ed eseguire DESEq2, come illustrato nella Figura S3B.
Nota : è imperativo che i dati di colonna immessi per "condizione" sia nello stesso ordine della colonna nella matrice di conteggio. - Per valutare la qualità dell'analisi, estrarre i conteggi normalizzati rlog utilizzati da DESeq2 come mostrato nella Figura S3B.
NOTA: Durante l'analisi, DESeq2 trasforma i conteggi con un "log regolarizzato", rlog, trasformazione per ridurre le differenze da campione a campione per i geni con conteggi bassi (informazioni basse) al fine di preservare le differenze nei geni con conteggi più elevati tra i campioni (informazioni elevate). - Quando si estraggono i risultati per ciascun profilo trascrizionale dai risultati di DESeq2, eseguire confronti a coppie in riferimento alla condizione di knockdown o al vettore vuoto della linea di base, come mostrato nella Figura S3C. Modificare ulteriormente questi risultati con i simboli del gene HGNC come mostrato nella Figura S3D.
- Come mostrato nella Figura S3E, estrarre i dati dai risultati DESeq2. Esporta come singolo file con l'ID del gene Ensembl, il simbolo HGNC, l'espressione media di base e i dati di espressione differenziale per tutti i costrutti con log2FoldChange e valori p grezzi e rettificati.
NOTA: l'utilizzo di un valore p rettificato < 0,05 è il limite consigliato per l'espressione differenziale. - Valutare il successo della normalizzazione dei lotti e la somiglianza all'interno del campione. Controllare il clustering del campione con PCA e i grafici a distanza da campione a campione utilizzando i conteggi normalizzati rlog utilizzando il codice illustrato nelle figure S4A-B.
- Immettere il progetto dell'esperimento per DESeq2 come mostrato nella Figura S3B. Costruire un DESeqDataSet (dds) utilizzando la funzione DESeqDataSetFromMatrix, stimare i fattori di dimensione ed eseguire DESEq2, come illustrato nella Figura S3B.
- Utilizzare i profili di espressione differenziale per generare grafici vulcanici utilizzando il codice nella Figura S4C. Valutare i cambiamenti nell'espressione genica tra i costrutti.
- Utilizzare i conteggi normalizzati rlog e il clustering gerarchico per identificare le firme geniche univoche per i diversi costrutti. Utilizzare il codice riportato nella Figura S4D.
- Estrarre i 1000 geni più variabili in tutti i costrutti in una matrice. Utilizzare pheatmap per eseguire il clustering gerarchico non supervisionato dei campioni in base a questi geni.
- Estrarre i cluster di interesse dal dendrogramma decidendo a quale livello dei cluster di dendrogramma di interesse appaiono. Impostare "k" uguale al numero di cluster a quel livello. Riplot la mappa di calore ordinata per cluster per determinare quali cluster sono di interesse, come mostrato nella Figura S5.
- Esportare l'elenco dei geni associati a ciascun cluster come illustrato nella Tabella S1. Utilizzare queste informazioni per determinare i geni nei cluster di interesse.
- Identificare i ruoli biologici per i diversi cluster di geni identificati e confrontare tra le classi. Questo può essere eseguito utilizzando una varietà di strumenti bioinformatici. ToppGene24 è utilizzato qui ed è liberamente disponibile online.
NOTA: Ci sono molti strumenti gratuiti che richiedono solo un elenco di geni da copiare e incollare in un campo su un sito web. Scegli gli strumenti analitici più appropriati per le domande di ricerca in esame. - Facoltativamente, se ci sono dati disponibili sul legame genomico che guida l'output trascrizionale per il fattore di trascrizione di interesse, confrontare la risposta trascrizionale ai geni associati a diversi elementi di legame per valutare ulteriormente la funzione mutante.
6. Confronto con fenotipi rilevanti
- Confrontare i fenotipi correlativi con i dati del profilo trascrittomico generati e interpretarli come appropriato.
Risultati
I dati preliminari di qRT-PCR hanno suggerito che un mutante EWS/FLI chiamato DAF, con specifiche mutazioni da tirosina ad alanina nella regione ripetitiva e disordinata di EWS, ha mantenuto la capacità di attivare i geni bersaglio EWS/FLI, ma non è riuscito a reprimere i geni bersaglio critici23. Al fine di comprendere meglio la relazione tra questi residui nel dominio EWS e la funzione EWS/FLI, è stato utilizzato il protocollo sopra descritto e delineato nella Figura 1. Le cellule del sarcoma di Ewing A673 sono state trasdotte viralmente con uno shRNA mirato al 3'UTR di FLI1,con conseguente esaurimento di EWS/FLI endogeni. Dopo quattro giorni di selezione, la funzione EWS/FLI è stata salvata con la trasduzione virale di diversi costrutti mutanti EWS/FLI marcati 3XFLAG, con vettore vuoto come controllo per nessun salvataggio. Un mutante non funzionale privo del dominio EWS, chiamato Δ22, è stato usato come controllo negativo e EWS/FLI wild-type, chiamato wtEF, è stato usato come controllo positivo (Figura 2A). DAF è stato utilizzato come costrutto di test, anche se è possibile utilizzare più di un costrutto di test, se lo si desidera. Le cellule sono state selezionate per altri 10 giorni per consentire l'espressione del costrutto per stabilizzarsi e quindi raccolte per l'RNA (con una fase di rimozione del gDNA), proteine e saggi di formazione di colonie. Sono state raccolte quattro repliche e qRT-PCR rappresentativi e western blot che mostrano un efficace abbattimento e salvataggio sono mostrati nella Figura 2B-D. Va notato che le cellule salvate da DAF non sono riuscite a formare colonie come mostrato nella Figura 2E,suggerendo una ridotta trasformazione oncogenica.
Dopo il completamento della convalida della replica e dei saggi fenotipici, l'RNA è stato presentato all'Istituto di Medicina Genomica presso il Nationwide Children's Hospital per la preparazione della biblioteca e il sequenziamento di prossima generazione con ~ 50 milioni di letture accoppiate da 150 bp raccolte. I dati sono stati restituiti come file fastq.gz. Le letture di bassa qualità sono state tagliate da questi file con TrimGalore e STAR è stato utilizzato per allineare le letture al genoma umano hg19 e contare le letture per gene. hg19 è stato utilizzato ai fini della compatibilità con gli altri set di dati curati per EWS / FLI utilizzati nell'analisi a valle. Questi conteggi di lettura sono stati combinati in un'unica matrice di conteggio per tutti i campioni, le prime 6 righe delle quali sono mostrate nella Figura 3.
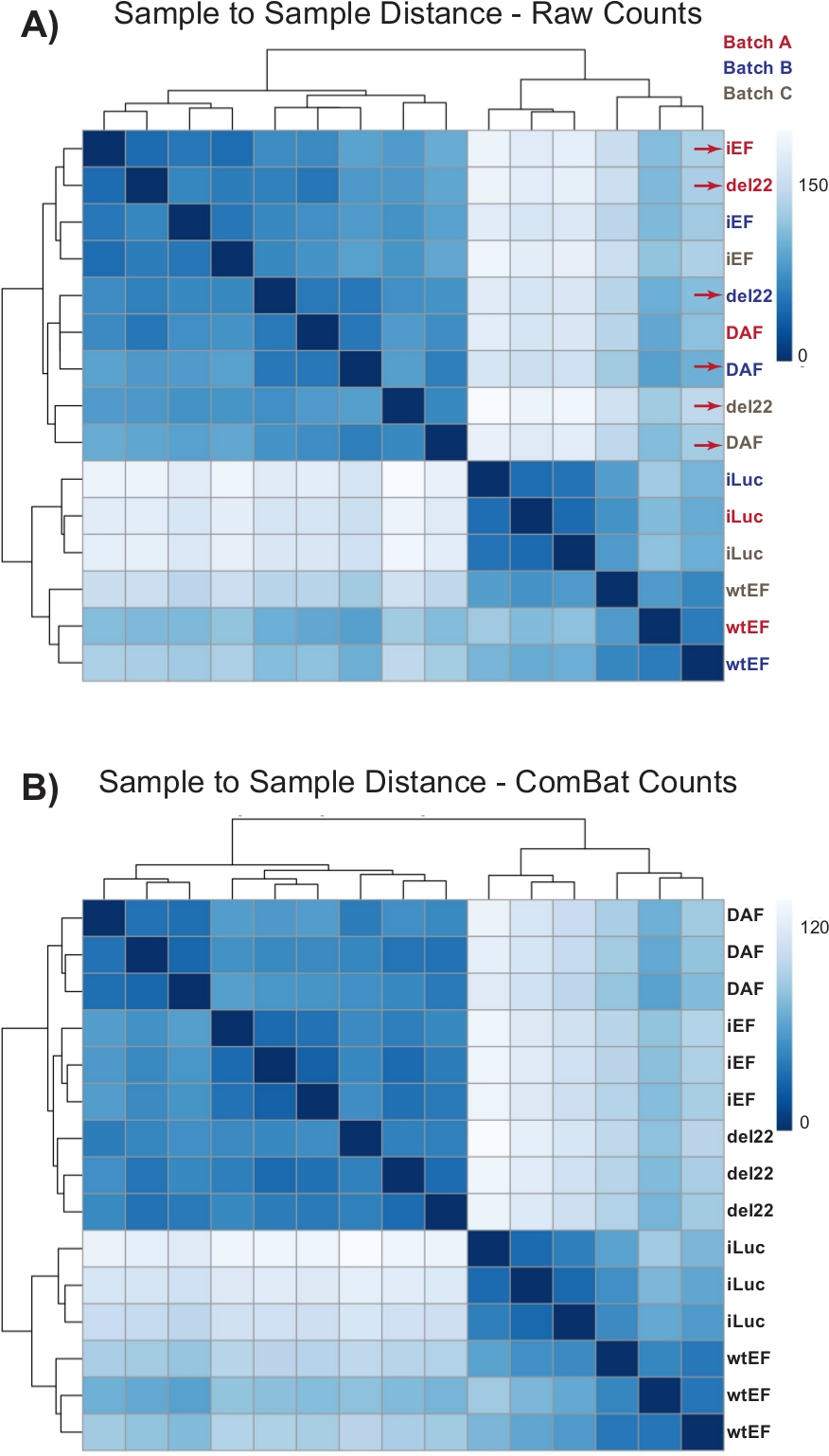
I conteggi sono stati inizialmente eseguiti attraverso DESeq2 senza normalizzazione del lotto, tuttavia, l'ispezione visiva della distanza da campione a campione ha mostrato potenziali effetti di confusione del lotto come mostrato evidenziato con frecce rosse nella Figura 4A. Ciò probabilmente è sorto a causa della variabilità biologica introdotta dal passaggio delle cellule in coltura e delle differenze nella lavorazione di ciascun lotto. La normalizzazione per gli effetti batch è stata eseguita con ComBat ed è generalmente raccomandata. Le distanze da campione a campione dei dati normalizzati dal lotto sono mostrate nella Figura 4B. Dopo la normalizzazione del batch, DESeq2 è stato utilizzato per generare profili trascrizionali per i tre costrutti (wtEF, Δ22 e DAF) relativi alla linea di base. Si noti che mentre le cellule A673 "parentali" (mock knockdown e mock rescue, chiamate "iLuc" qui) sono state incluse nell'analisi differenziale, il riferimento per questo esperimento sono le cellule con EWS / FLI-depleted, chiamate cellule iEF. Il profilo trascrizionale può essere generato per la proteina endogena qui confrontando il campione iLuc con iEF, e questo può essere utile per capire come funziona il sistema di salvataggio, ma questo non è l'obiettivo di questa particolare analisi. I profili trascrizionali generati per i mutanti includono controlli positivi (wtEF) e negativi (Δ22), rispetto all'iEF, in modo tale che questi dovrebbero funzionare come parametri di riferimento per altri mutanti. Questo è importante, poiché il controllo positivo in questo esempio non ha completamente ricapitolato la funzione di EWS/FLI endogena come discusso altrove7,23.
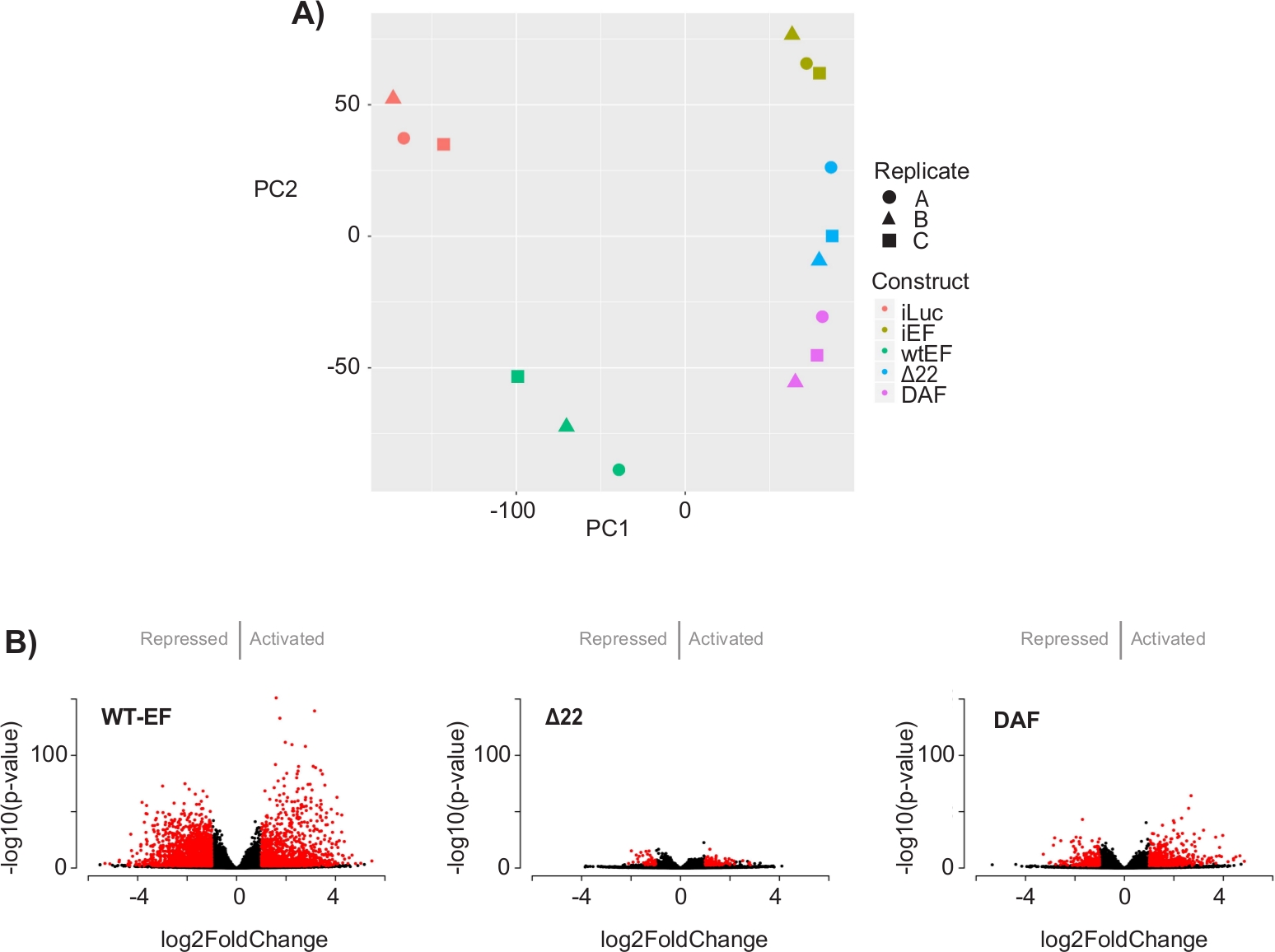
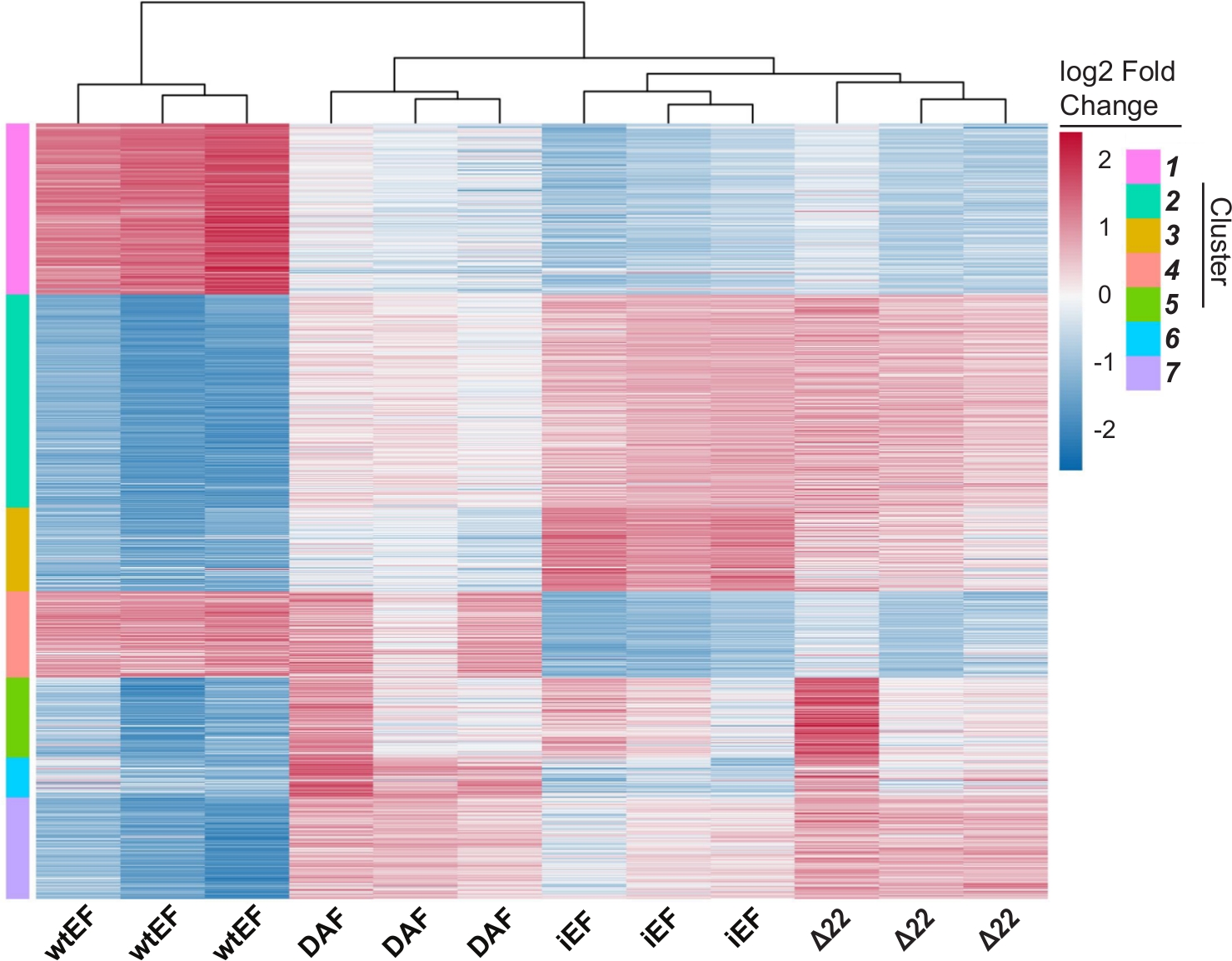
L'analisi dei componenti principali (PCA) nella Figura 5 suggerisce che il profilo trascrizionale del DAF è intermedio tra wtEF e Δ22, confermando la funzione parziale. Inoltre, il raggruppamento gerarchico dei 1000 geni più variabili tra i campioni ha mostrato che DAF non è riuscito a reprimere i geni bersaglio EWS / FLI e ha mantenuto solo parzialmente l'attività di attivazione genica come mostrato in Figura 6A e Figura S5. L'analisi di ToppGene ha suggerito che le classi di geni attivate da DAF sono funzionalmente distinte da quei bersagli attivati da EWS/FLI in cui DAF non è funzionale (Figura 6B). È interessante notare che la funzione dei geni attivati salvati da wtEF, ma non da DAF, sembra essere correlata al controllo trascrizionale e alla regolazione della cromatina. Sulla base dei risultati dei test di formazione delle colonie, i geni di questa firma genica di base dovrebbero essere ulteriormente analizzati per il loro ruolo nell'oncogenesi mediata da EWS / FLI. L'importanza della repressione genica mediata da EWS/FLI è stata precedentemente descritta17.
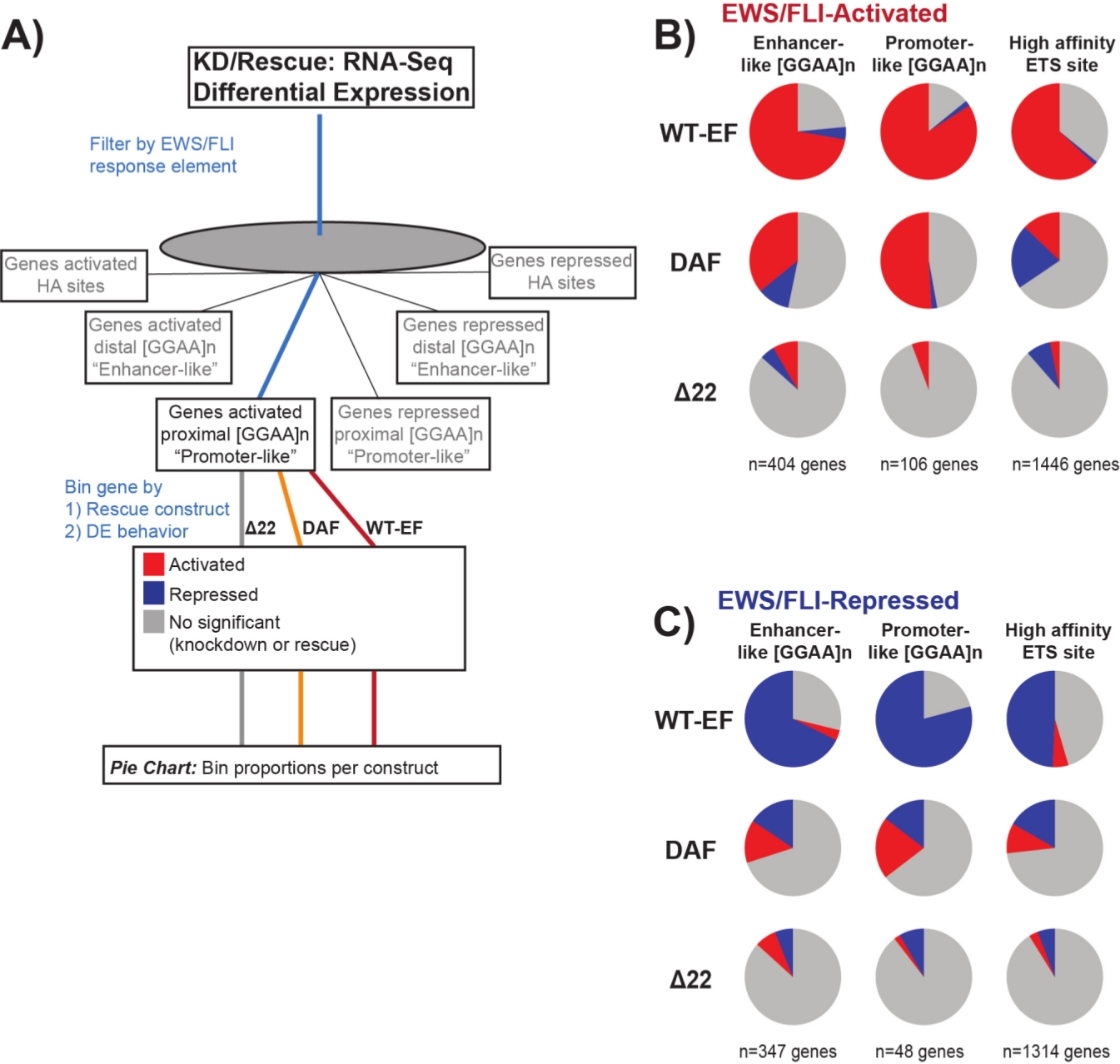
È noto che EWS/FLI possiede un'affinità di legame unica per gli elementi di ripetizione GGAA-microsatellite19,22,e che il legame a questi elementi guida la regolazione genica a valle11,15,18,20,22. Questi microsatelliti sono stati caratterizzati come associati all'attivazione o alla repressione e prossimali a (< 5 kb) TSS o distali a (> 5 kb) TSS25. Inoltre, ci sono geni regolati da EWS / FLI con motivi ETS ad alta affinità (HA) prossimali a TSS23. Al fine di analizzare ulteriormente le caratteristiche della funzione DAF e quali tipi di geni attivati da EWS / FLI DAF è stato in grado di salvare, è stata analizzata l'espressione differenziale dei geni associati a queste diverse classi. È interessante notare che DAF è stato più in grado di salvare i geni attivati dai microsatelliti GGAA, ma non è stato in grado di salvare i geni attivati vicino a un sito HA come visto nella Figura 7. Come si è visto con il clustering gerarchico, DAF non riesce a salvare la repressione mediata da EWS / FLI tra le classi di motivi. Questi dati suggeriscono che DAF mantiene caratteristiche strutturali sufficienti di EWS per legarsi e attivarsi dai microsatelliti GGAA, sia prossimali che distali a TSS. Questo probabilmente deriva dal dominio SYGQ intatto ritenuto importante per l'attività EWS / FLI alle ripetizioni GGAA11. Questi dati suggeriscono anche che le tirosine specifiche mutate in DAF svolgono ruoli importanti, ma poco compresi, nella regolazione genica mediata da EWS / FLI dai siti HA, nonché nella repressione genica, evidenziando un'importante area di ulteriori indagini.

Figura 1: Flusso di lavoro. Rappresentazione della procedura passo-passo per eseguire la mappatura struttura-funzione mediante trascrittomica. Le celle sono state preparate per la prima volta per esprimere la suite di costrutti necessari per la mappatura struttura-funzione. Dopo l'espressione, le cellule sono state raccolte per RNA e proteine e analizzate per fenotipi correlativi. L'espressione dei costrutti è stata convalidata e questo processo è stato ripetuto 3-4 volte per raccogliere repliche biologiche indipendenti. L'RNA è stato quindi sottoposto al sequenziamento di nuova generazione (NGS). Quando i dati sono stati ricevuti, i dati sono stati tagliati per la qualità, allineati e sono stati calcolati i conteggi per trascrizione. Gli effetti batch sono stati controllati e le firme trascrittomiche e l'espressione differenziale sono state determinate utilizzando DESeq2. È possibile incorporare il clustering gerarchico e l'analisi a valle che integrano altri set di dati -omics e diversi percorsi o analisi funzionali. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Validazione dell'espressione del costrutto e dei saggi correlativi. (A) Schema raffigurante i costrutti testati in questo esempio. (B) Validazione del knockdown di EWS/FLI endogeno ed espressione di costrutti marcati 3X-FLAG mediante immunoblot. (C,D) Validazione dell'attività di costruzione in un gene bersaglio attivato EWS/FLI(C), NR0B1,e(D)gene bersaglio represso, TGFBR2,mediante qRT-PCR. I dati sono presentati come deviazione media +/- standard. I valori P sono stati calcolati con un test di significato onesto di Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005 (E) Conteggi delle colonie da saggi soft-agar eseguiti per valutare l'attività di trasformazione dei costrutti. I valori P sono stati calcolati con un test di significato onesto di Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005. Questa figura è adattata da Theisen, et al.23Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Dati finali di conteggio raccolti per l'analisi. Screenshot delle prime 6 righe del file di conteggio con conteggi genetici per tutti i campioni da normalizzare e analizzare in batch. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Mappe di calore della distanza da campione a campione. (A) Grafico della distanza da campione a campione che mostra il raggruppamento del campione dei dati grezzi del conteggio. I campioni che vengono raggruppati sia per batch che per campione sono indicati con frecce rosse. (B) Diagramma di distanza da campione a campione dopo la normalizzazione del lotto con ComBat. Qui, i campioni di tutte le repliche si raggruppano insieme, indipendentemente dal batch. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Risultati dell'analisi delle espressioni differenziali. (A) Il grafico dell'analisi delle componenti principali (PCA) delle firme trascrittomiche generate per tutti i campioni mostra un forte raggruppamento intracampionario e dimostra che il DAF è intermediato tra i controlli positivo (wtEF) e negativo (Δ22). (B) Grafici vulcanici che mostrano il -log(p-value) tracciato contro il log2FoldChange per i geni in ogni costrutto. Geni con un valore p aggiustato < 0,05 e un |log2(FoldChange)| > 1 sono considerati significativi e sono mostrati in rosso. Il pannello 5B è adattato da Theisen, et al.23Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Clustering gerarchico per identificare le classi geniche. (A) Il clustering gerarchico dei primi 1000 geni più variabili in tutti i costrutti e la linea di base, iEF, mostra che DAF salva parzialmente l'attivazione genica mediata da EWS / FLI. (B)L'ontologia genica (funzione molecolare) è il risultato di ToppGene che mostra l'arricchimento funzionale dei geni attivati da EWS/FLI che vengono salvati o non salvati dal DAF. Il pannello 6B è adattato da Theisen, et al.23Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 7: Analisi dettagliata di diversi elementi di risposta del fattore di trascrizione a diversi costrutti: (A) Schematico raffigurante l'elaborazione dei dati utilizzata per generare pannelli (B) e (C) incorporando altri set di dati disponibili con i profili trascrittomici qui. (B,C) Compilazione che mostra il salvataggio di diverse classi di bersagli diretti EWS/FLI- (B) attivati e (C) repressi. I geni inclusi erano solo quei geni con espressione differenziale rilevabile da EWS/FLI endogena. In ogni grafico a torta, il grigio raffigura la porzione di geni che non vengono salvati dal costrutto. Il rosso raffigura la porzione di geni che sono attivati in modo differenziale e il blu raffigura la porzione di geni che sono differenzialmente repressi. Questa figura è adattata da Theisen, et al.23Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Figura S1: Caricamento dei file fastq.gz nell'ambiente HPC, ritaglio e allineamento. Fare clic qui per scaricare questa figura.
{kind=link}
Figura S2: Confronto dei conteggi di lettura tra i campioni ed esecuzione della normalizzazione batch con ComBat. Fare clic qui per scaricare questa figura.
{kind=link}
Figura S3: Esecuzione di DESeq2 ed estrazione dei risultati dell'analisi delle espressioni differenziali. Fare clic qui per scaricare questa figura.
{kind=link}
Figura S4: Analisi dell'output. Fare clic qui per scaricare questa figura.
{kind=link}
Figura S5: Clustering gerarchico per identificare le classi geniche: Raggruppamento gerarchico dei primi 1000 geni più variabili in tutti i costrutti e la linea di base, iEF, ordinati in k cluster. In questo caso k=7, ma questo parametro viene impostato dall'utente come mostrato nella Figura S4D. Fare clic qui per scaricare questa figura.
{kind=link}
Tabella S1: Elenco dei geni (ID gene Ensembl) con annotazione a cluster. Fare clic qui per scaricare questa tabella.
Discussione
Studiare i meccanismi biochimici dei fattori di trascrizione oncogenici è di fondamentale importanza per comprendere le malattie che causano e per progettare nuove strategie terapeutiche. Ciò è particolarmente vero nelle neoplasie maligne caratterizzate da traslocazioni cromosomiche con conseguente fattori di trascrizione della fusione. I domini inclusi in queste proteine chimeriche possono mancare di interazioni significative con i domini regolatori presenti nelle proteine wild-type, complicando la capacità di interpretare le informazioni struttura-funzione nel contesto della fusione26,27,28. Inoltre, molte di queste fusioni oncogeniche sono caratterizzate da domini intrinsecamente disordinati a bassa complessità10,13,29,30.
Il dominio EWS è un esempio di un dominio intrinsecamente disordinato che è coinvolto in una varietà di fusioni oncogeniche10. La natura intrinsecamente disordinata e ripetitiva ha ostacolato gli sforzi per comprendere i meccanismi molecolari impiegati dal dominio EWS. Gli sforzi precedenti per studiare la struttura-funzione hanno in gran parte fatto ricorso all'uso di diversi mutanti nel contesto di saggi genici reporter o in background cellulari che non riescono a ricapitolare il contesto cellulare pertinente, o mancano di variazioni strutturali che producono una funzione parziale significativa11,17,25. Il metodo qui presentato affronta questi problemi. La mappatura struttura-funzione viene eseguita in un contesto cellulare rilevante per la malattia e il sequenziamento di nuova generazione consente la profilazione trascrittomica per valutare la funzione del fattore di trascrizione nel setting della cromatina nativa. Nel caso specifico del mutante DAF di EWS/FLI, DAF è stato segnalato per mostrare poca attività nei saggi reporter utilizzando elementi di risposta isolati, ma per mostrare attività nel contesto del promotore genetico completo, sia in un test reporter che in cromatina nativa, suggerendo un fenotipointeressante 23. L'uso del metodo qui descritto risolve più direttamente la questione di quale tipo di elementi regolatori attraverso il genoma sono più reattivi nel contesto della malattia. Testando contemporaneamente tutti i geni bersaglio candidati nel loro contesto nativo di cromatina, un approccio trascrittomico ha maggiori probabilità di identificare costrutti con funzione parziale.
La forza intrinseca dell'utilizzo di uno sfondo cellulare rilevante per la malattia è forse il più grande limite di questa tecnica. Uno dei fattori più importanti è la scelta del sistema cellulare appropriato per questi esperimenti. Molte linee cellulari derivate da tumori maligni con fattori di trascrizione patognomonici non tollerano prontamente l'abbattimento di quel fattore di trascrizione, e in molti casi, in particolare per i tumori pediatrici, la vera cellula di origine rimane controversa e l'espressione dell'oncogene in altri background cellulari è proibitivamente tossica31,32 . In questi casi, può essere utile eseguire esperimenti in un diverso background cellulare, purché il ricercatore eserciti cautela nell'interpretazione dei risultati e convalidi in modo appropriato qualsiasi risultato rilevante in un tipo di cellula più rilevante per la malattia.
È di fondamentale importanza convalidare attentamente la stabilità e le conseguenze fenotipiche dell'espressione dell'oncogene e presentare solo campioni per il sequenziamento che soddisfano criteri rigorosi. Qui, questo includeva western blot per confermare l'abbattimento e il salvataggio e qRT-PCR di un piccolo numero di geni bersaglio noti per convalidare il controllo positivo (Figura 2). Allo stesso modo è fondamentale ridurre la massima variabilità possibile dei lotti eseguendo attentamente i preparati cellulari e di RNA nel modo più simile possibile attraverso ciascun lotto.
Il metodo qui descritto diventa particolarmente potente se abbinato ad altri tipi di dati genomici che parlano della funzione genome-wide del fattore di trascrizione in studio. Le direzioni future per questo tipo di analisi struttura-funzione si espanderebbero per includere ChIP-seq e ATAC-seq per determinare il legame del fattore di trascrizione e qualsiasi cambiamento indotto nell'accessibilità della cromatina. Come suite, questo tipo di dati può indicare dove diversi componenti strutturali di un fattore di trascrizione oncogenico contribuiscono a diversi aspetti della funzione (cioè legame al DNA vs modifica della cromatina vs reclutamento co-regolatore). Nel complesso, l'utilizzo di approcci basati su NGS per mappare le relazioni struttura-funzione dei fattori di trascrizione della fusione può rivelare nuove intuizioni nei determinanti biochimici della funzione oncogenica di queste proteine. Questo è importante per approfondire la nostra comprensione delle malattie che causano e per consentire lo sviluppo di nuove strategie terapeutiche.
Divulgazioni
SLL dichiara un conflitto di interessi in qualità di membro del comitato consultivo e azionista di Salarius Pharmaceuticals. SLL è anche un inventore elencato su United States Patents No. US 7.393.253 B2, "Metodi e composizioni per la diagnosi e il trattamento del sarcoma di Ewing", e US 8.557.532, "Diagnosi e trattamento del sarcoma di Ewing resistente ai farmaci". Ciò non altera la nostra aderenza alle politiche di JoVE sulla condivisione di dati e materiali.
Riconoscimenti
Questa ricerca è stata supportata dalla High Performance Computing Facility presso l'Abigail Wexner Research Institute del Nationwide Children's Hospital. Questo lavoro è stato supportato dal National Institutes of Health National Cancer Institute [U54 CA231641 a SLL, R01 CA183776 a SLL]; Alex's Lemonade Stand Foundation [Young Investigator Award a ERT]; Pelotonia [Fellowship to ERT]; e il National Health and Medical Research Council CJ Martin Overseas Biomedical Fellowship [APP1111032 a KIP].
Materiali
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
Riferimenti
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932(2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275(2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805(2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552(2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800(2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati