
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Abilitazione della compensazione in tempo reale nelle ossidazioni fotochimiche veloci delle proteine per la determinazione dei cambiamenti della topografia proteica
In questo articolo
Riepilogo
L'ossidazione fotochimica rapida delle proteine è una tecnica emergente per la caratterizzazione strutturale delle proteine. Diversi additivi solventi e ligand hanno varie proprietà di scavenging radicale idrossile. Per confrontare la struttura della proteina in diverse condizioni, è necessaria una compensazione in tempo reale dei radicali idrossili generati nella reazione per normalizzare le condizioni di reazione.
Abstract
L'ossidazione fotochimica rapida delle proteine (FPOP) è una tecnica di biologia strutturale basata sulla spettrometria di massa che sonda l'area superficiale delle proteine accessibile dal solvente. Questa tecnica si basa sulla reazione delle catene laterali di amminoacidi con radicali idrossili che si diffonde liberamente in soluzione. FPOP genera questi radicali in situ con fotolisi laser di perossido di idrogeno, creando una raffica di radicali idrossili che si esaurisce nell'ordine di un microsecondo. Quando questi radicali idrossili reagiscono con una catena laterale di amminoacidi accessibile al solvente, i prodotti di reazione presentano uno spostamento di massa che può essere misurato e quantificato dalla spettrometria di massa. Poiché il tasso di reazione di un amminoacido dipende in parte dalla superficie media accessibile al solvente di tale amminoacido, i cambiamenti misurati nella quantità di ossidazione di una determinata regione di una proteina possono essere direttamente correlati ai cambiamenti nell'accessibilità del solvente di tale regione tra diverse conformazioni (ad esempio, legante-bound contro ligando-free, monomer vs. aggregato, ecc.) FPOP è stato applicato in una serie di problemi in biologia, tra cui interazioni proteina-proteina, cambiamenti conformazionali delle proteine e legame proteina-ligando. Poiché la concentrazione disponibile di radicali idrossili varia in base a molte condizioni sperimentali nell'esperimento FPOP, è importante monitorare la dose radicale efficace a cui è esposto l'alyte proteico. Questo monitoraggio viene ottenuto in modo efficiente incorporando un dosimetro in linea per misurare il segnale dalla reazione FPOP, con fluenza laser regolata in tempo reale per ottenere la quantità desiderata di ossidazione. Con questa compensazione, i cambiamenti nella topografia delle proteine che riflettono i cambiamenti conformazionali, le superfici leganti i ligando e/o le interfacce proteina-proteina possono essere determinati in campioni eterogenei utilizzando quantità di campioni relativamente basse.
Introduzione
L'ossidazione fotochimica rapida delle proteine (FPOP) è una tecnica emergente per la determinazione dei cambiamenti topografici delle proteine mediante modifica covalente ultraveloci della superficie delle proteine esposta al solvente seguita dal rilevamento da parte di LC-MS1. FPOP genera un'alta concentrazione di radicali idrossili in situ dalla fotolisi laser UV del perossido di idrogeno. Questi radicali idrossili sono molto reattivi e di breve durata, consumati su una scala cronologica di circa un microsecondo in condizioni FPOP2. Questi radicali idrossili si diffondono attraverso l'acqua e ossidano vari componenti organici in soluzione a velocità cinetiche generalmente che vanno da veloce (106 M-1 s-1) a diffusione controllata3. Quando il radicale idrossile incontra una superficie proteica, il radicale ossiderà le catene laterali degli amminoacidi sulla superficie proteica, con conseguente spostamento di massa di tale aminoacido (più comunemente l'aggiunta netta di un atomo di ossigeno)4. Il tasso della reazione di ossidazione a qualsiasi aminoacido dipende da due fattori: la reattività intrinseca di tale aminoacido (che dipende dalla catena laterale e dal contesto di sequenza)4,5 el'accessibilità di tale catena laterale al radicale idrossile di diffusione, che è strettamente correlata alla superficie accessibile del solventemedio 6,7. Tutti gli aminoacidi standard, ad eccezione della glicina, sono stati osservati come etichettati da questi radicali idrossili altamente reattivi negli esperimenti FPOP, anche se a rese ampiamente diverse; in pratica, Ser, Thr, Asn e Ala sono raramente visti come ossidati nella maggior parte dei campioni tranne che sotto alte dosi radicali e identificati da un'attenta e sensibile frammentazione ETDmirata 8,9. Dopo l'ossidazione, i campioni vengono sfociati per rimuovere il perossido di idrogeno e gli ossidanti secondari (superossido, ossigeno singlet, idroperossido di peptidile, ecc.) I campioni spagolati vengono quindi digeriti proteolyticallymente per generare miscele di peptidi ossidati, dove le informazioni strutturali vengono congelate come "istantanea" chimica nei modelli dei prodotti di ossidazione dei vari peptidi (Figura 1). La cromatografia liquida accoppiata alla spettrometria di massa (LC-MS) viene utilizzata per misurare la quantità di ossidazione degli amminoacidi in un dato peptide proteolitico in base alle intensità relative delle versioni ossidizzate e nonidizzate di quel peptide. Confrontando questa impronta ossidativa della stessa proteina ottenuta in diverse condizioni conformazionali (ad esempio, legante-bound contro ligando-free), differenze nella quantità di ossidazione di una determinata regione della proteina possono essere direttamente correlate con le differenze nella superficie accessibile al solvente di quella regione6,7. La capacità di fornire informazioni topografiche proteiche rende FPOP una tecnologia attraente per la determinazione della struttura di ordine superiore delle proteine, anche nella scoperta terapeutica delle proteinee nello sviluppo 10,11.

Figura 1: Panoramica di FPOP. La superficie della proteina è covallentamente modificata dai radicali idrossili altamente reattivi. I radicali idrossili reagiranno con catene laterali di amminoacidi della proteina ad un tasso fortemente influenzato dall'accessibilità del solvente della catena laterale. I cambiamenti topografici (ad esempio, a causa del legame di un ligando come mostrato sopra) proteggeranno gli aminoacidi nella regione di interazione dalla reazione con i radicali idrossili, con conseguente diminuzione dell'intensità del peptide modificato nel segnale LC-MS. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Diversi costituenti presenti nella soluzione FPOP (ad esempio, ligando, escipienti, tamponi) hanno un'attività di scavenging diversa verso i radicali idrossili generati sulla fotolisi laser del perossido di idrogeno3. Allo stesso modo, un piccolo cambiamento nella concentrazione di perossido, nella fluenza laser e nella composizione del buffer può cambiare la dose radicale efficace, rendendo la riproduzione dei dati FPOP impegnativa tra i campioni e tra diversi laboratori. Pertanto, è importante essere in grado di confrontare la dose radicale idrossile disponibile per reagire con proteine in ogni campione utilizzando uno dei diversi dosimetri radicali idrossilidisponibili 12,13,14,15,16. I dosimetri radicali idrossili agiscono competendo con l'alyte (e con tutti gli spazzini in soluzione) per la piscina di radicali idrossili; la dose efficace di radicali idrossili viene misurata misurando la quantità di ossidazione del dosimetro. Si noti che "dose efficace idrossile radicale" è una funzione sia della concentrazione iniziale di idrossile radicale generato e l'e half-life del radicale. Questi due parametri sono parzialmente dipendenti l'uno dall'altro, rendendo la modellazione cinetica teorica piuttosto complessa (Figura 2). Due campioni potrebbero avere esigete radicali iniziali molto diverse pur mantenendo la stessa dose radicale efficace modificando la concentrazione iniziale di idrossilo radicale formato; genereranno ancora impronte identiche17. Adenine13 e Tris12 sono dosmetri radicali idrossili convenienti perché il loro livello di ossidazione può essere misurato dalla spettroscopia UV in tempo reale, consentendo ai ricercatori di identificare rapidamente quando c'è un problema con dose radicale idrossile efficace e di risolvere il loro problema. Per risolvere questo problema, un dosimetro in linea situato nel sistema di flusso direttamente dopo il sito di irradiazione in grado di monitorare il segnale da cambiamenti di assorbimento dell'adenina in tempo reale è importante. Questo aiuta a effettuare esperimenti FPOP in buffer o qualsiasi altro excipient con livelli ampiamente diversi di capacità di scavenging radicale idrossile17. Questa compensazione radicale dosaggio può essere eseguita in tempo reale, producendo risultati statisticamente indistinguibili per lo stesso conformere regolando la dose radicale efficace.
In questo protocollo, abbiamo procedure dettagliate per l'esecuzione di un tipico esperimento FPOP con compensazione radicale del dosaggio utilizzando l'adenina come dosimetro radicale ottico interno. Questo metodo consente agli investigatori di confrontare le impronte tra condizioni FPOP che hanno capacità di scavenging diverse eseguendo una compensazione in tempo reale.

Figura 2: simulazione cinetica della compensazione basata sulla dosimetria. La risposta di dosimetro dell'adenina da 1 mM è misurata in un alyte di 5 M di lysozyme con una concentrazione iniziale di idrossile radicale di 1 mM (▪OH t1/2x53 ns) e impostata come risposta dosimetrica target (nero). Con l'aggiunta di 1 mM dell'estito di scavenger, la risposta dosimetro (blu) diminuisce insieme alla quantità di ossidazione proteica in modo proporzionale (ciano). Anche l'emita vita del radicale idrossile diminuisce (▪OH t1/2-39 ns). Quando la quantità di idrossile radicale generato viene aumentata per dare una resa equivalente di dosimetro ossidato nel campione con scavenger istidina di 1 mM come raggiunto con 1 mM idrossile radicale in assenza di scavenger (rosso), la quantità di ossidazione proteica che si verifica in modo simile diventa identica (magenta), mentre l'idryl radicale half-life diminuisce ancora di più (▪OH t1/2= Adattato con il permesso di Sharp J.S., Am Pharmaceut Rev 22, 50-55, 2019. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Protocollo
1. Preparare la panca ottica e il capillaro per FPOP
CAUTION: I laser ad escimera KrF sono ostacoli agli occhi estremi e la luce diretta o riflessa può causare danni permanenti agli occhi. Indossare sempre un'adeguata protezione oculare, evitare la presenza di oggetti riflettenti vicino al percorso del fascio quando possibile e utilizzare i controlli di ingegneria per impedire l'accesso non autorizzato a un laser attivo e per frenare eventuali riflessi vaganti.
- Preparare il banco ottico FPOP.
- Accendere il laser per riscaldarsi. Impostare il laser su Trigger esterno, Energia costante, Nessuna sostituzione del gas. Impostare l'energia laser per impulso (in genere tra 80-120 mJ/impulso).
- Impostare il banco ottico con l'obiettivo plano-convesso (30 mm Dia. x 120 mm FL non rivestito) direttamente nel percorso del fascio laser e un backstop non riflettente per assorbire la luce come mostrato nella figura 3A.

Figura 3: Banco ottico per l'esperimento FPOP. (A) Il campione viene mescolato con H2O2, dosimetro radicale dell'adenina e spazzino della glutammina e caricato nella siringa. Il campione viene spinto attraverso il capillare di silice fuso attraverso il percorso del fascio focalizzato di un laser UV KrF excimer. La luce UV fotolizza H2O2 in radicali idrossili, che ossidano la proteina e il dosimetro dell'adenina. Il flusso della siringa spinge il campione illuminato fuori dal percorso del laser prima del successivo impulso laser, con un volume di esclusione non illuminato tra le regioni illuminate. Immediatamente dopo l'ossidazione, il campione viene passato attraverso uno spettrofotometro UV in linea, che misura l'assorbimento UV dell'adenina a 265 nm. Il campione viene quindi depositato in un buffer di quench per eliminare i restanti H2O2 e gli ossidanti secondari. (B) La dimensione del punto viene misurata dopo l'irradiazione di una nota adasi colorata apposta dietro il capillare con il laser a 248 nm. La larghezza del punto viene utilizzata per calcolare la frequenza di flusso del campione, e la silhouette del capillare al centro del punto viene utilizzata per allineare il banco ottico. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Tagliare una lunghezza appropriata del capillare di silice fusa (diametro esterno di 360 m e diametro interno di 100 m) e utilizzando un manicotto, collegare alla siringa a gas-stretto utilizzando un connettore a basso volume morto.
- Bruciare delicatamente il rivestimento in poliimide del capillare con una torcia di butano nel punto in cui il dosimetro in linea legge il segnale di assorbimento a 265 nm dopo l'esposizione laser dei campioni. Pulire delicatamente i detriti sul capillare usando il metanolo su una salvietta senza lanugine. Il rivestimento in poliimide nel sito di incidenza laser può essere bruciato in modo simile con la torcia butano o bruciato con la cottura laser ad escimero a bassa potenza.
NOTA: Attendere che il capillare si raffredderà in quanto è un pericolo di incendio usare il metanolo sul capillare caldo. - Posizionare questo capillare attraverso il percorso del raggio del laser e nel dosimetro in linea.
- Premere la leva sulla parte superiore del dosimetro in linea per aprire la cerniera. Rimuovere i supporti magnetici. Posizionare il capillare nella scanalatura computerata del dosimetro in linea, utilizzando i supporti magnetici per mantenere il capillare in posizione. Chiudere la cerniera dosimetro sopra il capillare, premendolo fino a quando la leva si blocca in posizione.
- Utilizzando il software dosimetry, fare clic sul pulsante Start Flash per iniziare a sparare il laser escimero. Impostare la potenza laser preimpostata tra 50-100 mJ/pulse sul software di controllo laser stesso e impostare la velocità di ripetizione preimpostata tra 10-20 Hz nella scheda Impostazioni del software dosimetria.
- Mettere a fuoco il raggio laser utilizzando una lente plano convessa montata su uno stadio motorizzato lineare. Misurare la larghezza e l'altezza dello spot laser nella posizione del capillare su una nota adesa utilizzando con precisione una pinza per calcolare la fluenza incidente (mJ/mm2), come illustrato nella figura 3B.
- Posizionare un'apertura opaca vicino al capillare per garantire una larghezza illuminata costante del capillare indipendentemente dai cambiamenti nella dimensione del fascio a causa del movimento della lente o cambiando l'energia per impulso del laser18.
- Con la cottura laser, spostare lo stadio motorizzato attraverso la sua gamma di movimento. Assicurarsi che il fascio rimanga centrato sull'apertura e che la sagoma del capillare possa essere osservata in tutto. Il diametro dell'apertura deve essere inferiore alla larghezza del fascio focalizzato in ogni punto della gamma dello stadio motorizzato.
- Scorrere l'acqua attraverso il capillare a 20 L/min per almeno un minuto per lavare il capillare.
- Fare clic sul pulsante Start Data sulla barra degli strumenti Strumenti di stampa per eseguire la raccolta dei dati.
NOTA: se il sistema di buffer per FPOP ha un'assorbimento UV significativa a 265 nm, il sistema FPOP deve essere azzerato sul buffer, non sull'acqua.
- Fare clic sul pulsante Start Data sulla barra degli strumenti Strumenti di stampa per eseguire la raccolta dei dati.
- Impostare la velocità di flusso calcolata sulla pompa di siringa.
- La velocità di flusso del campione proteico dipende dal volume irradiato per colpo (VIrr), dal numero di colpi laser al secondo (R) e dalla frazione del volume di esclusione nonrradiata desiderata (FEx) per correggere gli effetti del flusso laminare e la diffusione del campione (0,15-0,30 consigliato)2,19,20. Calcolare il VIrr (in L) in base al diametro interno del capillare in mm (d) e alla larghezza dello spot laser che impatte sul capillare (cioè la larghezza dell'apertura) in mm (w) utilizzando la seguente equazione:
VIrr : π (d/2)2w - Calcolare la velocità di flusso desiderata (in L/min) in base alla seguente equazione:
Flusso 60 R[VIrr (1 - FEx)]
- La velocità di flusso del campione proteico dipende dal volume irradiato per colpo (VIrr), dal numero di colpi laser al secondo (R) e dalla frazione del volume di esclusione nonrradiata desiderata (FEx) per correggere gli effetti del flusso laminare e la diffusione del campione (0,15-0,30 consigliato)2,19,20. Calcolare il VIrr (in L) in base al diametro interno del capillare in mm (d) e alla larghezza dello spot laser che impatte sul capillare (cioè la larghezza dell'apertura) in mm (w) utilizzando la seguente equazione:
2. Preparazione della soluzione proteica per FPOP
- Preparare la proteina in due o più condizioni diverse da confrontare (ad esempio, legante-bound e ligand-free; aggregato e monomero; da solo e con un partner vincolante proteina-proteina; ecc.) per rilevare i cambiamenti di conformazione.
- Impostare il volume totale utilizzato per FPOP in base alle esigenze dell'esperimento. Il limite minimo di solito dipende dal volume del capillare di irradiazione e dal materiale necessario per un rilevamento robusto e da una quantificazione relativa, e varia in gran parte a seconda del sistema LC-MS/MS utilizzato e del metodo di elaborazione del campione di post-etichettatura. Il volume totale per le soluzioni FPOP comunemente utilizzate nel nostro gruppo è di 20 L dopo l'aggiunta di perossido di idrogeno. La concentrazione finale della proteina è comunemente 1-10 M, con 17 mM di glutammina (per limitare la durata del radicale idrossilo), 1 mM adenina (per agire come dosimetro radicale)13,17 e10 mM buffer fosfato (un buffer che è un povero spazzino di radicali idrossili). I campioni sono generalmente preparati con più repliche per consentire la modellazione statistica dei risultati.
- Per scopi più generali, preparare campioni in triplicato in entrambi gli stati, più almeno un campione da utilizzare come controllo senza laser per misurare l'ossidazione dello sfondo. Preparare 18 L di questa combinazione di soluzioni FPOP.
NOTA: Molti tamponi e additivi comunemente utilizzati nella biochimica sono spazzini radicali idrossili. Questi additivi e buffer possono essere utilizzati; tuttavia, possono verificarsi riduzioni dell'ossidazione dovute allo scavenging radicale idrossile del buffer. In generale, mantenere tutti gli additivi al minimo richiesto dal sistema biologico per massimizzare la resa di ossidazione delle proteine. Il solfoxide di dimetile dovrebbe essere evitato a causa della propensione a generare radicali secondari; dimethylformamide è stata un'alternativa utile nelle nostre mani. Quando si utilizzano buffer che sono forti spazzini radicali idrossili, la glutammina può spesso essere esclusa dal mix di soluzioni FPOP.
- Per scopi più generali, preparare campioni in triplicato in entrambi gli stati, più almeno un campione da utilizzare come controllo senza laser per misurare l'ossidazione dello sfondo. Preparare 18 L di questa combinazione di soluzioni FPOP.
- Preparare 1 M di perossido di idrogeno immediatamente prima dell'esperimento FPOP.
NOTA: il 30% di perossido di idrogeno, comunemente venduto dai fornitori, include uno stabilizzatore, che aumenta la durata di conservazione. Una volta diluito, il perossido di idrogeno deve essere utilizzato rapidamente, sicuramente entro lo stesso giorno. Anche il perossido di idrogeno deve essere regolarmente testato per la decomposizione mediante FPOP utilizzando un dosimetro radicale idrossile. - Preparare tubi di microcentrifuge contenenti 25 L di soluzione di quench di 0,5 g/L di methionina amide e 0,5 g/L catalase. Se per FPOP viene utilizzato un volume campione superiore a 20 L, aumentare proporzionalmente il volume della soluzione di quench.
3. Eseguire l'esperimento FPOP
- Aggiungere 2 L di perossido di idrogeno nel 18 L del mix di soluzioni FPOP. Mescolare delicatamente il contenuto con una pipetta e far scorrere rapidamente la soluzione sul fondo dei tubi microcentrifuge. Raccogliere immediatamente con una siringa gastight e caricare nella pompa di siringa.
- Avviare il flusso sulla pompa di siringa con la velocità di flusso determinata al punto 1.8.1 (in genere tra 8-16 l/min) facendo clic sul pulsante Avvia pompa sul software dosimeter.
- Monitorare la lettura in tempo reale dell'adenina utilizzando il dosimetro in linea (vedere Tabella deimateriali ) e raccogliere il campione nei rifiuti. Attendere che il segnale Abs265 si stabilizzi.
- Fare clic sul pulsante Start Flash nel software dosimeter per avviare il laser alla velocità di ripetizione preimpostata e l'energia.
- Monitorare la lettura in tempo reale dell'adenina utilizzando un dosimetro in linea (vedere Tabella dei materiali); la differenza in Abs265 con il laser spento e il laser su è la lettura di265 dollari.
NOTA: La comparsa di letture Abs265 altamente instabili al momento del lancio del laser in presenza di perossido di idrogeno è dovuta alla generazione di bolle in soluzione. Ridurre la fluenza del laser e/o la concentrazione di perossido di idrogeno per eliminare le bolle.
4. Eseguire la compensazione
NOTA: diversi ligand, buffer, ecc. possono avere una capacità di scavenging diversa verso i radicali idrossili. È importante garantire che dosi radicali idrossili efficaci comparabili siano disponibili per reagire con proteine attraverso campioni diversi. Ciò si ottiene garantendo una risposta di dosimetro radicale idrossile equa tra i campioni. Utilizzando la dosimetria dell'adenina, il cambiamento nell'assorbimento UV a 265 nm (Aabs265) riflette l'effettiva dose radicale idrossile; più grande è la zAbs265, maggiore è la dose efficace di idrossile radicale.
- Confrontate la letturadi 265 dollari ottenuta con il dosimetro in linea con la lettura desideratadi 265 dollari ottenuta da esperimenti o controlli precedenti. Una lettura di265 dollari inferiore alla lettura desiderata indica una dose efficace insufficiente di radicali idrossili; una lettura di265 dollari indica una dose radicale efficace che è troppo alta. Se la lettura di265 dollari è al livello desiderato, raccogliere il campione immediatamente dopo l'irradiazione laser nel buffer di quench17.
- Compensare la dose radicale efficace per equalizzare il Abs265. Questa compensazione può essere eseguita in tre modi: modificare la concentrazione di perossido di idrogeno, aumentare la fluenza laser cambiando l'energia laser per impulso o aumentare la fluenza laser cambiando il piano focale della lente di messa a fuoco.
- Per apportare una grande modifica (>10 mAU) nella lettura di .aAbs265, rifare il campione con più o meno perossido di idrogeno ed eseguire nuovamente il campione secondo la sezione 3.
- Per fare un piccolo cambiamento nella lettura di265 dollari in tempo reale, regolare il piano focale del fascio incidente regolando la posizione dell'obiettivo di messa a fuoco utilizzando lo stage Motorizzato da 50 mm. Avvicinando il piano focale alla posizione del capillare si aumenterà la lettura di265 euro; portando il piano focale più lontano dalla posizione del capillare diminuirà la lettura di265 dollari.
- Monitorare l'adenina265 per misurare la quantità effettiva di idrossile radicale presente nel campione dopo l'irradiazione laser13. Il monitoraggio in tempo reale con un rilevatore capillare UV in linea consente una compensazione in tempo reale, come descritto al punto 4.2.2; regolare la posizione dell'obiettivo utilizzando lo stage motorizzato fino a quando la letturadi 265 dollari non è uguale alla lettura desiderata. Anche le misurazioni dell'assorbimento post-sperimentale con uno spettrofotometro UV sono accurate, ma richiedono l'uso di nuovi campioni per ogni dose radicale efficace.
5. Digerire i campioni proteici
NOTA: La trypsina è più comunemente usata per digerire campioni di proteine per FPOP ed è la proteasi utilizzata in questo protocollo. Si tratta di una proteasi affidabile che genera peptidi con siti di base sia al N- e C-terminus, promuovendo la moltiplicazione degli ioni peptidi caricati nella SM. Inoltre, si fende dopo l'lino e l'arginina, due aminoacidi che sono solo moderatamente reattivi ai radicali idrossili; pertanto, i cambiamenti nel modello di digestione a causa dell'ossidazione degli alyti sono rari. Altre proteasi sono state utilizzate con successo con FPOP21, ma si dovrebbe fare attenzione a garantire che i modelli di digestione siano comparabili tra campioni nonxidized e ossidati.
- Misurare il volume finale del campione FPOP smava. Aggiungere 500 mM Tris, pH 8.0 con 10 mM CaCl2 contenente 50 mM dithiothreitol (DTT) alla soluzione proteica dopo aver spento ad una concentrazione finale di 50 mM Tris, 1 mM CaCl2 e 5 mM DTT.
- Riscaldare il campione proteico a 95 gradi centigradi per 15 minuti.
- Raffreddare immediatamente il campione sul ghiaccio per 2 minuti.
- Aggiungere il rapporto peso di prova/proteina 1:20 ai campioni.
- Digerire la proteina durante la notte a 37 gradi centigradi con la miscelazione.
- Fermare la reazione di digestione con l'aggiunta di acido formico dello 0,1% e/o riscaldare il campione a 95 gradi centigradi per 10 min.
- Aggiungere 2 mM DTT ai campioni e riscaldare a 60 gradi centigradi per 15 minuti immediatamente prima di LC-MS/MS.
NOTA: Mentre altri gruppi hanno segnalato l'alchilazione dei tioli negli esperimenti FPOP, nelle nostre mani abbiamo notato prodotti collaterali su alchilazione di proteine ossidate (probabilmente a causa della reazione con carbonile nucleofilo formato come un prodotto di ossidazione minore). Pertanto, scegliamo di evitare l'alchilazione dei tioli quando possibile.
6. Eseguire la spettrometria di massa cromatografia liquida (LC-MS/MS)
- Preparare la fase mobile A costituita da acqua contenente acido formico dello 0,1% e fase mobile B costituita da acetonitrile con acido formico dello 0,1%.
- Caricare il campione prima su una colonna di intrappolamento C18 (300 M I.D. x 5 mm 100 dimensioni dei pori, dimensioni di particelle di 5 m) e lavare con 2% solvente B per 3 minuti ad una velocità di flusso di 5,0 L/min per rimuovere sali e piccole molecole idrofili.
- Quindi separare i peptidi sulla nanocolonna C18 (0,75 mm x 150 mm, dimensione delle particelle di 2 m, dimensione dei pori 100) ad una velocità di flusso di 300 nL/min. Il gradiente consiste in un aumento lineare dal 2 al 35% del solvente B oltre 22 min, rampato al 95% del solvente B oltre 5 min e tenuto per 3 minuti per lavare la colonna, quindi restituito al 2% B oltre 3 min e tenuto per 9 min per ri-equilibrare la colonna.
NOTA: Questo gradiente è sufficiente per LC-MS/MS della maggior parte delle miscele FPOP a una e due proteine che cercano di effettuare la quantificazione a livello di peptidi. Potrebbe essere necessario modificare la percentuale di solvente B per aumentare la risoluzione dei peptidi in rari casi in cui i peptidi interferiscono tra loro a causa di tempi di ritenzione simili e valori m/z. I progetti FPOP22 in scala proteoma o sperimentali che cercano di separare gli irrimesi prodotti di ossidazione peptidi1,23,24,25 possono richiedere gradienti LC più lunghi e non rientrano nell'ambito della presente relazione. - Elute i peptidi direttamente nella sorgente nanospray di uno spettrometro di massa ad alta risoluzione utilizzando un emettitore di nanospray conduttivo.
- Acquisire i dati in modalità ioni positivi. Impostare la tensione dello spruzzo a 2400 V e la temperatura del tubo di trasferimento degli ioni a 300 gradi centigradi.
- Acquisire le scansioni MS complete da m/z 250 a 2000 con una risoluzione nominale a m/z 200 di 60.000, seguite da otto successive scansioni MS/MS di ioni a ioni lineari dipendenti dai dati sui primi otto ioni peptidi più abbondanti utilizzando la dissociazione indotta da collisione al 35% di energia normalizzata per identificare i peptidi. Frammentare i peptidi fino a cinque volte entro 30 s e poi trasferire in una lista di esclusione per 60 s.
7. Trattamento dei dati e calcolo dell'ossidazione media dei peptidi
- Determinare la copertura della sequenza della proteina, i valori m/z e i tempi di ritenzione dei peptidi nonxidizzati utilizzando il motore di ricerca proteomica MS/MS.
- Impostare la tolleranza di massa precursore su 10 ppm e consentire fino a due siti di scissione persi per i campioni digeriti di trypsin, utilizzando la specificità standard della scissione della trypsina.
- Impostare la tolleranza di massa del frammento di massa peptide su 0,4 Dalton.
- Sulla base del rapporto m/z dei peptidi non modificati rilevati e dei noti spostamenti di massa dei principali prodotti di ossidazione, calcolare la m/z dei vari prodotti teorici di ossidazione di ogni peptide4,26,27,28,29.
- Identificare il cromogramma iono estratto di questi valori m/z utilizzando il software per visualizzare la corsa spettrometrica di massa (Figura 4). Identificare i prodotti di ossidazione dei peptidi in base al loro m/z, al loro stato di carica e alla somiglianza nel tempo di eluizione con il peptide non modificato. Nelle nostre mani, i prodotti di ossidazione dei peptidi elute tra 240 secondi prima a 180 secondi dopo il peptide non modificato utilizzando il gradiente LC sopra. Poiché l'ossidazione spesso si tradurrà in più prodotti di ossidazione isomeri, è comune osservare più picchi parzialmente risolti nei cromogrammi iono estratti dei prodotti di ossidazione peptidi, come illustrato nella Figura 4. I prodotti di ossidazione dei peptidi vengono quantificati in base all'area dei picchi nei cromatogrammi ioni estratti.

Figura 4: Cromatogramma ione estratto di un peptide e dei suoi prodotti di ossidazione dopo FPOP. La m/z dei prodotti di ossidazione peptidi sono calcolati in base alla m/z del peptide nonidizzato e ai prodotti di ossidazione noti; e le aree di questi prodotti peptidi sono determinate. L'area dei prodotti peptidi viene quindi utilizzata per il calcolo degli eventi di ossidazione medi per peptide. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
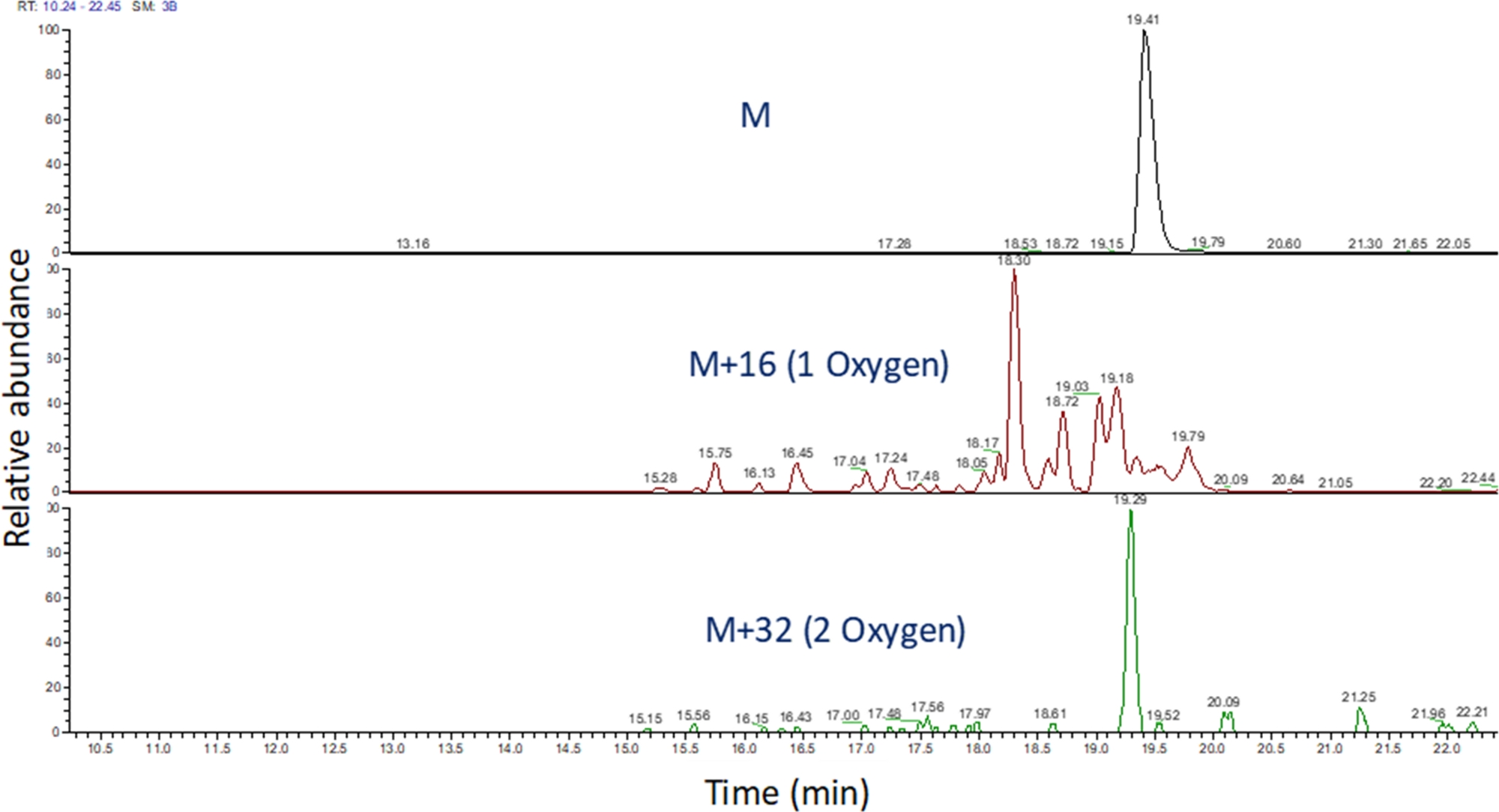{kind=link}
- Calcolare l'ossidazione media dei peptidi utilizzando la seguente equazione.

dove P indica il numero medio di eventi di ossidazione per molecola di peptidi, e I rappresenta l'area di picco del peptide nonidizzato (Iunoxidized) e il peptide con n eventi di ossidazione. Si noti che I(singly oxidized) includerebbe non solo le aggiunte di un singolo atomo di ossigeno, ma anche altri eventi di ossidazione singola meno comuni che lo sperimentatore può scegliere di misurare (ad esempio, decarboxilazione ossidativa, formazione di carbonilo, ecc.) 4,26,27,28,29.
Risultati
Il confronto dell'impronta di peptide a catena pesante del biosimilare adalimumab nel tampone di fosfati e quando riscaldato a 55 gradi centigradi per 1 h mostra risultati interessanti. Il t-test dello studente viene utilizzato per l'identificazione di peptidi che vengono modificati in modo significativo in queste due condizioni (p ≤ 0,05). I peptidi 20-38, 99-125, 215-222, 223-252, 260-278, 376-413 e 414-420 mostrano una protezione significativa dal solvente quando la proteina viene riscaldata per formare aggregati (<...
Discussione
Le tecniche strutturali basate sulla spettrometria di massa, tra cui lo scambio di idrogeno-deuterio, il collegamento chimico incrociato, l'etichettatura covalente e la spettrometria di massa a spruzzo nativo e la mobilità degli ioni, sono in rapida crescita grazie alla loro flessibilità, sensibilità e capacità di gestire miscele complesse. FPOP vanta diversi vantaggi che hanno aumentato la sua popolarità nel settore delle tecniche strutturali basate sulla spettrometria di massa. Come la maggior parte delle strategi...
Divulgazioni
Joshua S. Sharp rivela un significativo interesse finanziario in GenNext Technologies, Inc., una piccola azienda che cerca di commercializzare tecnologie per l'analisi della struttura di ordine superiore delle proteine, tra cui l'impronta delle proteine radicali idrossile.
Riconoscimenti
Riconosciamo il finanziamento della ricerca da parte del National Institute of General Medical Sciences grant R43GM125420-01 per sostenere lo sviluppo commerciale di un dispositivo FPOP benchtop e R01GM127267 per lo sviluppo di protocolli di standardizzazione e dosimetria per FPOP ad alta energia.
Materiali
| Name | Company | Catalog Number | Comments |
| Adenine | Acros Organics | 147440250 | Soluble in water upto 3.5 mM |
| Aperture | Edmund Optics | 39-905 | 1000 μm Aperture Diameter, Gold-Plated Copper Aperture |
| Aperture holder | Edmund Optics | 53-287 | 25.8mm Outer Diameter, Precision Pinhole Mount |
| Catalse | Sigma Aldrich | C-40 | Catalase from bovine liver, lyophilized powder, ≥10,000 units/mg protein |
| COMPex Pro laser | Coherent | 1113836 | COMPexPRO 102, F-Vversion, KrF laser, No XeCl |
| Dithiotheitol (DTT) | Promega | V3151 | DTT, Molecular Grade (DL-Dithiothreitol) |
| Fraction collector | GenNext Technologies, Inc. | N/A | Automated fraction collector |
| Fused silica capillay | Molex | 1068150023 | Polymicro Flexible Fused Silica Capillary Tubing, Inner Diameter 100 µm, Outer Diameter 375 µm, TSP100375 |
| Glutamine | Acros Organics | 119951000 | L(+)-Glutamine, 99% |
| Holder for lens | Edmund Optics | 03-668 | 53 mm Outer Diameter, Three-Screw Adjustable Ring Mount |
| Hydrogen peroxide | Fisher Scientific | H325-100 | Hydrogen Peroxide, 30% (Certified ACS), Fisher Chemical |
| LC-MS/MS system | Thermo Scientific | IQLAAEGAAPFADBMBCX | Dionex Ultimate 3000 coupled to Orbitap Fusion Tribrid mass spectrometer |
| Mas spec grade Acetonitrile | Fisher Scientific | A955-1 | Acetonitrile, Optima LC/MS Grade, Fisher Chemical |
| Mass spec grade formic acid | Fisher Scientific | A117-50 | Formic Acid, 99.0+%, Optima™ LC/MS Grade, Fisher Chemical |
| Mass spec grade water | Fisher Scientific | W6-4 | Water, Optima LC/MS Grade, Fisher Chemical |
| MES buffer | Sigma Aldrich | M0164 | MES hemisodium salt |
| Methionine amide | Bachem | 4000594.0005 | H-met-NH2.HCl |
| Micro V clamp | Thor Labs | VK250 | Micro V-clamp with stainless steel blades |
| Motorized stage | Edmund Optics | 68-638 | 50mm Travel Motorized Stage System with Manual Control |
| Nano C18 colum | Thermo Scientific | 164534 | Acclaim PepMap 100 C18 HPLC Columns |
| Optical bench | Edmund Optics | 56-935 | 18" x 18" breadboard |
| Pioneer FPOP Module System | GenNext Technologies, Inc. | N/A | Inline FPOP Radical Dosimetry System |
| Post holder | Edmund Optics | 58-979 | 3" Length, ¼-20 Thread, Post Holder |
| Sodium phosphate dibasic | Fisher Scientific | BP331-500 | Sodium Phosphate Dibasic Heptahydrate (Colorless-to-White Crystals), Fisher BioReagents |
| Sodium phosphate monobasic | Fisher Scientific | BP330-500 | Sodium Phosphate Monobasic Monohydrate (Colorless-to-white Crystals), Fisher BioReagents |
| Syringe | Hamilton | 81065 | 100 µL, Model 1710 RN SYR, Small Removable NDL, 22s ga, 2 in, point style 3 |
| Syringe pump | KD Scientific | 788101 | Legato 101 syringe pump |
| Trap C18 column | Thermo Scientific | 160454 | Thermo Scientific Acclaim PepMap 100 C18 HPLC Columns |
| Tris | Sigma Aldrich | 252859 | Tris(hydroxymethyl)aminomethane |
| Trypsin | Promega | V5111 | Sequencing Grade Modified Trypsin |
| UV plano convex lens | Edmund Optics | 84-285 | 30 mm Dia. x 120 mm FL Uncoated, UV Plano-Convex Lens |
Riferimenti
- Kaur, P., Kiselar, J., Yang, S., Chance, M. R. Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Molecular & Cellular Proteomics. 14 (4), 1159-1168 (2015).
- Hambly, D. M., Gross, M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. Journal of the American Society for Mass Spectrometry. 16 (12), 2057-2063 (2005).
- Buxton, G. V., Greenstock, C. L., Helman, W. P., Ross, A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O- in Aqueous Solution. Journal of Physical and Chemical Reference Data. 17 (2), 513 (1988).
- Xu, G., Chance, M. R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Analytical Chemistry. 77 (14), 4549-4555 (2005).
- Sharp, J. S., Tomer, K. B. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Analytical Chemistry. 78 (14), 4885-4893 (2006).
- Huang, W., Ravikumar, K. M., Chance, M. R., Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophysical Journal. 108 (1), 107-115 (2015).
- Xie, B., Sood, A., Woods, R. J., Sharp, J. S. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Scientific Reports. 7 (1), 4552 (2017).
- Li, Z., et al. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. Journal of Biological Chemistry. 290 (17), 10729-10740 (2015).
- Li, X., et al. Structural analysis of the glycosylated intact HIV-1 gp120-b12 antibody complex using hydroxyl radical protein footprinting. Biochemistry. 56 (7), 957-970 (2017).
- Li, K. S., Shi, L., Gross, M. L. Mass spectrometry-based fast photochemical oxidation of proteins (FPOP) for higher order structure characterization. Accounts of Chemical Research. 51 (3), 736-744 (2018).
- Li, J., Chen, G. The use of fast photochemical oxidation of proteins coupled with mass spectrometry in protein therapeutics discovery and development. Drug Discovery Today. 24 (3), 829-834 (2019).
- Roush, A. E., Riaz, M., Misra, S. K., Weinberger, S. R., Sharp, J. S. Intrinsic buffer hydroxyl radical dosimetry using Tris(hydroxymethyl)aminomethane. Journal of the American Society for Mass Spectrometry. 31 (2), 169-172 (2020).
- Xie, B., Sharp, J. S. Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Analytical Chemistry. 87 (21), 10719-10723 (2015).
- Niu, B., Zhang, H., Giblin, D., Rempel, D. L., Gross, M. L. Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). Journal of the American Society for Mass Spectrometry. 26 (5), 843-846 (2015).
- Niu, B., et al. Incorporation of a reporter peptide in FPOP compensates for adventitious scavengers and permits time-dependent measurements. Journal of the American Society for Mass Spectrometry. 28 (2), 389-392 (2017).
- Garcia, N. K., Sreedhara, A., Deperalta, G., Wecksler, A. T. Optimizing hydroxyl radical footprinting analysis of biotherapeutics using internal standard dosimetry. Journal of the American Society for Mass Spectrometry. 31 (7), 1563-1571 (2020).
- Sharp, J. S., Misra, S. K., Persoff, J. J., Egan, R. W., Weinberger, S. R. Real time normalization of fast photochemical oxidation of proteins experiments by inline adenine radical dosimetry. Analytical Chemistry. 90 (21), 12625-12630 (2018).
- Zhang, B., Cheng, M., Rempel, D., Gross, M. L. Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods. 144, 94-103 (2018).
- Konermann, L., Stocks, B. B., Czarny, T. Laminar flow effects during laser-induced oxidative labeling for protein structural studies by mass spectrometry. Analytical Chemistry. 82 (15), 6667-6674 (2010).
- Gau, B. C., Sharp, J. S., Rempel, D. L., Gross, M. L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Analytical Chemistry. 81 (16), 6563-6571 (2009).
- Li, K. S., et al. Hydrogen-Deuterium exchange and hydroxyl radical footprinting for mapping hydrophobic interactions of human bromodomain with a small molecule Inhibitor. Journal of the American Society for Mass Spectrometry. 30 (12), 2795-2804 (2019).
- Espino, J. A., Jones, L. M. Illuminating biological interactions with in vivo protein footprinting. Analytical Chemistry. 91 (10), 6577-6584 (2019).
- Charvatova, O., et al. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. Journal of the American Society for Mass Spectrometry. 19 (11), 1692-1705 (2008).
- Gau, B., Garai, K., Frieden, C., Gross, M. L. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 50 (38), 8117-8126 (2011).
- Gau, B. C., Chen, J., Gross, M. L. Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: Data processing and application to barstar. Biochimica et Biophysica Acta. 1834 (6), 1230-1238 (2013).
- Garrison, W. M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chemical Reviews. 87 (2), 381-398 (1987).
- Xu, G., Chance, M. R. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Analytical Chemistry. 77 (8), 2437-2449 (2005).
- Xu, G., Chance, M. R. Radiolytic modification of acidic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 76 (5), 1213-1221 (2004).
- Xu, G., Takamoto, K., Chance, M. R. Radiolytic modification of basic amino acid residues in peptides: probes for examining protein-protein interactions. Analytical Chemistry. 75 (24), 6995-7007 (2003).
- Misra, S. K., Orlando, R., Weinberger, S. R., Sharp, J. S. Compensated hydroxyl radical protein footprinting measures buffer and excipient effects on conformation and aggregation in an adalimumab biosimilar. AAPS Journal. 21 (5), 87 (2019).
- Simmons, D. A., Konermann, L. Characterization of transient protein folding intermediates during myoglobin reconstitution by time-resolved electrospray mass spectrometry with on-line isotopic pulse labeling. Biochemistry. 41 (6), 1906-1914 (2002).
- Vahidi, S., Konermann, L. Probing the time scale of FPOP (fast photochemical oxidation of proteins): radical reactions extend over tens of milliseconds. Journal of the American Society for Mass Spectrometry. 27 (7), 1156-1164 (2016).
- Chance, M. R. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochemical and Biophysical Research Communications. 287 (3), 614-621 (2001).
- Xu, G., Chance, M. R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical Reviews. 107 (8), 3514-3543 (2007).
- Zhang, Y., Rempel, D. L., Zhang, H., Gross, M. L. An improved fast photochemical oxidation of proteins (FPOP) platform for protein therapeutics. Journal of the American Society for Mass Spectrometry. 26 (3), 526-529 (2015).
- Cornwell, O., Radford, S. E., Ashcroft, A. E., Ault, J. R. Comparing hydrogen deuterium exchange and fast photochemical oxidation of proteins: a structural characterisation of wild-type and ΔN6 β(2)-microglobulin. Journal of the American Society for Mass Spectrometry. 29 (2), 2413-2426 (2018).
- Xie, B., Sharp, J. S. Relative Quantification of sites of peptide and protein modification using size exclusion chromatography coupled with electron transfer dissociation. Journal of the American Society for Mass Spectrometry. 27 (8), 1322-1327 (2016).
- Srikanth, R., Wilson, J., Vachet, R. W. Correct identification of oxidized histidine residues using electron-transfer dissociation. Journal of Mass Spectrometry. 44 (5), 755-762 (2009).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Improved identification and relative quantification of sites of peptide and protein oxidation for hydroxyl radical footprinting. Journal of the American Society for Mass Spectrometry. 24 (11), 1767-1776 (2013).
- Li, X., Li, Z., Xie, B., Sharp, J. S. Supercharging by m-NBA Improves ETD-Based Quantification of Hydroxyl Radical Protein Footprinting. Journal of the American Society for Mass Spectrometry. 26 (8), 1424-1427 (2015).
- Khaje, N. A., Sharp, J. S. Rapid quantification of peptide oxidation isomers from complex mixtures. Analytical Chemistry. 92 (5), 3834-3843 (2020).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati