
Method Article
Una procedura ChIP-Seq semiautomatica per studi epigenetici su larga scala
In questo articolo
Erratum Notice
Riepilogo
Questo articolo descrive un protocollo ChIP-Seq semiautomatico e microscalato di regioni del DNA associate a una specifica modificazione degli istoni (H3K27ac) utilizzando una piattaforma di gestione dei liquidi ChIP, seguita dalla preparazione della libreria utilizzando la tagmentazione. La procedura comprende la valutazione del controllo ai fini della qualità e della quantità e può essere adottata per altre modificazioni istoniche o fattori di trascrizione.
Abstract
L'immunoprecipitazione della cromatina seguita dal sequenziamento (ChIP-Seq) è un approccio potente e ampiamente utilizzato per profilare il DNA della cromatina associato a specifiche modificazioni istoniche, come H3K27ac, per aiutare a identificare gli elementi del DNA cis-regolatori. Il processo manuale per completare un ChIP-Seq è laborioso, tecnicamente impegnativo e spesso richiede numeri di celle di grandi dimensioni (> 100.000 celle). Il metodo qui descritto aiuta a superare queste sfide. Viene descritta in dettaglio una procedura completa semiautomatica, microscalata H3K27ac ChIP-Seq che include la fissazione cellulare, il taglio della cromatina, l'immunoprecipitazione e la preparazione della libreria di sequenziamento, per un lotto di 48 campioni per input di numero cellulare inferiori a 100.000 cellule. La piattaforma semiautonoma riduce la variabilità tecnica, migliora il rapporto segnale-rumore e riduce drasticamente la manodopera. Il sistema può quindi ridurre i costi consentendo volumi di reazione ridotti, limitando il numero di reagenti costosi come enzimi, perline magnetiche, anticorpi e tempo pratico richiesto. Questi miglioramenti al metodo ChIP-Seq si adattano perfettamente a studi epigenetici su larga scala di campioni clinici con numero limitato di cellule in modo altamente riproducibile.
Introduzione
L'ampio uso di saggi ChIP-Seq per determinare frammenti di DNA associati a specifiche modificazioni istoniche è in parte dovuto alla sua capacità di identificare elementi del DNA cis-regolatori, inclusi potenziatori attivi, promotori, silenziatori, eterocromatina e altri1,2,3,4. L'identificazione di regioni regolatorie non codificanti in tutto il genoma ha mostrato preziose informazioni per comprendere meglio la regolazione genica nella salute e nelle malattie4. Precedenti lavori del laboratorio hanno utilizzato ChIP-Seq per dimostrare che gli elementi cis-regolatori possono svolgere ruoli importanti in diversi tipi di cellule5. I saggi ChIP del fattore di trascrizione (TF) sono stati utilizzati per mostrare i polimorfismi a singolo nucleotide a rischio associato alla malattia6.
L'uso di ChIP-Seq con campioni clinici umani è impegnativo, principalmente a causa della limitazione del numero di cellule o del campione di tessuto desiderato. Di conseguenza, c'è stato uno sforzo concertato sul campo per migliorare e microscalare queste tecniche e, di conseguenza, sono emersi diversi saggi, come CUT & TAG5,7,8,9,10,11,12. Questo test utilizza una transposasi per il tagment e isola le regioni genomiche legate da un anticorpo specifico9. Questa tecnica è stata in grado di ridurre il numero di cellule fino a 1.000 e in alcuni casi a una singola cellula, tuttavia, l'uso di questa tecnica nella ricerca traslazionale e nell'impostazione clinica ha mostrato limitazioni dovute ai requisiti di utilizzo di cellule vive per questo metodo9,12. Il requisito delle cellule vive rende i campioni clinici logisticamente difficili da gestire e può introdurre effetti batch se i campioni non vengono elaborati contemporaneamente. Altri hanno ottimizzato le tecniche microscalate per le cellule fissate alla formaldeide, incluso lo sviluppo della ChIPmentation11, che è adattata qui in modo ad alto rendimento. L'uso di celle fisse consente di conservare i campioni fino alla raccolta e alla successiva elaborazione di tutti i campioni insieme per ridurre al minimo gli effetti del lotto.
Qui viene descritto un test ChIP-Seq microcalcato semiautomatico che riduce il tempo pratico sperimentale per profilare le modifiche degli istoni10. Il metodo semiautomatico consente saggi ChIP-Seq ad alto rendimento, consentendo di elaborare fino a 48 campioni completamente e pronti per il sequenziamento in appena 5 giorni, per un minimo di 10.000 cellule per campione utilizzando un gestore di liquidi ChIP. Il conduttore completa l'immunoprecipitazione (IP) e i successivi lavaggi in modo autonomo, il che aiuta a ridurre la variabilità tra i campioni. Il metodo semiautomatico riduce sia il tempo di intervento di oltre 15 ore per 48 campioni che la variabilità tecnica, consentendo di condurre studi epigenetici su larga scala in modo riproducibile e rapido per le cellule primarie o in coltura. Il protocollo spiega il processo dall'inizio alla fine per ChIP-Seq di alta qualità. Se le macchine specifiche non sono disponibili, il protocollo sarà comunque una risorsa utile per impostare e risolvere manualmente gli esperimenti ChIP-Seq.
Il test è stato eseguito con tre diversi tipi di cellule immunitarie umane primarie e una linea cellulare in coltura (HUT78 – ATCC: TIB-161). Per chiarezza, il protocollo è stato diviso in sette sezioni: fissazione cellulare, tosatura della cromatina tramite sonicazione, immunoprecipitazione automatizzata della cromatina, preparazione della libreria mediante tagmentazione del frammento di DNA, amplificazione della libreria, purificazione della biblioteca, seguita dalla quantificazione del DNA. Per le ricette tampone si rimanda alla Tabella supplementare 1.
Protocollo
L'Institutional Review Board (IRB) dell'Istituto La Jolla per l'allergia e l'immunologia (LJI; Protocollo IRB n. SGE-121-0714) ha approvato lo studio. Volontari sani sono stati reclutati e hanno fornito campioni di leucoaferesi dopo il consenso informato scritto.
1. Fissazione della cella
- Portare la concentrazione della sospensione cellulare a 1 a 2 x10 6 cellule/mL con terreno di coltura cellulare completo in un tubo da 15 mL (<10 mL di sospensione) o 50 mL di tubo (10-30 mL di cellule). Se <1 x 106 celle, utilizzare 0,5 mL del mezzo in un tubo da 1,5 ml.
- Ruotare delicatamente la sospensione cellulare, aggiungere 10x buffer di fissazione cellulare dropwise (1:10; vol:vol) e ruotare a bassa velocità per 10 minuti a temperatura ambiente (RT).
- Arrestare la reazione vorticando delicatamente e aggiungere 2,5 M di glicina nel rapporto di 1:20 (vol:vol). Capovolgere i tubi un paio di volte e incubare sul ghiaccio per almeno 5 minuti.
- Eseguire i passaggi rimanenti a 4 °C o sul ghiaccio. Ruotare i tubi a 800 x g per 5 minuti a 4 °C ed eliminare il surnatante.
- Sospendere delicatamente il pellet con 5 ml di PBS 1x PBS ghiacciato e incubare per 2 minuti sul ghiaccio.
- Ripetere 1,4 e 1,5 con 1 mL di PBS 1x ghiacciato e trasferire il campione in un tubo preraffreddato da 1,5 ml (etichettato per la conservazione a lungo termine). Se del caso, si raccomanda la preparazione di aliquote.
- Ruotare i tubi a 1.200 x g a 4 °C e rimuovere il più possibile il surnatante senza disturbare il pellet cellulare. Snap congelare il pellet in azoto liquido. Conservare a -80 °C.
ATTENZIONE: Prendere una protezione appropriata quando si maneggia azoto liquido.
2. Tosatura della cromatina
NOTA: Questo protocollo è ottimizzato per la tranciatura della cromatina di pellet con 0,3-3 x 106 celle in tubi a bassa legatura da 0,65 mL.
- Rimuovere il tubo dei campioni con pellet di cellule congelate da -80 °C e conservare su ghiaccio secco per evitare qualsiasi scongelamento del pellet prima di aggiungere il tampone di lisi. Questo passaggio è fondamentale.
- Aggiungere 70 μL di tampone di lisi rte al pellet fresco e conservare a RT per 1 minuto.
- Risospenare il pellet per 1 minuto senza l'introduzione di bolle e quindi incubare la sospensione cellulare a RT per 1 minuto prima di mettere il campione su ghiaccio.
- Trasferire il pellet risospeso in un tubo legante basso da 0,65 ml e mantenere il ghiaccio.
NOTA: Per ottenere una sonicazione riproducibile, preriscaldare il sonicatore facendolo funzionare solo con tubi vuoti per 3-6 cicli prima di sonicare i campioni. - Posizionare i campioni nel supporto del tubo del sonicatore e riempire eventuali spazi vuoti con tubi di bilanciamento riempiti con 70 μL di acqua. Lasciare i campioni a bagnomaria per circa 1 minuto prima di iniziare la sonicazione.
- Eseguire sonicazione per x cicli (a seconda del tipo di cella) con 16 s ON / 32 s OFF per ciclo.
NOTA: questo passaggio richiederà esperimenti di convalida per determinare il numero ottimale di cicli per una sonicazione efficiente. - Dopo ogni 3 cicli, rimuovere i campioni dal sonicatore, ruotare delicatamente e ruotare delicatamente i tubi prima di rimetterli nel supporto. Assicurarsi che non ci siano piccole goccioline all'esterno del tubo in quanto ciò può causare la formazione di bolle.
- Dopo aver completato i cicli necessari, filare i campioni a >14.000 x g per 15 minuti a 4 °C. Trasferire il surnatante in un nuovo tubo di rilegatura basso da 0,65 mL preraffeddato e a bassa legatura e tenerlo sul ghiaccio.
- Scollegare una frazione dei campioni sonicati per verificare l'efficienza di sonicazione.
- Trasferire 1-7 μL del surnatante (equivalenti a circa 250 ng di cromatina tranciata) in un tubo PCR da 0,2 ml e portare il volume fino a 10 μL con tampone di lisi a breve termine a RT.
- Aggiungere 1 μL di RNasi A e incubare per 30 minuti a 37 °C a 800 giri/min, quindi aggiungere 1 μL di proteinasi K.
- Incubare per 2 ore a 55 °C con agitazione a 1.000 giri/min.
- Rimuovere 2 μL del campione decollegato per la quantificazione del DNA utilizzando il saggio di quantificazione fluorescente10 (uno spettrofotometro non è raccomandato in quanto il sapone e le proteine degradate possono produrre distorsioni nei risultati).
- Eseguire il campione rimanente su un gel di agarosio all'1,2% per 1 ora a 70 V. Macchiare con colorante acido nucleico (1:20.000) e leggere il gel utilizzando un transilluminatore UV.
- Preparare aliquote di riserva di cromatina per la conservazione (diluire il campione a 25 ng/μL in 20 μL con tampone di lisi completo). Conservare tutta la cromatina tranciata a -80 °C.
3. ChIP-Seq automatizzato per la modifica degli istoni
NOTA: questo protocollo è progettato per l'esecuzione su un gestore di liquidi ChIP. Sebbene il sistema possa utilizzare buffer personalizzati, tutti i buffer sono forniti con il kit ChIP. Le strisce ChIP con 8 tubi utilizzati in questa sezione sono specifiche per il gestore di liquidi ChIP.
- Trasferire 16 aliquote di campione con 500 ng di cromatina tranciata in 20 μL da -80 °C e metterle sul ghiaccio per scongelare lentamente la cromatina. Una volta scongelato completamente, vortice brevemente e pulse-spin.
- Preparazione della cromatina
- Pipetta 100 μL di tampone tC1 integrato con 1x inibitore della proteasi e 20 mM di butirrato di sodio (tampone tC1 completo) in due strisce ChIP a 8 tubi.
- Trasferire 20 μL di ciascun campione di cromatina in un tubo appropriato delle strisce ChIP a 8 tubi contenenti i 100 μL del tampone tC1 completo. Lavare i tubi di cromatina aggiungendo un tampone tC1 completo da 80 μL ai tubi di cromatina e quindi trasferirli nuovamente nel tubo appropriato delle strisce ChIP a 8 tubi per un volume finale di 200 μL.
- Preparazione dell'anticorpo
- Calcolare il volume di anticorpi in modo tale che 0,5 μg di anticorpo sia in ogni tubo.
Volume di anticorpi = (numero di campioni x anticorpo per reazione) / concentrazione di anticorpi - Aggiungere la quantità calcolata di anticorpi in 500 μL di tampone tBW1. Vortice rapido e pulse-spin.
- Pipettare 70 μL di tBW1 in ciascuna delle due strisce ChIP a 8 tubi e aggiungere 30 μL dell'anticorpo + tBW1 a ciascuna delle provette. Ciò porterà il volume totale in ciascuno dei tubi a 100 μL.
- Calcolare il volume di anticorpi in modo tale che 0,5 μg di anticorpo sia in ogni tubo.
- Preparazione del tallone magnetico
- Vortice completo della soluzione di perline della proteina A. Per 0,5 μg di anticorpi, pipettare 5 μL di perline in un nuovo set di strisce ChIP a 8 tubi e spin a impulsi.
- Riempire l'ultima fila del gestore di liquidi ChIP con strisce ChIP a 8 tubi etichettate e vuote.
- Seguire le specifiche del programma ChIP-16-IPure-200D per il posizionamento di tutte le strisce nella macchina per il trattamento dei liquidi ChIP. Aggiungere i buffer nella posizione corretta ma utilizzare tW4 anziché il buffer tE1.
NOTA: Organizzare la giornata in modo tale che il gestore di liquidi ChIP esegua il ChIP durante la notte. Il programma durerà circa 16 ore per 16 campioni. Questo segna la fine del Giorno 1.
4. Integrazione transposasi di adattatori di libreria per la preparazione di librerie
- Preimpostare un bimbyler a 37 °C e 500 giri/min. Raffreddare un magnete per strisce di tubo da 0,2 ml su ghiaccio.
- Per 16 campioni, preparare 440 μL di tampone di tagmentazione sul ghiaccio. Pipettare 53 μL in un'unica nuova striscia a 8 tubi e mantenere sul ghiaccio.
- In una nuova striscia a 8 tubi da 0,2 mL, aggiungere 220 μL di tampone tC1 freddo e mantenere sul ghiaccio. Gli 8 tubi a striscia possono contenere questo volume ed essere ancora tappati.
- Rimuovere il tubo di striscia "IP samples" dalla macchina per il trattamento dei liquidi ChIP (riga 12) e tappare i tubi prima della rotazione degli impulsi. Cattura le perline usando il magnete per strisce a 8 tubi per 2 minuti e rimuovi con cura il surnatante.
- Trasferire 25 μL del tampone di tagmentazione alle perline con un multicanale, rimuovere dal magnete e mescolare delicatamente fino a quando le perline sono omogenee (circa 5 volte su e giù con la pipetta impostata a 20 μL).
- Tappare i tubi e metterli nel termomiscelatore preriscaldato e incubare per 3 minuti. Aumentare il tempo diminuirà l'efficienza della preparazione della libreria.
- Trasferire i tubi in una griglia metallica refrigerata e aggiungere 100 μL di tampone tC1 refrigerato a ciascun campione. Impostare una pipetta multicanale a 80 μL e mescolare il campione fino a quando le perline sono omogenee, interrompendo la reazione di tagmentazione.
- Riposizionare i campioni nel liquido-handler ChIP e procedere con la procedura di lavaggio Washing_for_IP-reacts_16_Ipure. Assicurarsi che il lavaggio venga eseguito due volte con tampone tC1 e due volte con tW4. L'eluizione deve essere completata come indicato dal layout del programma, con buffer tE1.
- Decrosslinking del DNA
- Rimuovere le strisce ChIP a 8 tubi nell'ultima fila del gestore di liquidi ChIP e aggiungere 2 μL RNasi A a ciascun campione.
- Tappare i tubi, ruotare a impulsi, mescolare delicatamente le perline con una pipetta multicanale fino a quando la miscela non è omogenea e richiudere i tubi.
- Incubare i campioni in un bimbyxer per 30 minuti a 37 °C e 900 giri/min.
- Rimuovere i campioni dal bimbyer, aggiungere 2 μL di proteinasi K. Seguire la stessa procedura di 4.9.2 dopo l'aggiunta.
- Incubare i campioni in un bimbyler per 4 ore a 55 °C e 1.250 giri/min, seguiti da 65 °C a 1.000 giri/min durante la notte.
NOTA: Questa è la fine del Giorno 2.
5. Purificazione dei frammenti di DNA tagmentati
- Etichettare sedici provette da 1,5 mL con il numero di campione appropriato e aggiungere 400 μL di tampone legante il DNA dal kit di pulizia del DNA a ciascuna.
- Rimuovere le strisce a 8 tubi dal bimbyxer e ruotare le strisce per garantire che qualsiasi prodotto evaporato venga trattenuto. Posiziona le strisce su un magnete a 8 strisce per catturare le perline.
- Trasferire 100 μL di DNA decollegato in ciascuno dei tubi da 1,5 mL. Aggiungere 100 μL del tampone legante il DNA alle strisce a 8 tubi per lavare le perline e quindi trasferirle nel tubo appropriato da 1,5 ml.
- Vortice per circa 10 s e pulse-spin i tubi da 1,5 ml.
- Caricare le colonne con i 600 μL contenenti il tampone legante il DNA e il campione ChIP.
- Ruotare i campioni per 20 s a 10.000 x g e ricaricare la colonna con il flow-through. Ruotare di nuovo con le stesse condizioni e scartare il flusso attraverso.
- Lavare le colonne due volte con un tampone di lavaggio da 200 μL (stessa centrifugazione del passaggio precedente) ed eliminare il flusso attraverso.
- Asciugare le colonne centrifugando per 2 minuti a 12.000 x g.
- Trasferire la colonna in un nuovo tubo di raccolta da 1,5 mL e aggiungere 9 μL di TE Buffer caldo (preriscaldato a 55 °C) direttamente alla matrice della colonna. Lasciare incubare la colonna per 1 minuto prima della centrifugazione per 1 minuto a 10.000 x g.
- Trasferire i 9 μL dell'elute in un nuovo set appropriato di strisce a 8 tubi.
- Completare nuovamente l'eluizione con 8 μL TE Buffer come prima. Trasferire l'elute nelle apposite strisce a 8 tubi (volume finale 17 μL per campione) e tenerlo sul ghiaccio.
6. Amplificazione e selezione dimensionale dei campioni purificati
- I seguenti passaggi utilizzano qPCR per determinare il numero di cicli necessari per un'amplificazione ottimale (CtD – Ct determinazione)
- Preparare la miscela CtD per tutti i campioni moltiplicando il contenuto del CtD Mix Buffer per il numero di campioni.
- Erogare 3,6 μL di miscela di CtD in una piastra qPCR e aggiungere 1,4 μL di campioni di DNA tagmentati (~10 % del volume totale). Eseguire il seguente qPCR: 98 °C per 3 min, 72 °C per 5 min, 98 °C per 30 s, 26 cicli di 98 °C per 10 s, 63 °C per 30 s e 72 °C per 30 s.
- Preparare l'Amp Mix per tutti i campioni moltiplicando il contenuto dell'AMP Mix Buffer per il numero di campioni. Erogare 14 μL di DNA tagmentato in pozzetti separati di una piastra PCR, quindi aggiungere 2,5 μL di due primer dell'indice di sequenziamento (25 μM) a ciascun campione (il volume della reazione finale è di 50 μL).
- Mescolare i campioni tramite pipetta multicanale ed eseguire il programma di amplificazione utilizzato nel CtD con il numero appropriato di cicli.
NOTA: Questo è un buon punto di arresto in quanto i campioni amplificati possono essere conservati a -20 °C per alcune settimane. Tuttavia, la purificazione può essere completata lo stesso giorno. Per 48 campioni, i passaggi 3 - 6.5 sono stati completati con altri due lotti separati e quindi amplificati in un unico lotto come descritto di seguito. - Eseguire la post-amplificazione, la selezione delle dimensioni e la quantificazione del DNA tagmentato come descritto di seguito. Questo di solito può essere completato con 48 campioni (può essere completato con meno campioni come desiderato).
- Aggiungere 90 μL di perline paramagnetiche (rapporto 1:1,8) in ciascun pozzetto, mescolare e lasciare incubare a RT per 2 minuti.
- Cattura le perline usando una piastra magnetica e scarta il surnatante. Lavare le perline 3 volte con 200 μL di etanolo fresco all'80% senza interrompere il pellet di perline.
- Rimuovere l'etanolo in eccesso con punte da 20 μL dopo il lavaggio finale e lasciare asciugare le perline per 10 minuti o fino a quando non compaiono crepe nei pellet di perline.
- Con la piastra ancora sul magnete, aggiungere 40 μL di acqua preriscaldata a ciascun pozzo. Sigillare la piastra, vorticare accuratamente e ruotare brevemente la piastra.
- Cattura le perline riposizionando la piastra sul magnete e trasferisci l'eluizione da 40 μL su una nuova piastra "campione". I campioni sono ora purificati e i passi successivi arricchiscono frammenti che vanno da 200-1.000 bp.
- Fase QC opzionale: rimuovere 4 μL dai campioni e trasferirli su una piastra QC. Aggiungere 4 μL di acqua ai campioni. Questo determina la percentuale di frammenti di grandi dimensioni.
- Aggiungere 22 μL di perline paramagnetiche (rapporto 1:0,55) ai campioni, mescolare accuratamente e incubare a RT per 2 minuti.
- Posizionare sul magnete per catturare le perline per 5 minuti e trasferire i supernatanti alle colonne 7-12 della piastra "campione". Rimuovere la piastra dal magnete e aggiungere 30 μL di perline (rapporto finale di 1:1.3). Mescolare con cura e lasciare riposare a RT per 2 minuti.
- Cattura le perline per 5 minuti e poi scarta il surnatante.
- Lavare tutte le perline 3 volte con 200 μL di etanolo fresco all'80 % come descritto in precedenza (fasi 6.6.2 – 6.6.3).
- Una volta che i pellet sono asciutti, eluire il DNA con un tampone TE preriscaldato da 8 μL per ogni pozzetto, mentre è ancora sul magnete.
- Rimuovere accuratamente la piastra dal magnete, sigillare e vortici. Lasciare incubare la piastra per 2 minuti a RT, rotazione dell'impulso e riposizionare la piastra sul magnete per 2 minuti. Trasferire il surnatante in una nuova piastra (Tavola 2).
- Per il massimo recupero, ripetere l'eluizione con un ulteriore 8 μL di tampone TE preriscaldato. Collocare i campioni nei pozzetti appropriati in modo tale che ogni campione abbia 16 μL di libreria finale.
NOTA: Alla fine di questo passaggio dovrebbero esserci due piastre (una se non è stata completata alcuna piastra QC). La piastra QC avrà i frammenti selezionati in base alle dimensioni e la seconda piastra dovrebbe avere 48 pozzetti di libreria finale (16 μL totali).
7. Quantificare le librerie finali e i campioni QC utilizzando un test di fluorescenza
- Quantificazione completa del DNA utilizzando un saggio di quantificazione della fluorescenza o un metodo simile.
- Se la quantificazione QC è stata completata, determinare la percentuale di perdita di campione che è stata < 1.000 bp. Non ci dovrebbe essere più di circa il 20% di perdita - se ce ne fosse di più, potrebbe esserci un problema con i rapporti di perline applicati.
- Determinare la dimensione dei frammenti di ciascun campione, preferibilmente utilizzando una macchina per elettroforesi capillare. Per calcolare la concentrazione molare utilizzare la seguente equazione: [Concentrazione della libreria (ng/μL) * 106]/[660 * Dimensione mediana del frammento (bp)]).
NOTA: le librerie sono pronte per essere raggruppate (quantità equimolari) e sequenziate seguendo le procedure standard di sequenziamento di nuova generazione.
Risultati
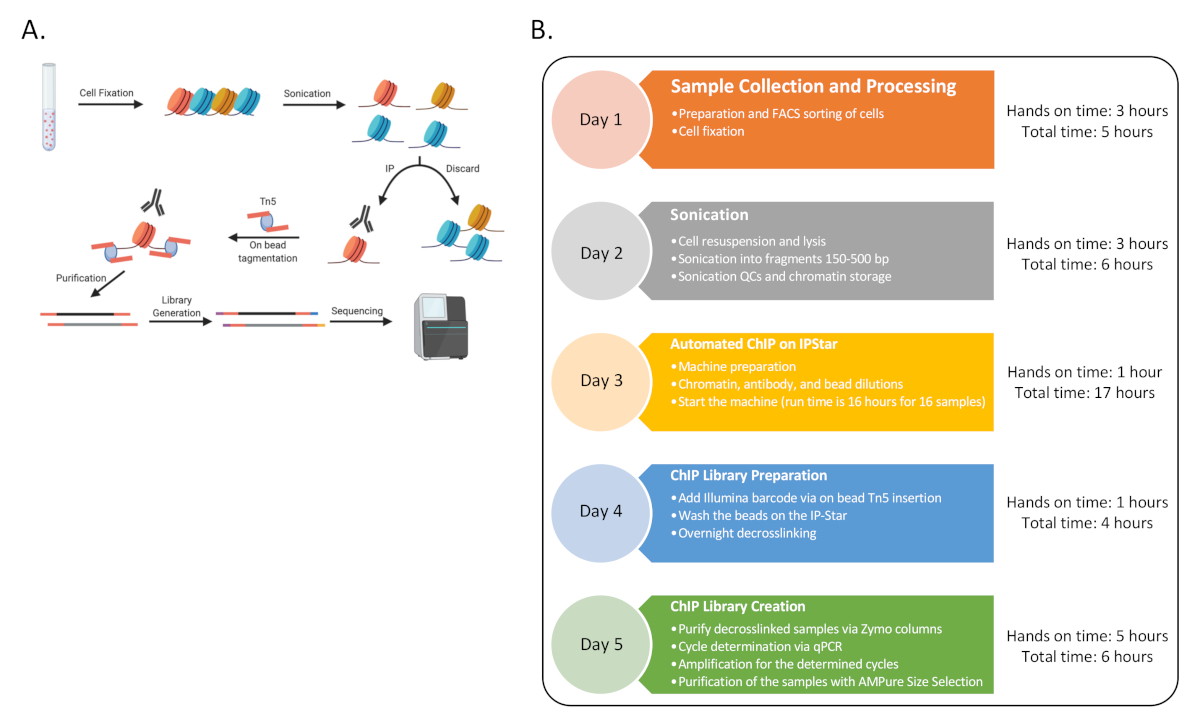
Come prova di concetto, ChIP-Seq è stato completato per sei donatori umani con tre set di tipi di cellule immunitarie: cellule T CD4 naïve (CD4), monociti classici (MO) e cellule natural killer (NK), arricchite dallo smistamento FACS come descritto prima13. La procedura sottolineata è costituita da nove procedure distinte come rappresentato nella Figura 1.

Figura 1: Diagramma di flusso generale per la procedura. (A) Un cartone animato della procedura complessiva (generato in BioRender). (B) Diagramma di flusso per tutte le fasi principali del protocollo e il tempo pratico e totale stimato associato a ciascun giorno. Il sequenziamento potrebbe avvenire alla fine del Giorno 5 o più tardi con più round. La sequenza temporale può anche essere scaglionata per tutta la settimana, dove il giorno sequenziale 3-4 può essere completato più volte in una settimana per generare 48 campioni ChIP. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Dopo l'isolamento cellulare mediante citometria a flusso13,le cellule selezionate sono state centrifugate e le cellule fissate e conservate come descritto sopra. Una volta raccolti tutti i campioni, i campioni sono stati lisati e preparati per la tosatura cromatica in lotti di 12 come descritto sopra. Per ogni campione, il numero di cicli per raggiungere la sonicazione ottimale è stato completato10. La misurazione quantitativa, così come le misurazioni delle dimensioni del frammento di cromatina tranciato, hanno mostrato una grande riproducibilità del nostro metodo sui tre insiemi di cellule immunitarie (Figura 2A). Le diverse cellule immunitarie umane sono state sonicate in lotti separati e hanno prodotto in modo molto coerente con > il 70% del campione tra 100 - 500 bp per 14 cicli (16 s ON, 32 s OFF per ciclo). A questo punto, i campioni con grandi frammenti dopo la sonicazione (< il 70% del campione tra 100 e 500 bp) sono stati considerati falliti. Questi campioni potevano essere sonicati per 1-2 cicli aggiuntivi o venivano scartati e sostituiti in seguito con celle di un altro pellet. Il nostro metodo ha mostrato che nessuno dei campioni richiedeva più sonicazione o è stato eliminato, suggerendo l'assoluta robustezza della procedura.

Figura 2: Esempi di QC pre-sequenziamento. (A) Gel di agarosio all'1,2% mostrano riproducibilità della sonicazione. Campioni di sonicazione per 6 donatori in tre tipi di cellule: cellule T CD4 naïve (CD4), monociti classici (MO) e cellule natural killer (NK). I campioni sono stati sonicati per 14 cicli (16 s ON, 32 s OFF per ciclo). Per ogni campione circa 200 ng di cromatina decollegata sono stati caricati su un gel di agarosio all'1%. I campioni sono stati considerati buoni se più del 70 % dei frammenti si trova entro 100-500 bp. (B) Top - Analisi qPCR curve di amplificazione per determinare il numero ottimale di cicli per l'amplificazione (Ct dove c'è 1/2 l'intensità massima). I campioni ideali hanno una Ct di circa 15 e l'amplificazione può essere completata fino a 2 cicli in più della Ct misurata. La freccia è un esempio di un cattivo esempio in cui la Ct è maggiore di 18. In basso - Viene mostrato un esempio di un insieme scadente di campioni che hanno una Tc maggiore di 18. Questi campioni hanno anche mostrato un'intensità di fluorescenza inferiore. (C) Sinistra - Le tracce di elettroforesi dell'analizzatore di frammenti hanno mostrato la distribuzione delle librerie tagmentate finali dopo l'amplificazione e la selezione delle dimensioni. I campioni con oltre l'85% della libreria di frammenti si trovano entro 200-1.000 bp sono stati considerati buoni campioni. Anche la misurazione dell'intensità di picco della fluorescenza è considerata un importante parametro QC, infatti se il segnale è basso, è improbabile che il campione si sequenzi bene. A destra - Vengono mostrati esempi di campioni positivi in CD4, MO e NK. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Dopo la quantificazione, i campioni sono stati eseguiti su un gestore di liquidi ChIP con anticorpi H3K27ac, seguito da tagmentazione con enzima trasposasi Tn5. Per determinare il numero appropriato di cicli di amplificazione mediante qPCR, è stato utilizzato il 10% dei campioni tagmentati. Per la determinazione del numero di cicli per l'amplificazione dei campioni, troviamo il ciclo in cui l'intensità del campione è la metà del massimo medio per la determinazione del ciclo (Figura 2B). I campioni con valori ct superiori a 18 non hanno eseguito bene dopo il sequenziamento e il loro valore Ct era quindi indicativo di un campione ChIP fallito. Questi campioni generalmente hanno anche prodotto una minore quantità di DNA dopo l'amplificazione. I campioni (100.000 celle in ingresso) con un valore Ct uguale o inferiore a 15 erano ideali e i campioni tra 15 e 18 erano accettabili ma meno coerenti dopo il sequenziamento. Per i campioni con meno di 100.000 celle di input, i valori ct sono stati solitamente trovati tra 15 e 18, ma non hanno avuto bisogno di più di 18 cicli per produrre abbastanza prodotto per il sequenziamento.
Dopo l'amplificazione con DNA-tag, le librerie sono state purificate e selezionate per ottenere una distribuzione dimensionale ideale, che va da 200 a 1.000 bp, per il sequenziamento NextGen. La valutazione della distribuzione delle dimensioni su ciascuna delle librerie è stata completata perché i migliori dati di sequenziamento sono stati ottenuti quando oltre l'85% dei frammenti di DNA variava tra 200 e 1.000 bp (Figura 2C). In particolare, poiché è stata caricata la stessa quantità di DNA (misurata mediante quantificazione di fluorescenza), è stato notato che i campioni con intensità di fluorescenza inferiore sono generalmente sequenziati male (Figura 2C).
Post sequenziamento, sono stati applicati controlli di qualità standard basati sulle linee guida ENCODE ChIP-Seq5,14,15.

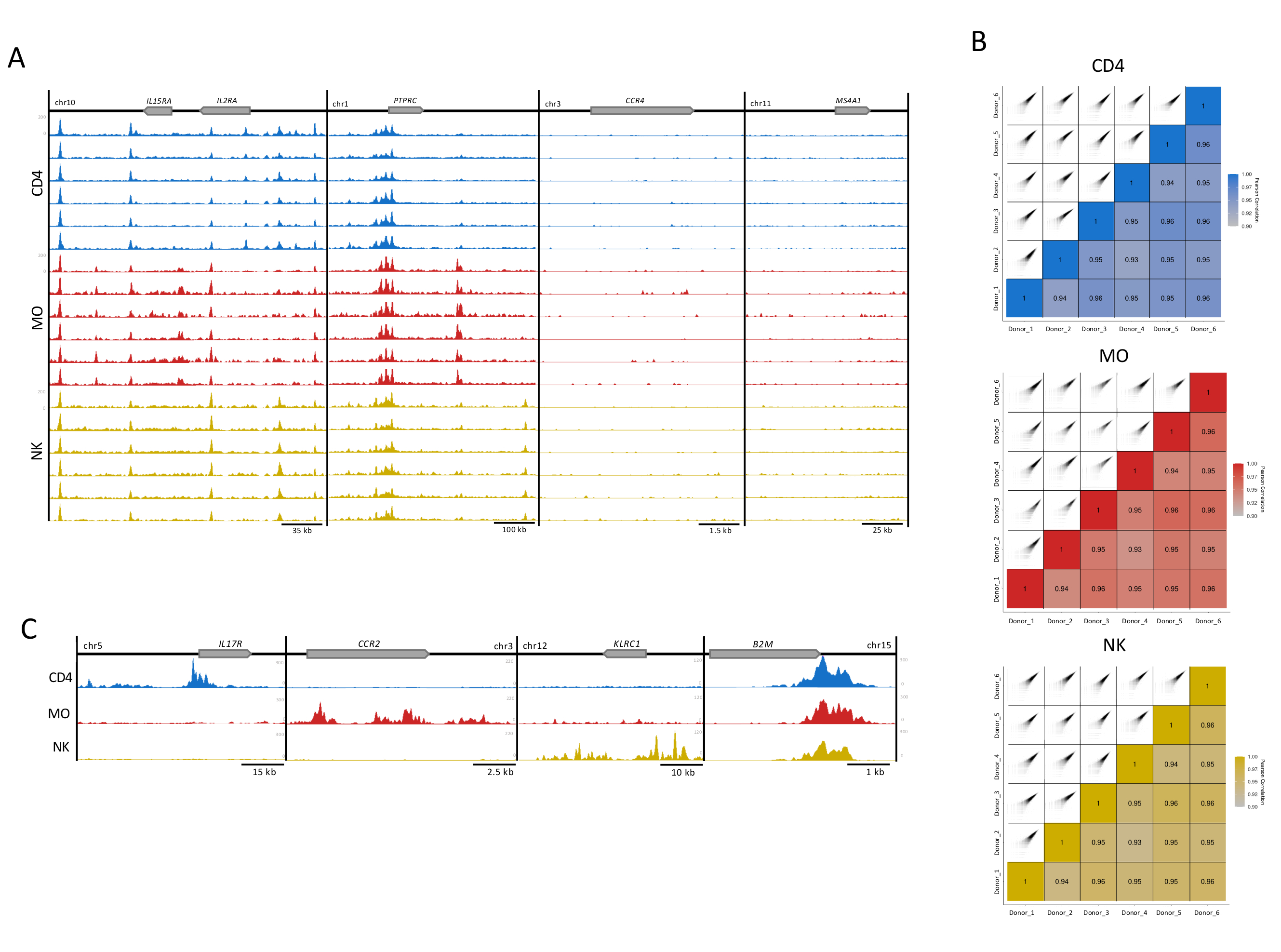
Figura 3: Riproducibilità dei campioni di cellule immunitarie. (A) Tracce H3K27ac (UCSC Genome Browser, intensità massima, funzione di levigatura di 4, tutte con asse Y ugualmente scalato) per 6 donatori (100.000 cellule per replicazione) in ogni tipo di cellula (CD4, MO e NK). Vengono mostrati quattro loci esemplari, due con (locus IL2RA e PTPRC) e due senza arricchimento per H3K27ac (CCR4 e MS4A1). (B) La correlazione di Pearson tra i donatori e i corrispondenti grafici di correlazione generati utilizzando un'estensione di 300 bp e una finestra di 500 bp all'interno del pacchetto MEDIPS per ciascuno dei tipi di cellule replica16. (C) File donatori uniti per ciascun tipo di cellula che mostrano tracce H3K27ac (intensità massima UCSC Genome Browser, funzione di levigatura di 4) in regioni specifiche del tipo cellulare (IL17R per CD4, CCR2 per MO e KLRC1 per NK) e il gene domestico B2M, presente in tutti i tipi di cellule. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Per il controllo della qualità visiva, sono state preparate tracce di arricchimento H3K27ac per la visualizzazione nel browser del genoma UCSC. Per quattro loci genetici, le singole tracce per ciascun campione hanno mostrato un'elevata qualità di mappatura e un rapporto segnale-rumore che riflette l'elevata coerenza e robustezza del nostro test (Figura 3A). I due loci a sinistra ospitano geni ben espressi in questi tipi di cellule, mentre i geni nei due loci a destra non sono espressi e servono come controlli di sfondo13 (Figura 3A). Inoltre, il pacchetto di analisi MEDIPS è stato utilizzato come variabile post-sequenziamento per valutare l'indice di correlazione tra repliche tecniche (Figura 3B)5,16,17, stabilendo il grado di correlazione per il livello di arricchimento delle letture per 500 bp bins16. Per la maggior parte dei confronti a coppie, gli indici di correlazione di Pearson hanno mostrato una correlazione superiore al 90% suggerendo un alto livello di coerenza tra le repliche biologiche (Figura 3B). Le repliche con correlazione accettabile sono state unite per aumentare il rapporto segnale-rumore. Mentre i loci specifici del tipo cellulare hanno mostrato un elevato arricchimento nelle cellule appropriate, un gene domestico (B2M) ha mostrato una modificazione degli istoni molto consistente (Figura 3C). Per l'analisi, la fusione di tracce da repliche aumenterà l'arricchimento, rafforzerà il segnale specifico, anche per importanti potenziatori specifici del tipo di cellula, e ridurrà la variabilità interindividuale inerente ai campioni umani5.
Sebbene per questo studio siano state utilizzate 100.000 cellule, c'era un'alta riproducibilità per un minimo di 10.000 cellule in una linea di cellule T in coltura umana (HUT78). L'analisi di correlazione tra set di dati ChIP-Seq eseguita da campioni con meno di 100.000 celle ha mostrato un'elevata riproducibilità e correlazione fino a 10.000 celle (Figura 4A).

Figura 4: Riproducibilità di campioni a basso input. (A) Esempi della consistenza di H3K27ac ChIP-Seq per cellule da 100.000 a 10.000 in cellule HUT-78 (una linea cellulare di linfoma a cellule T). Le tracce (UCSC Genome Browser, intensità massima, funzione di levigatura di 4, tutte con asse Y ugualmente scalato) mostrano il locus IL4. (B) Correlazioni di Pearson delle repliche utilizzando un'estensione di 300 bp e una finestra di 500 bp all'interno del pacchetto MEDIPS16. (C) Correlazioni di Pearson tra i diversi gruppi di numeri di cella (100.000, 50.000 e 10.000 celle) utilizzando gli stessi parametri MEDIPS di (B)16. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
L'analisi di correlazione di Pearson ha mostrato un alto indice di correlazione (dall'83% al 92%), suggerendo il mantenimento del segnale in campioni a basso numero di cellule. Tuttavia, c'è stato un aumento dello sfondo in quanto i numeri di celle sono stati ridotti e una caduta dei coefficienti di correlazione (Figura 4B). Per mantenere bassi i segnali di fondo, sono stati uniti i duplicati tecnici ed è stata testata la correlazione tra i gruppi (Figura 4C).
| Buffer di fissazione della cella 10X | |
| Composto | Concentrazione finale |
| Soluzione di formaldeide | 11% |
| NaCl | 100 metri |
| EDTA, pH 8,0 | 1 mM |
| EGTA, pH 8,0 | 0,5 mM |
| HEPES, pH 7,5 | 50 metri quadrati |
| Buffer di lisi completo | |
| Composto | Concentrazione finale |
| Tris-HCI, pH 8,0 | 50 metri quadrati |
| EDTA, pH 8,0 | 10 mM |
| SDS | 0.25% |
| Butirrato di sodio | 20 mM |
| Cocktail inibitore della proteasi | 1X |
| Tampone di lisi a breve termine | |
| Composto | Concentrazione finale |
| Tris-HCI, pH 8,0 | 50 metri quadrati |
| EDTA, pH 8,0 | 10 mM |
| SDS | 0.25% |
| Tagmentation Mix | |
| Composto | Concentrazione finale |
| Tris-HCI, pH 8,0 | 10 mM |
| MgCl2 | 5 mM |
| N,N-dimetilformammide | 10% |
| Enzima di tagmentazione Illumina | 1:24 vol:vol |
| CtD Mix | |
| Composto | Per campione (μL) |
| NextEra Index Primer A (25 μM) | 0.275 |
| NextEra Index Primer B (25 μM) | 0.275 |
| 2X KAPA HiFi HotStart Ready Mix | 2.75 |
| 1:1000 SYBR Colorante verde | 0.11 |
| Colorante passivo ROX | 0.11 |
| Acqua | Riempimento a 4 μL |
| AMP Mix | |
| Composto | Per campione (μL) |
| 2X KAPA HiFi HotStart Ready Mix | 27.5 |
| Acqua | Riempimento a 31 μL |
Tabella supplementare 1: Ricette tampone.
Tabella supplementare 2: correlazioni di campioni spearman e Pearson per i 6 donatori e ciascun tipo di cellula. Fare clic qui per scaricare questa tabella.
Discussione
Il metodo qui descritto espande la procedura di ChIPmentation11, che implementa un protocollo di preparazione della libreria di tagmentazione prima della purificazione del DNA, automatizzando e microscalando il protocollo. Dall'inizio di ChIP-Seq, i numeri di cellule richiesti sono stati ridotti drasticamente, da circa 20 milioni di cellule per istoni fino a centinaia e persino singole cellule1,7,10,12,18,19,20,21. Questi metodi di nuova concezione hanno permesso una comprensione più profonda di come i meccanismi di regolazione cisstanno lavorando nelle cellule aumentando la sensibilità e consentendo di testare rare popolazioni di cellule cliniche5,6,12,17. Ad esempio, una delle procedure più recenti e popolari, chiamata CUT & TAG, come alternativa ChIP-Seq robusta e sensibile9. Produce un eccellente rapporto segnale-rumore in quanto l'enzima Tn5 è legato covalentemente alla proteina A e riconosce la catena Fc dell'anticorpo ChIP con elevata specificità9. L'attività di fondo dell'enzima Tn5 è ridotta in quanto l'enzima non è funzionale prima di legarsi all'anticorpo bersaglio9. Tuttavia, l'implementazione di questo metodo in un contesto clinico è limitata poiché richiede cellule vive non fisse. Inoltre, la rimozione di frammenti di DNA dal nucleo ipotonico potrebbe avere effetti negativi sulla cromatina in quanto viene rimossa durante il test. Il requisito necessario per lavorare con cellule fresche e viventi è una fonte di problemi per campioni clinici rari e per grandi coorti di campioni, poiché grandi coorti possono richiedere numerosi anni per raccogliere5. Un altro tipo di metodo, drop-ChIP, utilizza elegantemente un dispositivo di microfluidica per generare tagmention basati su goccioline prima di elaborare il ChIP19. Tuttavia, utilizza un dispositivo microfluidico altamente specializzato e, mentre è possibile completare ChIP-Seq a cella singola, è anche limitato all'uso di cellule vive7,8,9,18,19. Metodi più recenti che si basano su ChIP-Seq come PLAC-Seq o HiChIP, tentano di comprendere le interazioni a 3 dimensioni (3D) tra i picchi ChIP-Seq22,23. Questi metodi 3D sono entusiasmanti in quanto identificano le interazioni cis-regolatorieo TF mediate attraverso il genoma e migliorano la comprensione della regolazione dell'espressione genica nei tipi di cellule di interesse, nei tessuti sani e nel contesto della malattia.
Ci sono alcuni passaggi critici da considerare affinché il protocollo abbia successo, come la qualità della cromatina sonicata e la qualità dell'anticorpo. L'efficienza di taglio è fondamentale, se la cromatina non è sonicata bene, l'efficienza del saggio diminuisce drasticamente24. La sonicazione è un aspetto impegnativo di ChIP-Seq a causa dei numeri di cella richiesti. Sul sonicatore utilizzato nel protocollo, l'efficienza è stata drasticamente ridotta sotto le 300.000 celle. Questo è un aspetto impegnativo in ChIP-Seq in quanto sonicare sotto quel livello richiederebbe spesso una frammentazione enzimatica, che è meno imparziale. Di conseguenza, la sonicazione è un fattore limitato importante per il vero ChIP-Seq microscalato. Altre piattaforme di sonicazione e kit disponibili in commercio sono stati testati per la cromatina sonicante, ma il sonicatore utilizzato qui ha avuto i risultati più robusti e riproducibili. Un altro vantaggio del sonicatore è non dover acquistare tubi specializzati per eseguire la sonicazione, il che riduce i costi quando si tratta di un gran numero di campioni. Per una sonicazione ottimale, in primo luogo, è importante preriscaldare il sonicatore come descritto sopra. In secondo luogo, per lisare il pellet, si consiglia di avere la punta della pipetta che tocca il fondo del tubo durante la lisi per rompere le celle con più vincoli fisici. In terzo luogo, qualsiasi formazione di bolle prima della sonicazione ostacola la capacità del campione di essere sonicato in modo uniforme. Se ci sono bolle formate durante la lisi, è importante rimuoverle con una pipetta. Questo può essere impegnativo senza rimuovere molto campione, ma se la punta viene leggermente premuta contro la bolla può essere lentamente disegnata senza perdita di molto campione. Infine, quando si determina il numero di cicli, completare un percorso temporale in cui ogni tre cicli, il campione viene rimosso, purificato e eseguito su un gel di agarosio. Evitare la sonicazione eccessiva / insufficiente dei campioni in quanto ciò riduce l'efficienza ChIP. Se il campione è sotto sonicato, i frammenti di grandi dimensioni possono avere un effetto negativo sulla qualità ChIP-Seq24. D'altra parte, se il campione è troppo sonicato, c'è il rischio che l'epitopo bersaglio si perda nel processo.
Un'altra parte essenziale di ChIP-Seq è la qualità dell'anticorpo. Prima di eseguire qualsiasi studio su larga scala, è necessario ottimizzare l'anticorpo che verrà utilizzato. L'obiettivo è quello di ottenere un rapporto segnale/rumore significativamente elevato delle regioni conosciute del genoma e un altro è la riproducibilità. Se l'anticorpo sta tirando un sacco di segnale di fondo, potrebbe essere consigliato di utilizzare un ingresso più grande o provare un lotto / fornitore diverso. Questo aggiungerà tempo prima di iniziare un esperimento su larga scala, ma è un passo essenziale. Per testare il segnale-rumore si consiglia di utilizzare qPCR con regioni note per essere un bersaglio del vostro anticorpo e un'altra regione nota per essere assente. È stato notato che le modifiche agli istoni sono più robuste e più facili da ottimizzare rispetto ai TF.
Il protocollo sopra descritto fornisce un metodo robusto per la modifica degli istoni ad alto rendimento ChIP-Seq in modo semiautonomo e microscalato. Il metodo limita la quantità di tempo pratico e aumenta la riproducibilità rispetto al ChIP-Seq manuale. Precedenti studi completati in laboratorio utilizzavano manualmente ChIP su repliche tecniche e ottenevano una media di correlazione di Spearman di0,50 5,tuttavia, con il sistema semiautomatico, la correlazione di Spearman tra diversi donatori con una media di cellule NK di 0,66 (Tabella supplementare 2). Anche questo è stato completato con circa il 40% in meno di tempo pratico. Il metodo qui descritto è stato ottimizzato per le modifiche degli istoni (H3K27ac mostrato qui, ma il protocollo non dovrebbe aver bisogno di alcuna modifica per gli altri) e richiederebbe solo piccole modifiche per essere implementato per TF ChIP-Seq. Nonostante la qualità dell'anticorpo, la modifica principale sarebbe per il tempo di sonicazione e potenzialmente i tamponi utilizzati durante l'IP. Di solito, per i saggi TF ChIP, il metodo può funzionare meglio con frammenti leggermente più lunghi di cromatina (con un intervallo di circa 350-800 bp) poiché i complessi TF: DNA sono probabilmente meno in grado di essere mantenuti attraverso una rigorosa sonicazione6. I buffer potrebbero anche dover passare a un mix personalizzato o ad altri kit disponibili nel settore, poiché i TF possono comportarsi in modo diverso rispetto alle modifiche degli istoni.
Sebbene il gestore automatico di liquidi ChIP sia stato testato per un minimo di 10.000 cellule, c'è stata una notevole diminuzione della riproducibilità a concentrazioni di cromatina più basse. A causa di ciò, il protocollo non è stato raccomandato a meno di 10.000 cellule, con 100.000 cellule che sono le condizioni ottimali. Il protocollo è stato anche completato utilizzando buffer ChIP del settore, che era una spesa aggiuntiva ma forniva dati di qualità superiore. Il protocollo potrebbe essere modificato per quanto riguarda le condizioni di sonicazione (purché la cromatina tranciata sia mantenuta all'interno dello stesso intervallo), i tamponi potrebbero essere personalizzati per l'immunoprecipitazione (IP; potrebbe essere necessaria l'ottimizzazione) o il gestore di liquidi ChIP potrebbe non essere utilizzato. Una limitazione del protocollo è l'uso del gestore di liquidi ChIP, che può essere un investimento costoso e può eseguire solo 16 campioni contemporaneamente. Il gestore di liquidi ChIP è limitato a reazioni su piccola scala e il numero di cellule superiore a un milione non è raccomandato. Tuttavia, il protocollo potrebbe essere completato senza di esso, completando l'IP e lavando manualmente i passaggi. Se l'IP e i lavaggi sono stati completati a mano, il tempo per completare il test aumenterà e la riproducibilità potrebbe diminuire, ma questa guida sarà comunque utile per eseguire un esperimento ChIP-Seq di alta qualità. Da notare, altri gestori di liquidi potrebbero essere adattati per eseguire reazioni ChIP semiautomatiche.
Per riassumere, i principali vantaggi di questo sistema sono la natura ad alta produttività, poiché l'IP e le fasi di lavaggio sono completate autonomamente. Pertanto, è possibile completare cicli sequenziali di esperimenti ChIP, consentendo di elaborare fino a 48 campioni e pronti per il sequenziamento in 5 giorni, con un tempo pratico limitato rispetto agli esperimenti Manuali ChIP-Seq. Un altro vantaggio è la maggiore riproducibilità poiché ChIP-Seq può essere difficile ottenere risultati altamente riproducibili. Altri metodi richiedono cellule vive, complessi sistemi di microtubaggio o il lavoro da completare tutto a mano. Questo sistema dovrà essere ottimizzato per campioni a basso input (<10.000 celle), consentendo in definitiva reazioni ChIP a cella singola. Il sistema è anche in grado di essere adattato per i nuovi metodi ChIP, come PLAC-Seq e HiChIP22,23.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Ringraziamo i membri del laboratorio Vijayanand per l'aiuto tecnico e le discussioni costruttive e il Dr. Sharron Squazzo e il Signor Geoffrey Berguet di Diagenode per l'assistenza tecnica con il sonicatore e la macchina e i protocolli chIP per il trattamento dei liquidi. Questo lavoro è stato supportato dalle sovvenzioni NIH AI108564, R01HL114093, S10RR027366 (BD FACSAria II) e S10OD016262 (Illumina HiSeq 2500).
Materiali
| Name | Company | Catalog Number | Comments |
| 200 µl tube strips (8 tubes/strip) + cap strips | Diagenode | C30020002 | Strip tubes for use on the IP Star; ChIP 8-tube strip |
| AMPure XP for PCR Purification | Beckman Coulter | A63880 | SPRI beads |
| Axygen 0.6 mL MaxyClear Snaplock Microcentrifuge Tube | Corning | MCT-060-C | 0.65 mL low binding tube |
| Bioruptor Pico Sonicator | Diagenode | B01060010 | Sonicator used in the lab but others can be used |
| ChIP DNA Clean & Concentrator (Capped Columns) | Zymo Research | D5205 | DNA clean-up kit |
| Dynabeads Protein A for Immunoprecipitation | ThermoFisher | 10001D | |
| EDTA (0.5 M), pH 8.0, RNase-free | ThermoFisher | AM9260G | |
| EGTA pH 8.0 | Millipore Sigma | E3889-25G | |
| Eppendorf ThermoMixer C | Eppendorf | 2231000667 | |
| Formaldehyde solution | Millipore Sigma | 252549-1L | |
| Glycine | Millipore Sigma | 50046-250G | |
| H3K27ac polyclonal antibody - Premium | Diagenode | C15410196 | |
| HEPES (1 M) pH 7.5 | ThermoFisher | 15630080 | |
| IDT for Illumina Nextera DNA Unique Dual Indexes | Illumina | 20027213 | |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | 20034197 | |
| IP-Star Compact Automated System | Diagenode | B03000002 | Automated system for ChIP-Seq studies; ChIP liquid handler |
| KAPA HiFi HotStart ReadyMix | Roche | KK2601 | PCR mix |
| Medium reagent container for SX-8G IP-Star Compact | Diagenode | C30020003 | |
| MgCl2 (magnesium chloride) (25 mM) | ThermoFisher | R0971 | |
| N,N-Dimethylformamide | Millipore Sigma | D4551-250ML | CAUTION - low flash point |
| NaCl (5 M), RNase-free | ThermoFisher | AM9760G | |
| PBS (10X), pH 7.4 | ThermoFisher | 70011044 | |
| PCR Flex-free 8-tube stripes, attached individual optical caps | USA Scientific | 1402-4700 | 8 strip tubes, 0.2 mL 8-tube strip |
| Proteinase Inhibitor Cocktail | Millipore Sigma | P8340 | |
| Proteinase K Solution (20 mg/mL), RNA grade | ThermoFisher | 25530049 | |
| PureLink RNase A (20 mg/mL) | ThermoFisher | 12091021 | |
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher | P7581 | Used in the flourescence quantification |
| QuantStudio 6 Flex Real-Time PCR System | ThermoFisher | 4485699 | qPCR |
| ROX Reference Dye | ThermoFisher | 12223012 | |
| Sodium butyrate | Millipore Sigma | 303410-100G | |
| SYBR Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) | ThermoFisher | S11494 | nucleic acid dye |
| SYBR Green I Nucleic Acid Gel Stain - 10,000X concentrate in DMSO | ThermoFisher | S7563 | |
| TE Buffer | ThermoFisher | 12090015 | |
| Tips (bulk) | Diagenode | C30040020 | Tips for the IP Star |
| True MicroChIP Kit | Diagenode | C01010130 | Contains all the buffers for the IP; ChIP kit |
| UltraPure 1M Tris-HCI, pH 8.0 | ThermoFisher | 15568025 | |
| UltraPure SDS Solution, 10% | ThermoFisher | 24730020 |
Riferimenti
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Johnson, D., Mortazavi, A., Myers, R., Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science. 316, 1497-1502 (2007).
- Mikkelsen, T. S., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 448 (7153), 553-560 (2007).
- Furey, T. S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nature Reviews Genetics. 13 (12), 840-852 (2012).
- Seumois, G., et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nature Immunology. 15 (8), 777-788 (2014).
- Schmiedel, B. J., et al. 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nature Communication. 7, 13426 (2016).
- Ai, S., et al. Profiling chromatin states using single-cell itChIP-seq. Nature Cell Biology. 21 (9), 1164-1172 (2019).
- Brind'Amour, J., et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature Communication. 6, 6033 (2015).
- Kaya-Okur, H. S., et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communication. 10 (1), 1930 (1930).
- Youhanna Jankeel, D., Cayford, J., Schmiedel, B. J., Vijayanand, P., Seumois, G. An Integrated and Semiautomated Microscaled Approach to Profile Cis-Regulatory Elements by Histone Modification ChIP-Seq for Large-Scale Epigenetic Studies. Methods Molecular Biology. 1799, 303-326 (2018).
- Schmidl, C., Rendeiro, A. F., Sheffield, N. C., Bock, C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nature Methods. 12 (10), 963-965 (2015).
- Skene, P. J., Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 6, 21856 (2017).
- Schmiedel, B. J., et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 175 (6), 1701-1715 (2018).
- Landt, S. G., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research. 22 (9), 1813-1831 (2012).
- Chen, L., et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell. 167 (5), 1398-1414 (2016).
- Lienhard, M., Grimm, C., Morkel, M., Herwig, R., Chavez, L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 30 (2), 284-286 (2014).
- Engel, I., et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nature Immunology. 17 (6), 728-739 (2016).
- Grosselin, K., et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature Genetics. 51 (6), 1060-1066 (2019).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Wang, Q., et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Molecular Cell. 76 (1), 206-216 (2019).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
- Diagenode. The Ultimate Guide for Chromatin Shearing Optimization with Bioruptor Standard and Plus. Diagenode. , (2012).
Erratum
Formal Correction: Erratum: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies
Posted by JoVE Editors on 9/14/2020. Citeable Link.
An erratum was issued for: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies. An author's name was updated.
The name was corrected from:
Pandurangan Vijayanad
to:
Pandurangan Vijayanand
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati