
Method Article
Ottimizzazione della crescita dei cristalli di endotiapepsina per esperimenti di cristallografia seriale
In questo articolo
Riepilogo
Lo scopo di questo articolo è quello di dare allo spettatore una solida comprensione di come trasformare il loro protocollo di diffusione del vapore di piccoli volumi, per la crescita di grandi cristalli proteici singoli, in un metodo di microcristallizzazione batch di grandi volumi per la cristallografia seriale.
Abstract
Qui viene presentato un protocollo per facilitare la creazione di grandi volumi (> 100 μL) di fanghi microcristallini adatti per esperimenti di cristallografia seriale sia a sincrotroni che a XFEL. Il metodo si basa sulla comprensione del diagramma di fase dei cristalli proteici e su come tale conoscenza può essere utilizzata. Il metodo è diviso in tre fasi: (1) ottimizzazione della morfologia dei cristalli, (2) transizione al batch e (3) ridimensionamento. La fase 1 prevede la ricerca di singoli cristalli ben diffusi, si spera ma non necessariamente, che si presentano in una morfologia simile a un cubo. Nella fase 2, la condizione della fase 1 è ottimizzata dal tempo di crescita dei cristalli. Questa strategia può trasformare i cristalli cresciuti dalla diffusione del vapore in batch. Una volta che la crescita dei cristalli può avvenire entro circa 24 ore, un morfogramma della miscela proteica e precipitante può essere tracciato e utilizzato come base per una strategia di ridimensionamento (Fase 3). Quando i cristalli possono essere coltivati in batch, è possibile tentare il ridimensionamento e ottimizzare la dimensione e la concentrazione del cristallo all'aumentare del volume. L'endotiapepsina è stata utilizzata come proteina dimostrativa per questo protocollo. Alcune delle decisioni presentate sono specifiche per l'endotiapepsina. Tuttavia, si spera che il modo in cui sono stati applicati ispiri un modo di pensare a questa procedura che altri possano adattare ai propri progetti.
Introduzione
La cristallografia macromolecolare a temperatura ambiente (RT) è di nuovo popolare all'interno della comunità della biologia strutturale. Lo sviluppo di sorgenti luminose XFEL (X-ray Free Electron Laser) ha stimolato lo sviluppo di approcci di consegna del campione RT 1,2,3,4, e questi metodi sono stati ora applicati ai sincrotroni 5,6,7,8. Non solo i metodi RT aprono la possibilità di strageties sperimentali pompa-sonda 9,10,11,12, ma ci sono anche prove crescenti che promuovono stati conformazionali alternativi all'interno delle proteine 13,14,15,16,17.
Tuttavia, la ragione principale per cui i criometodi hanno guadagnato trazione rispetto agli approcci RT alla fine del 1990 è stato il rallentamento del danno da radiazioni da temperature dei cristalli sotto zero18. I criometodi19 hanno iniziato a consentire la raccolta di un set di dati completo da un singolo cristallo proteico. I moderni metodi RT presso XFEL e sincrotroni hanno risolto il problema del danno da radiazione monocristallina mediante lo sviluppodi strategie di rilascio rapido (> 100 Hz) 1,2,3,4. Questi metodi consentono la raccolta di un set di dati completo da migliaia di cristalli esposti individualmente. Questi approcci di rilascio RT richiedono quindi la produzione di grandi quantità di soluzioni contenenti microcristalli omogenei (> 100 μL di cristalli < 50 μm). Tuttavia, poiché i criometodi tendono a richiedere solo cristalli singoli, i metodi per creare tali fanghi microcristallini non sono attualmente onnipresenti nei laboratori di cristallografia proteica.
Ci sono esempi in letteratura di come eseguire parti della procedura di ottimizzazione della microcristallizzazione per campioni di cristallografia seriale. Qui, si dovrebbe fare una distinzione tra proteine di membrana e proteine solubili. I protocolli per ottimizzare la crescita di cristalli proteici micro-membrana cresciuti in monooleina (o qualche altro lipide), per la fase cubica lipidica (LCP), sono stati ben descritti20,21,22. Tuttavia, i metodi per la microcristallizzazione delle proteine solubili, comprese le proteine di membrana coltivate in condizioni non LCP, sono generalmente carenti. Studi precedenti si sono concentrati su parti specifiche del processo, come lo screening dei microcristalli 23,24, il miglioramento della nucleazione24 e il ridimensionamento utilizzando la diffusione a interfaccia libera 25, ma non un metodo completo.
Tuttavia, è stato recentemente descritto un metodo26 che tenta di offrire un protocollo completo. Come molti aspetti della cristallografia delle proteine, non è una novità. Molte delle idee proposte erano già state descritte da Rayment (2002)27. Il metodo mira a mostrare ai cristallografi come eseguire la conversione da un singolo cristallo coltivato utilizzando la diffusione del vapore, a una metodologia batch per far crescere migliaia di microcristalli. Il metodo si concentra sulla diffusione del vapore come punto di partenza comune, poiché il 95% di tutte le deposizioni della Protein Data Bank (PDB) proviene da cristalli cresciuti in piastre di diffusione del vapore26. La diffusione del vapore non è, tuttavia, il metodo ideale per la microcristallizzazione26, quindi viene descritta una metodologia per convertire la diffusione del vapore in cristallizzazione batch. Una volta che i cristalli possono essere coltivati in batch, i percorsi di ridimensionamento a volumi più grandi diventano più praticabili. Dati i capricci della cristallizzazione delle proteine, gli autori sottolineano che questo metodo non è sicuro. Tuttavia, il protocollo dovrebbe, almeno, fornire una panoramica dello "spazio di cristallizzazione" di una proteina.
Questo metodo si basa sul diagramma di fase di cristallizzazione delle proteine e su come la comprensione di tale diagramma possa fungere da guida durante l'ottimizzazione della microcristallizzazione. Un diagramma di fase proteica è comunemente rappresentato come un grafico x/y con concentrazioni di precipitante e proteine rispettivamente sugli assi x e y (Figura 1A). Dal punto di acqua pura (angolo in basso a sinistra - Figura 1A), la concentrazione di proteine e precipitante aumenta fino a raggiungere la linea di solubilità. La linea di solubilità segna il punto di supersaturazione (linea viola - Figura 1A). Quando una proteina è supersatura, la soluzione diventa termodinamicamente instabile e inizierà a separarsi in due fasi: "ricca di proteine" e una soluzione satura stabile. Questa separazione può avvenire ovunque oltre la linea di solubilità e la sua cinetica dipende dalle proprietà della proteina e dai componenti della soluzione.
Quando le concentrazioni di proteine e precipitanti sono troppo grandi, la proteina si decompone in modo instabile dalla soluzione e provoca precipitato amorfo (regione rosa - Figura 1A). Tuttavia, la separazione di fase ordinata può avvenire nella regione di nucleazione [vedi Garcia-Ruiz (2003) 28 per una descrizione dettagliata] e i nucleant cristallini hanno la propensione a formarsi (regione verde - Figura 1A). La nucleazione e la crescita rimuovono le proteine dalla soluzione e spostano la goccia nella regione metastabile dove la crescita può continuare fino al raggiungimento della linea di solubilità [vedi McPherson e Kuznetsov (2014) 29 per una discussione dettagliata]. Il diagramma è, per la stragrande maggioranza delle condizioni di cristallizzazione, una grossolana semplificazione30. Indipendentemente da ciò, tuttavia, il diagramma è ancora di grande utilità per i microcristallografi in quanto la mappatura del diagramma consente di determinare la linea di solubilità e la cinetica della nucleazione.
In termini di creazione di microcristalli, i due fattori durante la cristallizzazione che devono essere ottimizzati sono il numero di cristalli (Xn) e la loro dimensione media più lunga (Xs). X n sarà proporzionale al numero di eventi di nucleazione (n ) (Eq. 1).
 Eq. 1
Eq. 1
X s è proporzionale alla concentrazione di proteine libere al di sopra della linea di solubilità (Ps) divisa per Xn (Eq. 2).
 Eq. 2
Eq. 2
In una situazione perfetta, ogni evento di nucleazione produrrebbe un possibile cristallo e ognuno di questi cristalli avrebbe uguale accesso alla proteina disponibile in soluzione. La Figura 2 è una rappresentazione grafica di uno scenario ideale della relazione tra Xn e Xs. In pratica, il controllo principale che un cristallografo ha su Xn e Xs è influenzando la quantità di nucleazione o con l'aggiunta di cristalli di semi. Il microcristallografo deve giudicare come aumentare Xn in modo tale da poter creare una concentrazione di cristallo adatta e una dimensione del cristallo.
La maggior parte delle tecniche di cristallizzazione richiede un "periodo transitorio" (Figura 1B). Ad esempio, in un esperimento di diffusione del vapore, dopo aver miscelato le proteine e le soluzioni precipitanti, le concentrazioni di ciascuna cambieranno man mano che la goccia si equilibra con la soluzione del pozzo. Si spera che questi cambiamenti trasferiscano gradualmente la goccia nella zona di nucleazione dove aumenterà la propensione alla cristallizzazione. Quando i cristalli iniziano a nucleane e crescere, la quantità di proteine in soluzione inizierà a diminuire, diminuendo la probabilità di un'ulteriore nucleazione. La quantità finale di nucleazione sarà specifica per proteine e condizioni, e dipenderà anche dalla profondità di penetrazione nella zona di nucleazione. Data la limitata penetrazione della zona di nucleazione dei metodi che richiedono una fase transitoria, il livello di nucleazione sarà in definitiva limitato alla velocità di nucleazione al limite della regione di nucleazione metastabile.
A causa dell'importanza di essere in grado di migliorare il livello di nucleazione per un microcristallografo, è importante passare a una metodologia di cristallizzazione batch. Il lotto può trarre maggior vantaggio dall'intera regione di nucleazione (Figura 1C). Nei metodi batch, l'idea è quella di mescolare la proteina e il precipitante insieme in modo tale da creare una soluzione supersatura senza la necessità di cambiamenti nelle concentrazioni dei componenti. La nucleazione dovrebbe essere possibile immediatamente dopo la miscelazione. I metodi batch consentono quindi di raggiungere teoricamente l'intera zona di nucleazione. Qualsiasi aumento della cinetica di nucleazione oltre il limite metastabile-nucleazione può quindi essere utilizzato.
Se il livello basale della nucleazione dei cristalli non è sufficiente per generare un grande Xn, è possibile utilizzare metodi di microsemina. Nella micro-semina, i cristalli pre-cresciuti vengono rotti per creare un impasto di frammenti cristallini che possono fungere da impalcatura per la crescita di cristalli freschi31,32. La microsemina è stata ampiamente utilizzata nella preparazione seriale di campioni cristallografici come un modo per aumentare Xn senza la necessità di aumentare la nucleazione dei cristalli (Figura 1C).
La transizione dalla diffusione del vapore al lotto può essere visualizzata su un diagramma di fase come lo spostamento del punto di partenza sperimentale dalle regioni non supersature o metastabili alla zona di nucleazione. Questo può essere fatto aumentando le concentrazioni di proteine e/o precipitanti e/o il rapporto tra i due all'interno della goccia (Figura 1D), e osservando quali condizioni producono cristalli che appaiono rapidamente (< 24 ore)26. L'equilibrio completo della caduta di diffusione del vapore può richiedere giorni o settimane33. Pertanto, cercando condizioni che mostrano cristalli che appaiono rapidamente, è possibile trovare condizioni batch senza dover passare a formati di screening di cristallizzazione alternativi come micro-batch34,35,36,37.
Una volta trovata la zona di nucleazione, è stata trovata una condizione batch e si può creare un morfogramma - qui, un diagramma di fase approssimativo. Il morfogramma è di grande utilità quando si considera se utilizzare un protocollo seeded-batch o straight batch. Tracciando l'Xn in funzione della concentrazione proteica e del precipitante, è possibile effettuare una valutazione della cinetica di nucleazione26. Se X n rimane basso in tutta la regione di nucleazione, può essere necessario un lotto seminato per rendere Xn abbastanza grande da limitare la crescita dei cristalli. Questa valutazione è il primo passo nel processo di scalabilità a volumi più grandi (> 100 μL).
Questo metodo è stato progettato in modo tale da poter essere condotto nella maggior parte dei laboratori di cristallizzazione utilizzando apparecchiature standard di cristallizzazione a diffusione del vapore. Sono stati condotti anche molti studi che descrivono tecniche per facilitare molte parti di questo processo, se l'attrezzatura è disponibile. Questi includono, ma non sono limitati a, diffusione dinamica della luce (DLS) 25,27, imaging non lineare 20,24,25, diffrazione delle polveri 20,24,27 e microscopia elettronica 26 [vedi Cheng et al. (2020) 40 per una bella recensione].
Lo scopo di questo lavoro è quello di fornire una dimostrazione visiva del metodo per passare dalla cristallizzazione a diffusione di vapore di piccolo volume (< 500 nL) alla cristallizzazione batch di grandi volumi (> 100 μL). Endothiapepsin da Cryphonectria parasitica è stato usato come sistema di esempio per dimostrare questa traduzione. Il tipo di esperimento e il metodo di consegna del campione per cui sono richiesti i microcristalli influenzeranno l'output ideale di Xs 26. Per esperimenti di miscelazione che richiedono una risoluzione di tempo di millisecondi41 o ugelli virtuali gas-dinamici42, può essere auspicabile un X sfinale di < 5 μm. In questo caso, l'obiettivo era quello di produrre cristalli proteici che diffrattassero a circa 1,5 Å, per un esperimento pompa-sonda attivato da fotoni e utilizzando un approccio di consegna a bersaglio fisso.
Per fornire un'illustrazione dei requisiti del campione di un tale esperimento di cristallografia seriale utilizzando endotiapepsina, la Tabella 1 mostra i parametri sperimentali di un ipotetico esperimento. Le informazioni di esempio erano basate sul protocollo descritto di seguito. Date alcune stime prudenti sui tassi di successo e sui requisiti di raccolta dei dati, 50 mg è la stima del consumo totale del campione per l'intero esperimento.
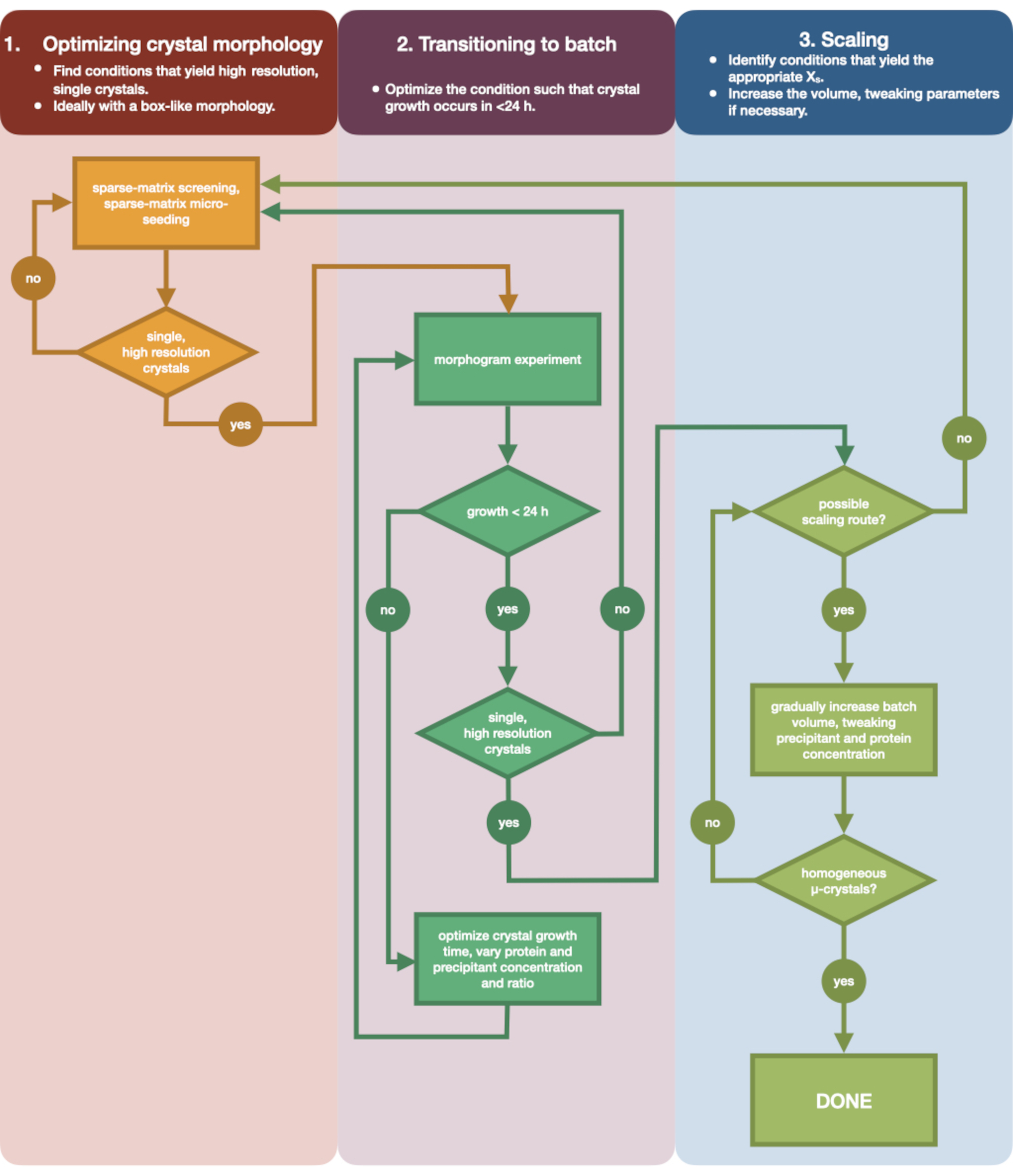
La Figura 3 mostra un diagramma di flusso del processo di ottimizzazione completo dalla cristallizzazione iniziale della diffusione del vapore di piccoli volumi al lotto su larga scala. Per la maggior parte dei progetti di cristallografia seriale, questo protocollo inizierà nella fase 2: "transizione al batch", poiché la proteina bersaglio sarà già stata cristallizzata. Tuttavia, il passaggio 1 è stato incluso per completezza e per ricordare ai lettori la sua importanza. Trovare una condizione che dia origine a un cristallo diffusore, singolo e grande è il miglior punto di partenza per l'ottimizzazione dei microcristalli. Nella fase 2, questa condizione può quindi essere ottimizzata dalla diffusione del vapore al lotto e può essere tracciato un morfogramma delle regioni di nucleazione e metastabilità. Una volta eseguita questa operazione, è possibile eseguire il ridimensionamento della condizione del batch a volumi più grandi nel passaggio 3. Alla fine del diagramma di flusso, un cristallografo avrà creato un protocollo batch ripetibile, di microcristallizzazione di grandi volumi (> 100 μL) per l'endotiapepsina. Questo metodo può quindi essere applicato alla loro particolare proteina di interesse.
Protocollo
NOTA: Tutti gli esperimenti di cristallizzazione a goccia da 96 pozzetti sono stati impostati utilizzando piastre a 2 o 3 gocce. Un robot per la gestione dei liquidi e un imager/hotel di cristallizzazione sono stati utilizzati per facilitare la preparazione e il monitoraggio di tutti gli schermi a 96 pozzi. Tutte le concentrazioni di reagenti per gli esperimenti di cristallizzazione sono indicate alle loro concentrazioni iniziali prima della miscelazione.
1. Ottimizzazione della morfologia dei cristalli
NOTA: Passaggi 1.1.1. e 1.1.6. descrivere come sono state trovate condizioni di cristallizzazione dell'endotiapepsina e come queste condizioni sono state ottimizzate per trovare una singola condizione che ha prodotto cristalli singoli e ben diffranti.
- Ottimizzazione a matrice sparsa
- Preparare una soluzione fresca di endotiapepsina.
NOTA: L'endotiapepsina, quando acquistata come Superan 600, deve essere trasferita con tampone dalla sua soluzione di stoccaggio e concentrata.- Preparare 3 L di 0,1 M Na acetato pH 4,6 a 4 °C.
- Tagliare 20 cm di tubo di dialisi e lavare brevemente nel tampone. Sigillare un'estremità del tubo usando una clip, posizionare 50 ml della soluzione di endotiapepsina nel tubo e quindi sigillare l'altra estremità.
- Lasciare la soluzione in dialisi per almeno 4 ore (o durante la notte) a 4 °C in 1 L del tampone di Na Acetato. A causa dei componenti del tampone di stoccaggio, la soluzione nella sacca per dialisi sarà ora di circa 100 ml.
- Trasferire la sacca per dialisi contenente l'endotiapepsina in un litro fresco di 4 °C, 0,1 M Na acetato pH 4,6. Ripetere ancora una volta questo passaggio in modo che il buffer originale sia stato diluito 2000x contro l'acetato di Na.
- L'endotiapepsina sarà ora a circa 10 mg / ml. Concentrare a 100 mg/mL utilizzando un concentratore centrifugo da 10 kDa e una centrifuga.
- Raffreddare la soluzione di endotiapepsina in azoto liquido in aliquote da 50 μL e conservare a -80 °C.
- Preparare uno schermo a matrice sparsa PACT Premier a 96 pozzetti.
- Utilizzando un robot per la manipolazione dei liquidi, erogare 100 nL di 70 mg/mL di endotiapepsina e 100 nL di soluzione di pozzetto in un singolo sottopozzetto per pozzetto. Mescolare la proteina e la soluzione del pozzo 3 volte dopo l'aggiunta del tampone di cristallizzazione.
- Sigillare la piastra e lasciare agire per 28 giorni a 20 °C scattando immagini ogni giorno per la prima settimana e successivamente ogni settimana per 4 settimane.
- Analisi a matrice sparsa
- Identificare i colpi che producono singoli cristalli di endotiapepsina. Dallo schermo PACT, le condizioni che contenevano MgCl2 sono cresciute come singoletti piuttosto che cluster di aghi.
- Ottimizzazione a matrice sparsa
- Dal MgCl2 contenente le condizioni identificate nel Passo 1.1.3.1, creare uno schermo a 96 pozzetti che combina e varia casualmente i diversi componenti del pozzo.
- Utilizzando un robot per la manipolazione dei liquidi, erogare 100 nL di 70 mg/mL di endotiapepsina e 100 nL di soluzione di pozzetto in un singolo sottopozzetto per pozzetto. Mescolare la proteina e la soluzione del pozzo 3 volte dopo l'aggiunta del tampone di cristallizzazione.
- Sigillare la piastra e lasciare agire per 28 giorni a 20 °C scattando immagini ogni giorno per la prima settimana e successivamente ogni settimana per 4 settimane.
- Analisi di ottimizzazione
- Utilizzando un software di foglio di calcolo adatto, classificare le condizioni di cristallizzazione che danno origine ai cristalli in base alla qualità del cristallo e al livello di precipitazione, senza cristalli (da 0) a ideali (5) e da basso (0) ad alto (5), rispettivamente. Per quanto riguarda la qualità dei cristalli, i criteri generali sono i cristalli singoli con una morfologia scatolare.
- Eseguire un'analisi di correlazione di Pearson tra il contenuto della condizione di cristallizzazione e la quantità di cristalli e il livello di precipitazione.
- Tracciare questi dati come una mappa di calore. Cerca componenti e condizioni correlati ai risultati preferiti.
- Analisi di diffrazione.
- Confermare che i cristalli cresciuti dalle condizioni identificate nel Passo 1.1.5 sono adatti per la cristallografia seriale eseguendo un esperimento di diffrazione a raggi X.
- Caricare un campione dei cristalli di endotiapepsina da ciascuna delle condizioni identificate su supporti che consentono la raccolta di dati a 100 o 293 K ed eseguire un esperimento di diffrazione a raggi X. Se si lavora sotto crio, utilizzare glicole etilenico al 25% come crio-protettivo.
- Elaborare questi dati tramite una suite software adeguata. I cristalli di endotiapepsina dovrebbero diffrattarsi oltre 1,5 Å. Verificare la presenza di gemellaggi, poiché i cristalli gemellati possono complicare significativamente l'elaborazione seriale dei dati cristallografici.
- Se i cristalli sono singoletti e si diffrattano a 1,5 Å, procedere al punto 2. In caso contrario, tornare al passaggio 1.1.2 e provare più schermi a matrice sparsa per identificare le condizioni promettenti. Dopo le analisi effettuate nelle fasi 1.1.5. e 1.1.6., una condizione di cristallizzazione del 25% (p/v) PEG 6.000, 0,1 M Tris-HCl pH 7,0 e 0,15 M MgCl2 avrebbe dovuto essere trovata come ideale approssimativo.
- Preparare una soluzione fresca di endotiapepsina.
2. Passaggio al batch
- Esperimento morfogramma
- Creare un ceppo di semi microcristallini.
NOTA: È buona norma quando si producono ceppi di semi, per produrre semi da cristalli appositamente coltivati per il compito. Questo aiuta notevolmente con la riproducibilità. Altre idee presentate nelle fasi da 2.1.1.1 a 2.1.1.11 sono di utilizzare sempre i cristalli cresciuti da un numero standard di pozzi - qui 5 - e di aliquotare le scorte una volta che sono state fatte per annullare i cicli di congelamento-disgelo.- Preparare una piastra di cristallizzazione a 96 pozzetti con pozzetti contenenti il tampone di cristallizzazione: 25% (p/v) PEG 6.000, 0,1 M Tris-HCl pH 7,0 e 0,15 M MgCl2.
- Utilizzando un robot per la manipolazione dei liquidi, erogare 200 nL di endotiapepsina scongelata da 70 mg/ml e 200 nL di soluzione di pozzetto in un singolo pozzetto per pozzetto. Mescolare la proteina e la soluzione del pozzo 3 volte dopo l'aggiunta del tampone di cristallizzazione.
- Sigillare la piastra e lasciare per 24 ore.
- Riempire una provetta da centrifuga da 1,5 ml con 250 μL di tampone di cristallizzazione e 10-15 sfere di vetro da 1 mm. Lasciare raffreddare il tubo della centrifuga sul ghiaccio per 5-10 minuti.
- Selezionare 5 pozzetti con cristalli, aprire i pozzetti con un bisturi e, usando una punta per pipetta, schiacciare i cristalli nei pozzetti.
- Aspirare 1 μL di tampone dalla provetta della centrifuga ghiacciata e utilizzarla per omogeneizzare il liquame cristallino frantumato. Una volta omogeneo, aspirare l'intero liquame e raccoglierlo nel tubo della centrifuga raffreddato.
- Ripetere il passaggio 2.1.2.6 per ciascuno dei 5 pozzetti secondari.
- Vortice il tubo della centrifuga contenente il tampone, i fanghi e le perline raggruppati a 1000 giri / min per 30 s.
- Riportare il tubo della centrifuga a ghiaccio per 30 s.
- Ripetere i passaggi 2.1.2.8 e 2.1.2.9 altre due volte.
- Il ceppo è ora pronto e può essere suddiviso in lotti da 10 μL e conservato a -20 °C.
- Eseguire l'esperimento del morfogramma.
- Preparare uno schermo a griglia a 96 pozzetti a 2 gocce. Variare la concentrazione di PEG 6.000 dal 5 al 40% (p/v) lungo le colonne della piastra, mantenendo il tampone e il sale a 0,1 M Tris-HCl pH 7,0 e 0,15 M MgCl2, rispettivamente.
- Preparare una diluizione sequenziale di endotiapepsina in 0,1 M Na Acetato pH 4,6 da 100 a 12,5 mg/ml in 8 fasi. Una diversa concentrazione di endotiapepsina verrà utilizzata per ogni fila del piatto.
- Utilizzando un robot per la gestione dei liquidi, erogare 150 nL di endotiapepsina in entrambi i sottopozzetti 1 e 2. Nel sottopozzetto 1, erogare 150 nL della soluzione del pozzetto. Nel pozzetto 2, aspirare 50 nL di ceppo scongelato e 100 nL di soluzione di pozzetto, quindi erogare entrambi nella soluzione proteica. Mescolare le soluzioni 3 volte dopo l'aggiunta del tampone di cristallizzazione.
- Sigillare la piastra e lasciare a 20 °C scattando immagini ogni 0, 3, 6, 12, 18, 24 h, poi ogni giorno per la prima settimana e ogni settimana per le successive quattro. Se l'imaging automatico non è possibile, non preoccuparti dell'imaging orario il giorno 1.
- Creare un ceppo di semi microcristallini.
- Analisi morfogramma
- Guardando le immagini scattate dopo 24 ore, stimare il numero di cristalli presenti in ciascun pozzetto e registrare queste stime nel foglio di lavoro "generatore di morfogrammi" fornito. Queste stime non devono essere precise; Contare individualmente migliaia di microcristalli, se presenti, non è pratico o necessario. Principalmente cercare di garantire che le stime siano coerenti su tutta la piastra.
NOTA: La regola delle 24 ore si basava sulle osservazioni fatte in Beale et al . (2019)26. Le condizioni di cristallizzazione a diffusione del vapore possono richiedere giorni o settimane per equilibrarsi. I cristalli che appaiono rapidamente hanno maggiori probabilità di essere cresciuti attraverso un processo batch piuttosto che dal graduale equilibrio dei componenti di goccia. Il criterio delle 24 ore è, quindi, alquanto arbitrario e un tempo limite esatto tra un lotto e un esperimento di diffusione del vapore dipenderà dalla miscela specifica della condizione [vedi Beale et al. (2019) 26 per tutti i dettagli]. - Inserire le concentrazioni iniziali di endotiapepsina e PEG 6.000 nelle caselle indicate.
- Il foglio di lavoro traccerà automaticamente i risultati nel formato di diagramma di fase tradizionale con precipitante e concentrazione proteica rispettivamente sugli assi x e y . Le condizioni che danno origine solo a cristalli nelle loro gocce seminate indicano la regione metastabile del diagramma (blu trasparente), mentre le condizioni che hanno cristalli sia nelle gocce seminate che in quelle non seminate indicano la zona di nucleazione (verde fisso).
NOTA: Idealmente, la maggior parte della zona di nucleazione dovrebbe essere presente sul diagramma (cioè, ci sono alcuni pozzi chiari sul fondo del diagramma e alcuni precipitati dovrebbero essere visibili ad alte concentrazioni proteiche e precipitanti). Se questo non è il caso, forse, ripetere l'esperimento ma aumentare la concentrazione di proteine e / o precipitanti (se possibile). - Se il cristallo è apparso in meno di 24 ore, procedere al punto 2.3.1. In caso contrario, procedere al passaggio 2.4 e continuare l'ottimizzazione verso il batch.
- Guardando le immagini scattate dopo 24 ore, stimare il numero di cristalli presenti in ciascun pozzetto e registrare queste stime nel foglio di lavoro "generatore di morfogrammi" fornito. Queste stime non devono essere precise; Contare individualmente migliaia di microcristalli, se presenti, non è pratico o necessario. Principalmente cercare di garantire che le stime siano coerenti su tutta la piastra.
- Analisi dei cristalli
- Come detto alla fine del passaggio 1, prima di passare alla fase successiva, assicurati che questi cristalli abbiano la morfologia e la qualità di diffrazione desiderate. Per quanto riguarda la morfologia, i cristalli sono osservabilmente non gemellati e si formano come singoletti piuttosto che strutture simili a palle d'ago o a ventaglio? Per quanto riguarda la diffrazione, raccogliere dati di diffrazione dai cristalli, se possibile. Se questi cristalli non diffrattono, è improbabile che i cristalli cresciuti in un volume maggiore si diffrattino.
- Caricare un campione dei cristalli di endotiapepsina dall'esperimento del morfogramma su supporti che consentono la raccolta di dati a 100 o 293 K ed eseguire un esperimento di diffrazione a raggi X. Se si lavora sotto crio, utilizzare glicole etilenico al 25% come crio-protettivo.
- Elaborare questi dati tramite una suite software adeguata. I cristalli di endotiapepsina dovrebbero diffrattarsi oltre 1,5 Å. Attraverso il campione di cristalli, osservare la dimensione della cella, il numero totale di osservazioni e la mosaicità; Queste misure daranno un'indicazione sull'omogeneità dei cristalli diffrattori.
- Se la morfologia del cristallo e la qualità della diffrazione sono sufficienti, procedere al passaggio 3.
- Ottimizza il tempo di crescita dei cristalli.
NOTA: L'analisi del morfogramma (Passo 2.2) avrà dato un'indicazione del punto di partenza della cristallizzazione (cioè la regione del diagramma di fase in cui si trova la goccia quando le soluzioni precipitante e proteica sono state miscelate). Il calo nella regione metastabile o al di sotto della linea di solubilità? La cristallizzazione del lotto inizia nella zona di nucleazione (Figura 1C). L'obiettivo di questa fase è spostare questo punto di partenza da sotto la linea di solubilità o la regione metastabile, nella zona di nucleazione (Figura 1D). Se il seme cade dal punto 2.2. hanno prodotto cristalli rapidamente, questa è un'indicazione che la miscela di gocce è già nella regione metastabile, in caso contrario, allora è probabile che la goccia non sia supersatura.- Ottimizzazione del tempo di crescita dei cristalli.
- Utilizzando lo stesso schermo del punto 2.1.3, preparare un esperimento di cristallizzazione a diffusione del vapore a 96 pozzetti in una piastra a 3 gocce.
- Aumentare la concentrazione proteica iniziale di endotiapepsina sull'asse y (cioè, concentrare ulteriormente la proteina, forse 120 mg / ml per endotiapepsina).
- Eseguire una diluizione seriale, come al punto 2.1.3.2, in modo tale che ogni riga della piastra contenga una concentrazione proteica sequenzialmente inferiore.
- Utilizzare rapporti di goccia diversi in ciascuna delle tre gocce sulla piastra: 1: 1, 1: 2 e 2: 1, proteina: precipitante.
- Visualizza o immagina la targa il primo giorno alle 0, 3, 6, 12, 18, 24 h e poi ogni giorno per la prima settimana e ogni settimana per le successive quattro. Se l'imaging automatico non è possibile, non preoccuparti dell'imaging orario il giorno 1.
- Identifica le gocce che producono i cristalli che appaiono più rapidamente e li rende i punti di partenza di ottimizzazioni ripetute fino a quando la crescita dei cristalli avviene entro 24 ore.
- Quando è stata identificata una condizione cristallina che appare rapidamente, tornare al Passo 2.1 per ridisegnare il morfogramma come preludio per iniziare la scalatura.
- Ottimizzazione del tempo di crescita dei cristalli.
3. Ridimensionamento
- Percorsi di ridimensionamento del rango. In questa fase, non è necessario decidere su un singolo percorso di scala, ma solo identificare e classificare le opzioni in modo che possano essere esplorate a turno. Man mano che il volume della miscela batch viene aumentato durante la procedura di ridimensionamento, si verificheranno cambiamenti nella velocità di nucleazione e nella gamma di dimensioni dei cristalli. Tuttavia, questi possono essere superati modificando attentamente le concentrazioni dei componenti man mano che il volume scalato aumenta.
NOTA: I passaggi 3.1.1 e 3.1.2 descrivono come discernere, dal morfogramma, se un protocollo batch o seeded-batch è più appropriato.- Protocollo batch semplice
- L'Xn nella zona di nucleazione è proporzionale alla concentrazione proteica e/o precipitante? Cioè, X n aumenta in funzione della concentrazione del precipitante e/o della proteina? -Sì? Andare al passaggio 3.1.1.2. No? Andare al passaggio 3.1.2.
- Individuare le condizioni che producono cristalli della dimensione richiesta e andare al passaggio 3.2.
- Protocollo seeded-batch
- La Xn piatta attraversa la zona di nucleazione? Cioè, Xn non aumenta in funzione della concentrazione di precipitanti e/o proteine.
- Individua le condizioni del seme che producono cristalli della dimensione richiesta e vai al passaggio 3.2. Se tutti i cristalli sono troppo grandi, andare al punto 3.1.2.3.
- Ripetere l'esperimento del morfogramma (Passo 2.1) ma questa volta aumentare la concentrazione del ceppo di semi utilizzato nei pozzetti seminati. Lo stock di semi può essere aumentato utilizzando più cristalli nella sua creazione. Ad esempio, invece di 5 pozzetti nel passaggio 2.1.1.5, utilizzare 10 pozzi.
- Visualizza o immagine la piastra sopra le prime 0, 3, 6, 12, 18, 24 h.
- L'Xn dovrebbe essere aumentato e l'Xs diminuito nelle gocce di semi. Ripetere questo ciclo se sono necessari cristalli più piccoli e quindi seguire un protocollo seeded-batch.
- Protocollo batch semplice
- Ridimensionamento graduale
- Ridimensionamento in piastre da 96 pozzetti. Dal morfogramma dell'endotiapepsina, è stato inizialmente selezionato per il ridimensionamento un metodo batch semplice che utilizza la condizione di cristallizzazione 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e PEG 6.000 al 30% (p/v). 100 mg/mL di endotiapepsina miscelati con il tampone di cristallizzazione in rapporto 1:1.
- Preparare 2-3 pozzetti in una piastra a goccia da 96 pozzetti con 100 μL di 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 30% (p/v) PEG 6.000.
- Utilizzando una soluzione di endotiapepsina appena scongelata da 100 mg/ml, erogare 0,5 μL di proteine e 0,5 μL di precipitante per pozzetto, sigillare e conservare a 20°C.
- Visualizza o immagine la piastra sopra le prime 0, 3, 6, 12, 18, 24 h. Notare eventuali cambiamenti nell'intervallo di Xs e Xn.
- Se si sono verificati cambiamenti, ripetere i punti da 3.2.1.1 a 3.2.1.2 ma aumentare o diminuire la concentrazione di proteine, precipitanti e/o semi per ripristinare eventuali modifiche nell'intervallo di Xs e Xn.
- Quando l'intervallo di Xs e Xn è accettabile, procedere al punto 3.2.2.
- Ridimensionamento in piastre a goccia sospesa a 24 pozzetti
- Preparare un singolo pozzetto di una piastra a goccia sospesa a 24 pozzetti ungendo i bordi del pozzetto con grasso sottovuoto.
- Preparare 0,5 ml di 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 30% (p/v) PEG 6.000 e riempire il pozzo unto.
- Utilizzando una soluzione di endotiapepsina appena scongelata, pipettare 1 μL di proteine sulla superficie di un vetrino. Pipettare 1 μL di tampone di cristallizzazione sulla goccia proteica e miscelare utilizzando la pipetta.
- Visualizza o immagine la piastra sopra le prime 0, 3, 6, 12, 24 h. Notare eventuali cambiamenti nell'intervallo di Xs e Xn.
- Se si sono verificati cambiamenti, ripetere i punti da 3.2.2.1 a 3.2.2.4 ma aumentare o diminuire la concentrazione di proteine, precipitanti e/o semi per ripristinare eventuali modifiche nell'intervallo di Xs e Xn.
- Quando/se l'intervallo di Xs e Xn è accettabile, procedere al punto 3.2.2.7.
- Ripetere i passaggi da 3.2.2.1 a 3.2.2.5, aumentando gradualmente il volume totale dell'esperimento fino a 10 μL.
- Una volta ad un volume pari o superiore a 10 μL, procedere con le provette da centrifuga al punto 3.2.3.
- Incrostazioni in provette da centrifuga
NOTA: Il perfezionamento della condizione del lotto di endotiapepsina è avvenuto principalmente al punto di volumi di 200 μL (vedere Risultati, Ridimensionamento). Il processo è iniziato con una condizione di cristallizzazione di 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 30% (p/v) PEG 6.000. Tuttavia, la concentrazione di PEG alla fine è cambiata al 40% (p / v). I semi erano anche necessari per controllare l'Xn e, per evitare che i cristalli crescessero troppo grandi, la crescita dei cristalli doveva essere estinta. I passaggi da 3.2.3.1 a 3.2.3.7 descrivono in dettaglio il processo di ottimizzazione delle condizioni. Passo 3.2.4. Descrivere il protocollo batch finale.- Preparare 1 mL di tampone di cristallizzazione: 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 30% (p/v) PEG 6.000.
- Utilizzando endotiapepsina da 100 mg/ml appena scongelata, aggiungere 25 μL di proteine in una provetta da centrifuga da 1,5 ml.
- Miscelare accuratamente il tampone di cristallizzazione con la soluzione proteica in rapporto 1:1 con una punta di pipetta. Posizionare il tubo in un revolver/rotatore ad alta agitazione a 20 °C.
- Prendere aliquote regolari (5, 10, 30, 60 min, 2, 5, 10, 24 h) 2,5 μL e visualizzare in un emocitometro. Registrare l'intervallo Xn e Xs .
- Se sono state apportate modifiche, ripetere i passaggi 3.2.3.1. al punto 3.2.3.4. ma aumentare o diminuire la concentrazione di proteine, precipitanti e/o semi per ripristinare eventuali modifiche all'intervallo di Xs e Xn
- Quando l'intervallo di X e Xn è accettabile, procedere al punto 3.2.3.7.
- Ripetere i punti da 3.2.2.1 a 3.2.2.5, aumentando gradualmente il volume totale dell'esperimento fino a 200 μL o superiore, secondo necessità.
- Protocollo finale seeded-batch
- Preparare il ceppo di semi.
- Preparare 2 mL di tampone di cristallizzazione: 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 40% (p/v) PEG 6.000.
- Utilizzando endotiapepsina appena scongelata, aggiungere 100 μL di proteine in una provetta da centrifuga da 1,5 ml.
- Miscelare accuratamente il tampone di cristallizzazione con la soluzione proteica in rapporto 1:1 con una punta di pipetta. Posizionare il tubo in un revolver/rotatore ad alta agitazione a 20 °C per 24 ore per consentire la crescita di cristalli da 50 μm.
- Aggiungere 10-15 perle di vetro da 1 mm al liquame di cristallo da 50 μm.
- Vortice il tubo della centrifuga contenente il liquame e le perline a 1000 giri / min per 30 s.
- Riportare il tubo della centrifuga a ghiaccio per 30 s.
- Ripetere i passaggi 3.2.4.1.5 e 3.2.4.1.6 altre 10 volte.
- Questo è ora 200 μL di un 1x seme-stock. Diluire il ceppo di semi 10 volte con l'aggiunta di 1,8 ml di tampone di cristallizzazione. Aliquotare il 10x stock di semi in lotti da 50 μL e conservare a -20 °C.
- Protocollo seeded-batch.
- Preparare il tampone di cristallizzazione: 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 40% (p/v) PEG 6.000.
- In una provetta da centrifuga, mescolare 100 μL di tampone di cristallizzazione con i 50 μL di 10x cementi appena scongelati.
- Utilizzando endotiapepsina 100 mg/mL appena scongelata, aggiungere 150 μL di proteine in una provetta da centrifuga da 1,5 ml.
- Miscelare accuratamente la miscela tampone di cristallizzazione/seme con la soluzione di endotiapepsina con una punta di pipetta e porre il tubo in un revolver/rotatore ad alta agitazione a 20 °C.
- Monitorare la cristallizzazione prendendo aliquote regolari da 2,5 μL e visualizzare i cristalli in un emocitometro. Registrare l'intervallo Xn e Xs .
- Dopo circa 80 minuti, quando i cristalli hanno raggiunto un Xs di 15 μm, spegnere la reazione aggiungendo 150 μL di 0,05 M Na Acetato pH 4,6, 0,05 M Tris-HCl pH 7,0, 0,075 M MgCl2 e 20% (p/v) PEG 6.000 (una soluzione composta da tampone endotiapepsina e tampone di cristallizzazione, miscelato 1:1).
- Conservare i cristalli a 20 °C.
- Il protocollo ha prodotto un intervallo di dimensioni e un numero di cristalli accettabili per l'esperimento previsto? Sì - FATTO- No - tornare al passaggio 3.1. e prova un'opzione di ridimensionamento alternativa. Ad esempio, un diverso rapporto proteina:precipitante può essere possibile o aggiungere semi se questo non è stato fatto in precedenza. Quando questi sono tutti esauriti, potrebbe essere necessario trovare una nuova condizione al passaggio 1.
- Preparare il ceppo di semi.
- Ridimensionamento in piastre da 96 pozzetti. Dal morfogramma dell'endotiapepsina, è stato inizialmente selezionato per il ridimensionamento un metodo batch semplice che utilizza la condizione di cristallizzazione 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e PEG 6.000 al 30% (p/v). 100 mg/mL di endotiapepsina miscelati con il tampone di cristallizzazione in rapporto 1:1.
Risultati
Ottimizzazione della morfologia dei cristalli
Il passaggio 1, l'ottimizzazione della morfologia del cristallo, è stato incluso per ricordare al lettore la sua importanza. Potrebbe essere possibile creare microcristalli perfetti da aghi scarsamente diffrazioni; Tuttavia, gli autori suggerirebbero che è meglio ottimizzare i due separatamente. In primo luogo, trovare condizioni che danno origine a un singolo cristallo ben diffuso tramite diffusione del vapore, e quindi convertire queste condizioni in batch piuttosto che cercare di combinare i due passaggi insieme. La scoperta di condizioni altamente nucleanti, in questa fase, non è necessaria; La morfologia e la qualità della diffrazione sono gli obiettivi principali.
Prima di iniziare la microcristallizzazione dell'endotiapepsina, è stata condotta un'analisi delle condizioni di cristallizzazione della struttura depositata dal PDB. Condizioni di cristallizzazione e protocolli approssimativi potrebbero essere ottenuti per 47 delle 48 deposizioni di entetiapepsina. Questi erano sostanzialmente tutti basati sulla prima cristallizzazione dell'endotiapepsina condotta da Moews e Bunn (1970) 46. Date le somiglianze di queste condizioni e la loro origine "classica", è stato eseguito uno schermo a matrice sparsa a 96 pozzetti, diffusione del vapore, per esplorare una più ampia varietà di condizioni di cristallizzazione. L'endotiapepsina è stata concentrata a 70 mg/ml e uno schermo PACT a matrice sparsa47 è stato eseguito in una piastra a goccia a 96 pozzetti a 20 °C mescolando 100 nL di proteine con 100 nL di soluzione di pozzetto. Ogni condizione di questo esperimento dopo 36 ore ha dato origine a cristalli. Tuttavia, un'analisi della morfologia dei cristalli ha indicato che alcune condizioni potrebbero rivelarsi migliori per l'ottimizzazione della microcristallizzazione.
La figura 4A mostra una goccia dallo schermo PACT che era ampiamente rappresentativa di quelle osservate nella maggior parte della piastra. A prima vista, si può essere tentati di pensare che questi cristalli potrebbero valere la pena di essere ulteriormente ottimizzati per la microcristallizzazione. I cristalli sono grandi e sembra esserci una nucleazione significativa. Tuttavia, la morfologia complessiva del cristallo non è ideale. In primo luogo, i cristalli non sono osservabili singoletti in quanto sembra che più cristalli stiano crescendo da singoli punti di nucleazione. In secondo luogo, la dimensione del cristallo è altamente asimmetrica con la crescita che si verifica principalmente lungo un singolo asse. Tali cristalli hanno teoricamente maggiori probabilità di allinearsi preferenzialmente quando vengono consegnati al fascio di raggi X. Entrambe le caratteristiche presentano problemi durante la raccolta e l'elaborazione di dati cristallografici seriali.
La figura 4B, tuttavia, mostra i cristalli di endotiapepsina cresciuti in presenza di MgCl2. Questa morfologia era coerente in tutte le condizioni che contenevano MgCl 2 e quindi suggeriva che la loro morfologia fosse dovuta a MgCl2. Le condizioni di MgCl2 hanno prodotto cristalli singoli, più simili a scatole, che rappresentavano un bersaglio migliore per gli esperimenti seriali finali.
C'erano quattro condizioni all'interno dello schermo PACT che conteneva MgCl2. Per comprendere meglio l'influenza di tutti i diversi componenti di queste condizioni sulla cristallizzazione dell'endotiapepsina, è stata eseguita un'ottimizzazione casuale. È stato creato uno schermo contenente una combinazione casuale di tamponi e precipitanti in un intervallo di concentrazioni e pH. Anche la concentrazione di MgCl2 è stata variata e quindi le gocce risultanti sono state arbitrariamente classificate da 0-5 (0 è assenza di cristalli o precipitazioni) in termini di qualità visiva dei cristalli e livello di precipitazione.
La Figura 5A mostra una mappa termica dei risultati di un'analisi di correlazione di Pearson tra il livello di precipitazione e la qualità dei cristalli e le variabili dello schermo (esempi delle gocce di questo esperimento sono mostrati in Figura 5B, C e D). I risultati hanno indicato che il pH della soluzione era altamente correlato al livello di precipitazione, con tamponi alcalini con conseguente maggiore precipitazione. La concentrazione di MgCl2 era leggermente correlata al livello di precipitazione, così come il pH e la concentrazione del precipitante alla qualità dei cristalli.
Sulla base di questi risultati, è stata presa la decisione di portare i cristalli cresciuti in 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2, 20% (w / v) PEG 6,000 alla fase successiva del protocollo - Transizione al batch. La morfologia dei cristalli era accettabile e un'analisi della diffrazione dei raggi X e delle metriche di qualità dei dati di questi cristalli ha suggerito che non vi era alcuna differenza significativa tra i cristalli cresciuti dentro e fuori dalla presenza di Mg2+ (Figura 9).
Transizione al batch
Per molte ottimizzazioni di microcristallizzazione seriale, la fase 2 sarà il punto di partenza. La proteina di interesse sarà già stata cristallizzata per la criocristallografia e il protocollo di cristallizzazione dovrà ora essere trasformato per creare fanghi microcristallini. Questo protocollo ha utilizzato solo piastre di diffusione del vapore a 96 pozzetti per eseguire la trasformazione in batch poiché la diffusione del vapore è il metodo di cristallizzazione utilizzato dal 95% delle voci PDB26. Il protocollo ha evitato di passare al microbatch34,35,37 poiché questa transizione potrebbe ancora comportare un'ottimizzazione simile. Questo non vuol dire che questo protocollo possa essere fatto solo in piastre di diffusione del vapore. Tutti i passaggi presentati funzionerebbero anche in microbatch se questo fosse il metodo di cristallizzazione originale.
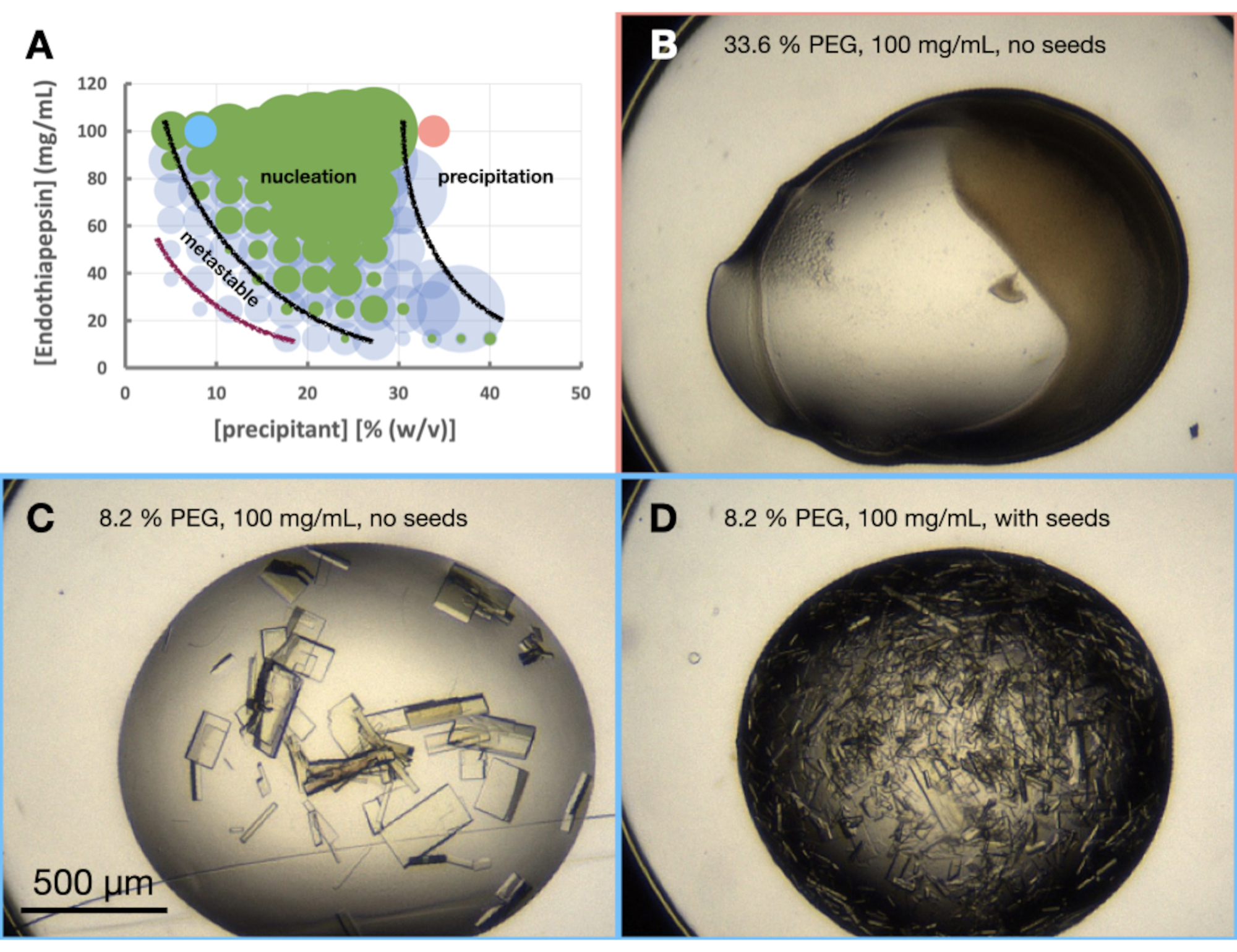
Per valutare la cristallizzazione dell'endotiapepsina nella condizione scelta, è stato creato un morfogramma o un diagramma di fase approssimativo. Lo scopo dell'esperimento del morfogramma è triplice. In primo luogo, un'analisi del morfogramma è di grande utilità quando si valutano i percorsi di ridimensionamento nella fase 3 - Scala. In secondo luogo, il morfogramma agisce come uno strumento di ottimizzazione, aiutando a scoprire le condizioni di diffusione del vapore che danno origine ai cristalli tramite batch [cioè cristalli che appaiono rapidamente (< 24 ore)]. In terzo luogo, se i cristalli non sono apparsi rapidamente, un'analisi delle gocce seminate può dare al cristallografo un'idea della posizione approssimativa della condizione corrente sul diagramma di fase. Ad esempio, se le condizioni seminate danno cristalli ma quelle non seminate no, è probabile che tali condizioni si trovino nella regione metastabile.
L'esperimento del morfogramma dell'endotiapepsina è stato eseguito sulla base della condizione 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, 20% (p/v) PEG 6.000. Le concentrazioni di proteine e PEG variavano rispettivamente da 100 a 12,5 mg/ml e dal 5 al 40% (p/v). Le gocce sono state analizzate e i risultati tracciati utilizzando il foglio di lavoro fornito (Figura 6A).
Era anche già chiaro dalla fase di ottimizzazione della morfologia dei cristalli che la crescita dei cristalli di endotiapepsina in questa condizione, e a queste concentrazioni proteiche, avrebbe portato a cristalli cresciuti in meno di 24 ore. Ciò indicava che la cristallizzazione stava avvenendo tramite un lotto piuttosto che un processo guidato dalla diffusione del vapore. Il cristallo cresciuto in queste condizioni era, quindi, adatto per il ridimensionamento a volumi maggiori.
Se i cristalli non fossero stati visibili nelle gocce non seminate dopo 24 ore, allora sarebbe stato probabile che la cristallizzazione dipendesse ancora da una transizione (Figura 1B) e, quindi, non in batch. In questo caso, i risultati dell'esperimento del morfogramma sono ancora interessanti. Forniscono un'indicazione del probabile punto di partenza per la cristallizzazione sul diagramma di fase e, quindi, come dovrebbe procedere la successiva ottimizzazione. Guarda le gocce seminate. I semi consentiranno la crescita dei cristalli nella regione metastabile indipendentemente dalla nucleazione. Ad esempio, se i cristalli appaiono entro 24 ore nelle gocce seminate ma non nelle gocce senza semi, ciò indica che è possibile osservare parte della regione metastabile. Se non si osservano cristalli nelle gocce seminate o non seminate, tutti i pozzetti rimangono sottosaturi.
Scalata
Osservando il morfogramma (Figura 6A), si potrebbero fare numerose osservazioni. La quantità di nucleazione sembra essere influenzata sia dalle concentrazioni proteiche che da quelle del precipitante. C'era anche una demarcazione molto chiara delle gocce che portano alla precipitazione proteica, con gocce contenenti: nulla, cristalli o precipitati (Figura 6B). Anche l'aggiunta di semi (Figura 6D) ha notevolmente aumentato Xn rispetto alle gocce senza semi (Figura 6C). Prendendo tutti questi risultati insieme, è stato deciso di tentare di scalare sia un protocollo batch che un protocollo seeded-batch al 30% (w / v) PEG 6.000 e 100 mg / mL endothiapepsin.
Il ridimensionamento iniziale del test è stato eseguito in piastre di caduta sospese a 24 pozzetti. I volumi di caduta sono stati gradualmente aumentati in modo da poter osservare eventuali cambiamenti nel comportamento di cristallizzazione (Figura 7). Come si può vedere, sia nelle gocce non seminate che in quelle seminate si è verificata la crescita dei cristalli. Tutte le gocce non seminate hanno sviluppato una gamma di dimensioni di cristalli, ma prevalentemente cristalli di grandi dimensioni (100-200 μm - dimensione più lunga). Le gocce seminate, tuttavia, producevano cristalli più piccoli (5 - 50 μm - dimensione più lunga). Questi test iniziali hanno suggerito che i semi sarebbero stati necessari per diminuire Xs, ma anche che questa condizione dovrebbe essere adatta per volumi più grandi.
Quando il volume è stato aumentato in 200 μL, il volume di cristallizzazione è stato continuamente agitato durante la crescita dei cristalli. La ragione principale di questa agitazione era quella di garantire che la soluzione di cristallizzazione rimanesse omogenea e che i cristalli in crescita non si depositassero sul fondo o sui lati dei tubi. L'assestamento dei cristalli può portare a una popolazione di cristalli eterogenea con cristalli sia molto grandi che piccoli. Agitare la soluzione di cristallizzazione può anche promuovere la nucleazione44,45.
Sfortunatamente, il PEG 6.000 non seminato al 30% (w / v) non ha prodotto cristalli, quindi la concentrazione di PEG è stata aumentata al 35% (w / v). Questo aumento ha migliorato notevolmente la cristallizzazione, con un intervallo finale Xn e Xs di 3,6 ± 1,2 x 106 cristalli·mL-1 e 42 ± 4,1 μm, rispettivamente (Figura 8A e B - nero). Sebbene un miglioramento significativo e una concentrazione di cristalli accettabile, i cristalli finali erano troppo grandi per l'esperimento pianificato, quindi sono state intraprese ulteriori ottimizzazioni. Per ridurre le dimensioni dei cristalli finali sono state esplorate due strade (Figura 1E): diminuendo la concentrazione proteica per cercare di limitare la crescita finale dei cristalli (Figura 8A e B - rosa caldo) e aumentando la concentrazione di PEG per cercare di aumentare la nucleazione (Figura 8A e B - verde).
La riduzione della concentrazione proteica purtroppo ha anche ridotto drasticamente l'Xn, che alla fine ha prodotto cristalli ancora più grandi. L'aumento della concentrazione di PEG al 40% ha prodotto un intervallo finale Xn e Xs di 3,1 ± 0,7 x 106 cristalli·mL-1 e 39 ± 2,3 μm, rispettivamente. Questi non erano significativamente diversi dal 35%, ma poiché la dimensione finale del cristallo è stata ridotta, questa condizione è stata mantenuta con ulteriori ottimizzazioni.
Per aumentare la Xn, sono stati aggiunti semi. Ciò ha aumentato drasticamente Xn (1,1 ± 1,8 x 108 cristalli ·mL-1) e ha portato a un X s più piccolo (4,2 ± 4,0 μm) (Figura 8A e B - viola tratteggiato). Questi cristalli, sebbene molto adatti per alcuni esperimenti di cristallografia seriale, sono stati ritenuti troppo piccoli, quindi la concentrazione dei semi aggiunti è stata modificata.
Questa messa a punto dello stock di semi aggiunto, tuttavia, si è rivelata difficile da ripetere in modo affidabile; Pertanto, è stata tentata la tempra. Dopo l'aggiunta di uno stock di semi, la dimensione del cristallo è stata monitorata e una volta raggiunta una dimensione del cristallo adatta (circa 10 - 20 μm), la cristallizzazione del lotto è stata spenta (Figura 8C e D). La tempra è stata proposta, per quanto riguarda la microcristallizzazione, in Kupitz et al. (2014)25. Anche se forse non è un metodo ideale, poiché la soluzione proteica alla fine verrà sprecata26, la tecnica è stata molto utile in questa situazione poiché la crescita dei cristalli era difficile da controllare. L'idea alla base della tempra è quella di riportare rapidamente la miscela di cristallizzazione in un punto appena sopra la linea di solubilità (Figura 1F). Una volta che la soluzione è tornata alla linea di solubilità, la soluzione è tornata ad una soluzione satura stabile e non si verificherà ulteriore crescita dei cristalli.
Il tentativo di estinguere una reazione di cristallizzazione non è privo di rischi. Se viene aggiunta una soluzione di tempra troppo grande, la proteina in soluzione potrebbe essere diluita così tanto da superare la linea di solubilità. In questo caso, la soluzione diventerà sottosatura e i cristalli inizieranno a dissolversi. Per evitare ciò, è possibile stimare la quantità di soluzione di tempra richiesta in base ai risultati del morfogramma. Al punto di tempra, prendi la concentrazione della soluzione proteica. Confrontando la concentrazione proteica sulla linea di solubilità e la concentrazione proteica in soluzione, è possibile effettuare una stima della diluizione richiesta.
La versione temprata dell'esperimento del 40% (p/v) PEG 6.000, 10 x semi diluiti ha fornito una concentrazione finale di cristalli e una gamma di dimensioni di 2,6 ± 3,1 x 106 cristalli·mL-1 e 15 ± 3,9 μm, rispettivamente.
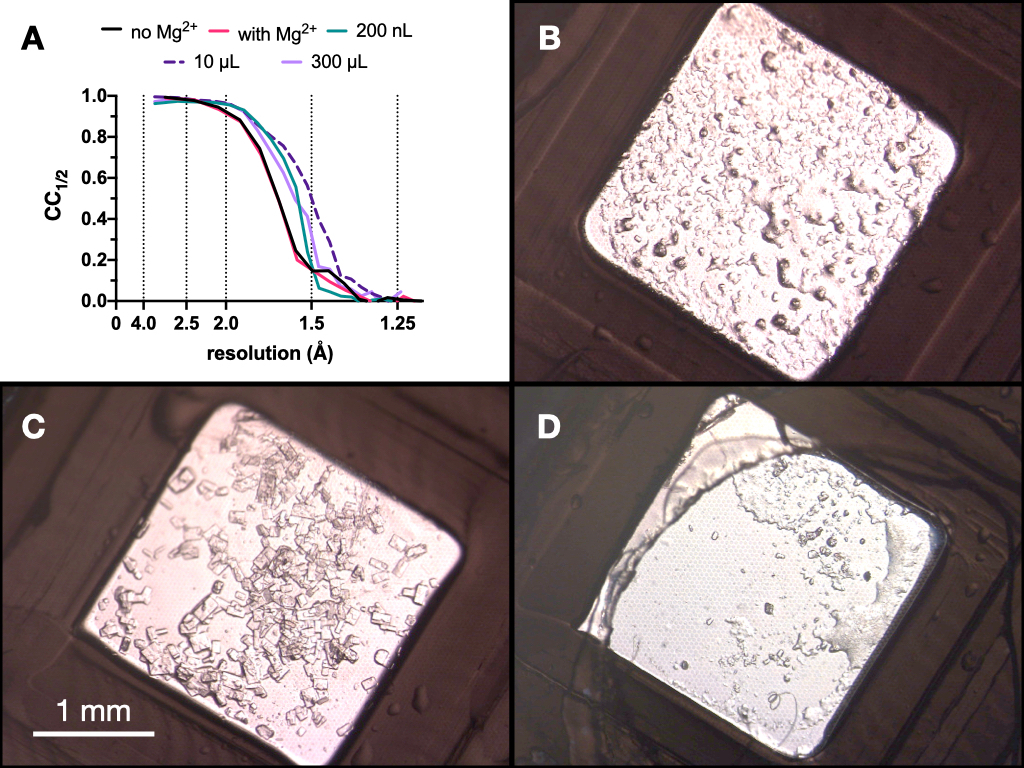
Durante l'intero processo, le raccolte di dati radiografici di prova dei cristalli di endotiapepsina sono state raccolte sulla linea di fascio PXII della sorgente luminosa svizzera utilizzando un fuoco di 10 x 30 μm, un'energia di 12,4 keV attenuata dell'80% e in condizioni crio-condizioni. I dati sono stati elaborati utilizzando quadranti e la figura 9 mostra un confronto di CC1/2. Nessun cambiamento drammatico in CC1/2 è stato osservato nel corso dell'ottimizzazione.

Figura 1: Una panoramica della cristallizzazione transitoria e batch e dei metodi di ridimensionamento mappati su un diagramma di fase. A. Le zone e i limiti del diagramma di fase di cristallizzazione della proteina archetipica. Le concentrazioni di precipitante e proteine sono tracciate rispettivamente sugli assi x e y , con il punto di acqua pura all'origine. La linea viola indica il confine di sovrasaturazione delle proteine e le zone metastabili, nucleazione e precipitazione sono mostrate rispettivamente in blu, verde e rosa. B. Un esempio dei limiti di penetrazione della zona di nucleazione di un metodo di cristallizzazione della "fase transitoria", come la diffusione del vapore. In questo esperimento teorico, il precipitante di caduta e le concentrazioni proteiche iniziano appena sotto la linea di solubilità - non ancora supersatura. Mentre la goccia si equilibra, le concentrazioni della componente goccia aumentano in modo tale che la goccia diventa supersatura e continua a spostarsi - o transizione - nella zona di nucleazione. Dopo la nucleazione dei cristalli, la concentrazione proteica in soluzione inizia a diminuire. La concentrazione continua a diminuire man mano che i cristalli crescono fino a fermarsi definitivamente alla linea di solubilità. La linea tratteggiata blu segna un limite teorico della transizione nella zona di nucleazione. Non appena inizia la nucleazione, la concentrazione proteica diminuirà, impedendo un'ulteriore penetrazione. C. Esempio di traiettorie di cristallizzazione batch e seeded-batch. In batch, la miscelazione della proteina e del precipitante deve creare una soluzione supersatura all'interno della zona di nucleazione in modo che possa verificarsi la crescita dei cristalli. Nel lotto di semi, non è strettamente necessario trovarsi nella zona di nucleazione a causa dell'aggiunta di microsemi, quindi è possibile esplorare anche le posizioni nella regione metastabile. D. Un'ipotetica ottimizzazione dell'esperimento di cristallizzazione mostrato in B dalla diffusione del vapore al lotto. Il punto di partenza originale della diffusione del vapore è passato, tramite il vettore di ottimizzazione risultante, alla nuova posizione di partenza; all'interno della zona di nucleazione. Il vettore risultante è il prodotto di due ottimizzazioni: un aumento delle concentrazioni sia di proteine che di precipitanti. E. Esempi di ottimizzazioni durante il ridimensionamento delle condizioni batch per personalizzare le X ne X s finali. F. Estinzione dell'esperimento di cristallizzazione mediante l'aggiunta di tampone di cristallizzazione. È essenziale che la tempra non porti la concentrazione proteica fuori dalla regione metastabile e, quindi, al di sotto del punto di sovrasaturazione proteica. Altrimenti, i cristalli inizieranno a dissolversi di nuovo in soluzione. B. e C . sono stati adattati da Beale et al. (2019)26 con il permesso degli autori. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

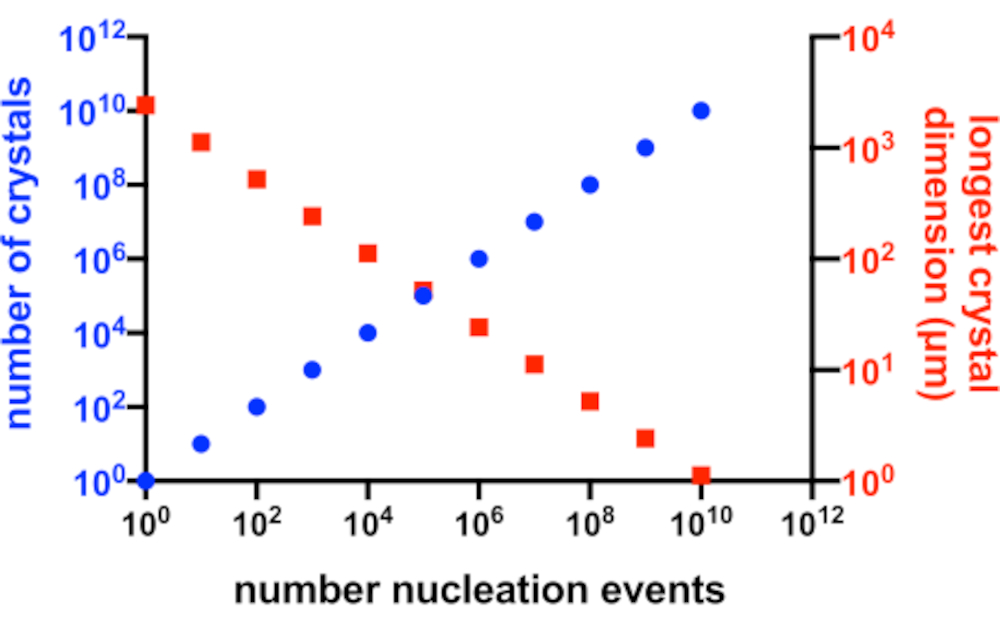
Figura 2: Aumento di Xn e diminuzione di Xs. La relazione idealizzata tra il numero di cristalli prodotti da un esperimento di cristallizzazione e la loro dimensione media più lunga. Per creare questo grafico, è stata utilizzata la cristallizzazione di un'ipotetica proteina modello da 10 kDa. La proteina cristallizzata ad una concentrazione di 10 mg/mL e ha prodotto P2 1 2 1211 cristalli con dimensioni di 49x50x51 Å. Si presumeva che ogni evento di nucleazione producesse un cristallo. Si presumeva che la crescita dei cristalli fosse omogenea da ogni faccia. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 3: Un diagramma di flusso che mostra i passaggi per ottimizzare un cristallo coltivato in un esperimento di diffusione del vapore di piccolo volume (<500 nL) in un esperimento batch di grandi volumi (> 100 μL). L'ottimizzazione dei cristalli è divisa in tre fasi: (1) Ottimizzazione della morfologia del cristallo. (2) Passaggio al lotto. (3) Ridimensionamento. Nella fase 1 è importante identificare i cristalli adatti per la microcristallizzazione. Alcune proteine sono presenti solo in una morfologia a singolo cristallo indipendentemente dalla condizione di cristallizzazione. Tuttavia, vale la pena cercare condizioni che danno origine a singoli cristalli simili a cubi, o il più vicino a questi umanamente possibile. Singoli cristalli simili a cubi, ipoteticamente e aneddoticamente, daranno generalmente origine a risultati migliori dagli esperimenti di cristallografia seriale. Una volta selezionata la morfologia di un cristallo e confermata la diffrazione è necessario spostare l'esperimento di cristallizzazione dalla diffusione del vapore al lotto (Fase 2). Qui, i cristalli dovrebbero essere ottimizzati dal loro tempo di nucleazione. L'obiettivo è quello di trovare condizioni che producano cristalli che appaiono rapidamente (> 24 ore) poiché è probabile che queste condizioni colpiscano immediatamente la zona di nucleazione e siano quindi batch. Una volta trovata una condizione nella zona di nucleazione, è possibile creare un morfogramma. Il morfogramma consente alla maggior parte della zona di nucleazione di mappare e potenziali percorsi di scala identificati per la fase 3. Il volume di una condizione di lotto identificata può quindi essere gradualmente o rapidamente scalato per produrre un volume finale di >100 μL. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Un'analisi delle condizioni di cristallizzazione dell'endotiapepsina da uno schermo a matrice sparsa PACT. A. e B. sono foto dopo 24 ore dei pozzetti A4 e C10, rispettivamente dallo schermo PACT . I componenti del buffer di cristallizzazione sono evidenziati nella figura. Il tampone SPG è acido succinico, sodio diidrogeno fosfato e glicina miscelati in un rapporto molare 2: 7: 7. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 5: Un'analisi dell'ottimizzazione della cristallizzazione dell'endotiapepsina dalle condizioni PACT MgCl2 . Una mappa termica dei risultati di un'analisi di correlazione di Pearson tra pH tampone, concentrazione di MgCl2 e concentrazione del precipitante e livello di precipitazione e qualità dei cristalli. Il livello di precipitazione e la qualità dei cristalli sono stati entrambi valutati arbitrariamente su una scala da 0 a 5 (dove 0 è assenza di cristalli o precipitazioni) dopo 24 ore B. C. e D . mostrano esempi di cristallizzazione e precipitazione in tre diverse gocce. Vengono inoltre mostrate le condizioni di cristallizzazione e le valutazioni del livello di precipitazione e della qualità dei cristalli. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 6: Morfogramma di endotiapepsina quando cristallizzato in 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e PEG 6.000. Un morfogramma creato dal foglio di calcolo "phase-diagram-generator" fornito. Il numero relativo di cristalli in ogni goccia è indicato dalla dimensione dei cerchi e i risultati della goccia 1 (proteine e precipitante) e della goccia 2 (proteine, precipitanti e semi) sono evidenziati rispettivamente in verde e blu. I valori delle concentrazioni di proteine e precipitanti, rispettivamente sull'asse x e y, indicano valori premiscelati di ciascun volume anziché finale. Sulla base dei risultati, sono state tracciate linee nere e una linea viola per mostrare i confini della zona di nucleazione e della zona metastabile, rispettivamente. a. C. e D. mostrano alcuni risultati di esempio dell'esperimento. I punti rossi e blu contrassegnati su A. indicano le posizioni di B. e C. e D., rispettivamente. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 7: Prove iniziali di ridimensionamento dell'endotiapepsina in piastre a goccia sospese a 24 pozzetti. Le stesse concentrazioni di proteine e precipitanti sono state utilizzate per tutte le tracce: 100 mg/mL di endotiapepsina in 0,1 M Na acetato pH 4,6 e 0,1 M Tris-HCl pH 7,0, 0,15 M MgCl2 e 30% (p/v) PEG 6.000, rispettivamente. Tutte le immagini visualizzate sono state scattate dopo 24 ore e i volumi finali di caduta sono etichettati su ogni immagine. Il pannello di sinistra (A, D e G) è un mix 1:1 di proteine e precipitante, il pannello centrale (B, E e H) è un mix 1:2:3 di semi, precipitante e proteine e il pannello di destra (C, F e I) sono immagini ingrandite del pannello centrale. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 8: Analisi della microcristallizzazione dell'endotiapepsina in volumi di 200-300 μL. A. e C. mostrano come Xn è cambiato nel tempo dell'esperimento. B. e D. mostrano come Xs (dimensione più lunga) è cambiato nel tempo. I risultati degli esperimenti sono stati separati per chiarezza. La linea tratteggiata rossa su C. e D. mostra il punto in cui è stata eseguita la tempra. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 9: Risultati CC1/2 e immagini dei cristalli ottenuti in ogni fase del processo di microcristallizzazione per valutare la qualità della diffrazione. CC1/2 tracciato rispetto alla risoluzione dai dati raccolti da cristalli cresciuti: con e senza Mg - parte dell'ottimizzazione Stage 1, in un volume di 200 nL, un volume di 10 μL e il volume finale di 300 μL. B.C. e D. mostrano i cristalli del volume di 200 nL, 10 μL e 300 μL, rispettivamente. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
| Informazioni sulle proteine | |
| Proteina | Endotiapepsina |
| Peso molecolare (kDa) | 33.8 |
| Gruppo spaziale | P12 11 |
| a, b, c (Å) | 45.2, 73.3, 52.7 |
| α, β, ɣ (°) | 90.0, 109.2, 90.0 |
| Parametri a destinazione fissa | |
| Volume caricato per chip (μL) | 150 |
| Aperature per chip | 25,600 |
| Concentrazione di cristalli richiesta (cristalli/ml) | 500,000 |
| Informazioni di esempio | |
| Massa proteica utilizzata per produrre 200 μL di campione (mg) | 10 |
| Dimensione più lunga del cristallo (μm) | 15 |
| Concentrazione di cristalli (cristalli/ml) | 2,500,000 |
| Variabili sperimentali | |
| Numero di punti temporali richiesti | 5 |
| Numero di immagini richieste per punto temporale | 50,000 |
| Hit rate (pattern integrati/immagini raccolte) | 0.3 |
| Obiettivi fissi richiesti per punto temporale (arrotondati per eccesso) | 7 |
| Requisiti di esempio | |
| Volume di campione richiesto per punto temporale (μL) | 1,050 |
| Volume totale del campione richiesto per l'esperimento (ml) | 5.25 |
| Massa totale di proteine richieste (mg) | 52.5 |
Tabella 1: Un esempio dei requisiti di campionamento per un ipotetico esperimento di pompa-sonda ottica eseguito utilizzando bersagli fissi. La proteina utilizzata in questo esperimento teorico era l'endotiapepsina. I parametri a bersaglio fisso erano basati su esperimenti riportati in Ebrahim et al. (2019) 48 e Davy et al. (2019) 49. Le informazioni campione provenivano dal protocollo riportato in questo articolo video e le variabili sperimentali erano stime conservative basate sull'esperienza vissuta. I seguenti requisiti di campionamento sono stati successivamente calcolati sulla base delle ipotesi precedenti.
Discussione
Il metodo presentato mostra come ottimizzare la cristallizzazione dell'endotiapepsina da cristalli di grandi dimensioni (≥ dimensione più lunga di 100 μm), cresciuti in schermi a 96 pozzetti a matrice sparsa, a microcristalli, cresciuti in provette da centrifuga (volume 300 μL) tramite lotto. L'idea alla base del protocollo è che le misure adottate per ottimizzare l'endotiapepsina potrebbero essere utilizzate anche per altre proteine. In definitiva, rispondere al problema della creazione di grandi volumi (>100 μL) di microcristalli (10-20 μm) per esperimenti di cristallografia seriale a XFEL e sincrotroni.
Il protocollo divide il compito della microcristallizzazione di grandi volumi in tre fasi: (1) ottimizzazione della morfologia dei cristalli, (2) transizione al batch e (3) ridimensionamento. Nella fase 1, la gamma di forme cristalline che una proteina può creare dovrebbe essere esplorata nelle piastre di diffusione del vapore. L'obiettivo dovrebbero essere le condizioni che danno origine a singoli cristalli simili a scatole che si diffrattano alla risoluzione richiesta. Nella fase 2, le condizioni selezionate possono quindi essere trasformate dalla diffusione del vapore in batch. Qui, il criterio di ottimizzazione è il tempo di crescita dei cristalli e trovare le condizioni che danno origine a cristalli proteici entro 24 ore. Un morfogramma può anche essere tracciato dando allo sperimentatore un'idea della posizione della linea di solubilità e dei confini della zona di nucleazione. Questo morfogramma è di grande utilità nel passaggio 3, scalabilità. Il morfogramma darà un'indicazione del fatto che la nucleazione da sola possa aumentare Xn e ridurre Xs. Man mano che il volume dell'esperimento aumenta, Xn e Xs possono essere continuamente valutati come criteri chiave per il successo del ridimensionamento.
Nel caso dell'endotiapepsina, la fase 1 ha portato alla luce quella che potenzialmente era una morfologia cristallina precedentemente sconosciuta per l'endotiapepsina. Questa morfologia aveva lo stesso gruppo spaziale di quelli precedentemente riportati ma, cosa importante per la cristallografia seriale, una forma più scatolare. Anche i singoli cristalli sembravano crescere da singoli punti di nucleazione, a differenza dei ventilatori creati da altre condizioni (Figura 4). Per la condizione selezionata, la fase 2 era già parzialmente soddisfatta poiché la crescita dei cristalli si è verificata in < 24 ore. Il morfogramma indicava che sia un protocollo lineare che un protocollo seeded-batch potrebbe avere successo durante il ridimensionamento nel passaggio 3. Il ridimensionamento iniziale in batch diritto, ha creato una condizione che ha prodotto cristalli con un intervallo Xn e Xs di 3,6 ± 1,2 x 106 cristalli·mL-1 e 42 ± 4,1 μm, rispettivamente. Questi cristalli, sebbene accettabili per alcuni esperimenti di cristallografia seriale, sono stati ritenuti troppo grandi. Quindi sono state eseguite ulteriori ottimizzazioni. Il protocollo finale ha prodotto cristalli con una gamma di concentrazioni e dimensioni di 3,1 x 106 cristalli·mL-1 e 15 ± 3,9 μm, rispettivamente. Questo era più che ideale per gli esperimenti pianificati.
Il metodo si concentra sulla trasformazione di cristalli proteici "solubili" cresciuti in piastre di diffusione del vapore in lotti. La ragione di questa attenzione è che la stragrande maggioranza dei cristalli proteici solubili viene coltivata attraverso la diffusione del vapore26. Tuttavia, i concetti presentati potrebbero essere applicati anche a cristalli proteici solubili coltivati utilizzando altri metodi, come il micro-batch. I concetti possono anche essere applicabili a cristalli proteici di membrana coltivati in LCP; poiché anche questo è un processo di cristallizzazione batch.
Un aspetto chiave del protocollo è il processo di trasformazione delle condizioni dei cristalli cresciuti in piastre di diffusione del vapore in modo tale che possano essere coltivati in batch. Per questa trasformazione, il metodo utilizza il criterio proposto da Beale et al. (2019)26. I cristalli cresciuti attraverso un processo batch, anche in piastre di diffusione del vapore, si formeranno rapidamente (< 24 ore). Questo criterio è un'approssimazione basata sulla velocità di equilibrio della caduta di diffusione del vapore, ed è più vero per le condizioni precipitanti basate su PEG. Tuttavia, le condizioni di cristallizzazione conterranno un'ampia varietà di composti che influenzeranno il tempo di equilibrio. L'equilibrio delle condizioni di cristallizzazione a base di sale, ad esempio cloruro di ammonio altamente concentrato, può avvenire in 1-2 giorni. Pertanto, il criterio delle 24 ore potrebbe non essere vero per le condizioni a base di sale. Le condizioni a base di sale possono anche avere diagrammi di fase più complessi26,30 che potrebbero non essere conformi all'archetipo presentato in questo protocollo. Una riduzione del criterio temporale per le condizioni a base salina a 12 o 6 ore può essere necessaria se il ridimensionamento in volumi più grandi si rivela impossibile.
Un'altra limitazione di questo metodo è la sua apparente complessità. Il protocollo che è stato seguito per ottimizzare la microcristallizzazione dell'endotiapepsina ha effettivamente cambiato relativamente poco la condizione originale dallo schermo a matrice sparsa. Il primo colpo osservato nello schermo PACT è stato 0,1 HEPES pH 7,0, 0,2 M MgCl2 e 20% (w / v) PEG 6.000. Il tampone di cristallizzazione scalato finale era 0,1 Tris-HCl pH 7,0, 0,15 M MgCl2 e 40% (p/v) PEG 6.000. È anche molto probabile che il cambiamento del tampone da HEPES a Tris-HCl e la concentrazione di MgCl2 abbiano contribuito poco al successo del processo. Lasciando l'aumento della concentrazione di PEG 6.000 l'unica ottimizzazione, e quella che avrebbe potuto essere raggiunta molto semplicemente.
Questa valutazione, tuttavia, è anche troppo semplicistica. Non solo sconta i problemi incontrati durante il ridimensionamento (cioè l'uso di semi e tempra), ma anche il fatto che solo perché questa proteina si è rivelata semplice, non vi è alcuna garanzia che anche la prossima si rivelerà tale. I passaggi consigliati nel protocollo sono stati ideati perché ottimizzare il ridimensionamento dei volumi di cristallizzazione delle proteine può essere molto costoso dal punto di vista proteico. Nel corso dei sette studi di ridimensionamento dell'endotiapepsina che sono mostrati, sono stati consumati 100 mg di proteine. È vero che alcuni di questi passi sono stati eseguiti per mostrare le loro conseguenze alla luce di questo protocollo. Anche così, 100 mg di una proteina, più potenzialmente altri 50 mg per le proteine consumate durante un esperimento (Tabella 1), possono essere un investimento significativo in termini di tempo o denaro.
Fortunatamente, non è chiaro se questa massa di campioni richiesti sia onnipresente in tutte le proteine. L'endotiapepsina era altamente solubile e quindi richiedeva una grande concentrazione proteica per raggiungere la supersaturazione. In altri (attualmente in fase di ottimizzazione), la supersaturazione può essere raggiunta a 10 o anche 5 mg / ml. Tali variabili sono specifiche della proteina e devono essere abbracciate quando appaiono.
Altre limitazioni del metodo includono la sua dipendenza da apparecchiature complesse come robot di gestione dei liquidi per la creazione di schermi e lastre e imager per visualizzare automaticamente le lastre quando richiesto. Sono state offerte routine alternative per limitare la necessità di alcune di queste apparecchiature, ma il protocollo richiederà più tempo da seguire senza di esse. Il protocollo suggerisce anche di testare la diffrazione di cristalli ottimizzati. Per i cristallografi senza accesso regolare a un sincrotrone, questi test potrebbero rivelarsi impegnativi. I controlli in ogni fase potrebbero non essere necessari, ma questi test sono fortemente raccomandati una volta identificato un hit e pre e post-ridimensionamento. I cristalli non diffrattori in un XFEL non sono, purtroppo, un evento raro. Detto questo, è meglio sbagliare sul lato della cautela per quanto riguarda le ipotesi sulla diffrazione dei cristalli.
In definitiva, questo protocollo e i risultati presentati qui offriranno una guida, idee e un esempio a coloro che lottano con la produzione di campioni per esperimenti di cristallografia seriale. Si spera che, man mano che la cristallografia seriale viene ulteriormente sviluppata, le richieste di campioni della tecnica saranno ridotte in modo tale da ridurre la necessità di protocolli come questo. Tuttavia, anche in questo caso, le strategie qui presentate saranno comunque utili a coloro che desiderano esplorare lo spazio di cristallizzazione della loro proteina.
Divulgazioni
Gli autori non hanno conflitti di interesse da rivelare.
Riconoscimenti
Questo progetto ha ricevuto finanziamenti dal programma di ricerca e innovazione Horizon 2020 dell'Unione europea nell'ambito dell'accordo di sovvenzione Marie Skłodowska-Curie n. 701647. Mille grazie per l'assistenza e il supporto degli scienziati della beamline della Swiss Light Source beamline X10SA-PXII.
Materiali
| Name | Company | Catalog Number | Comments |
| Swissci 96-well 2-Drop plates | Molecular Dimensions | MD11-002 | 96-well 2-drop crystallisation plate |
| Swissci 96-well 3-Drop plates | Molecular Dimensions | MD11-003 | 96-well 3-drop crystallisation plate |
| mosquito LCP liquid handling robot | sptlabtech | mosquito LCP | Crystallisation robot |
| ClearVue Sheets | Molecular Dimensions | MD6-015 | 96-well crystallization plate seals |
| Safe-Tube 1.5 mL | Eppendorf | 30120086 | 1.5 mL centrifuge tubes |
| Scaple | Swan and Morton | No. 3 scalple and No. 3 handle | Scalple for cutting open plate seals |
| MS 3 Vortex | IKA | 3319000 | Vortex for mixing solution and making seed stocks |
| 24-well XRL Plate | Molecular Dimensions | MD3-11 | 24-well hanging-drop plates |
| Tube revolver/rotator | Thermo Fischer Scientific | 88881001 | Tube revolver for mixing solution during scaling |
| Eppendorf Research plus pipettes | Eppendorf | Range of manual pipettes, 0.5-10, 1-20, 10-100, 100-1000 µL | |
| Eppendorf pipette tips | Eppendorf | Range of tip sizes for manual pipettes | |
| Suparen 600 | Prochem AG | Suparen 600 | Endothiapepsin solution |
| Sodium Acetate | Sigma-Aldrich | 241245-1KG | Sodium Acetate |
| Tris | Merck | 8382T014 | Tris |
| Magnesium Chloride | Sigma-Aldrich | M2670-1kg | Magnesium Chloride |
| PEG 6,000 | Sigma-Aldrich | 81255-1kg | PEG 6,000 |
| Ethelyene glycol | Sigma-Aldrich | 324558-1L | Ethelyene glycol for cyro-protecting the crystals |
| PACT Premier HT screen | Molecular Dimensions | MD1-36 | PACT Premier 96-well crystal screen |
| DOW CORNING high vacuum grease | Molecular Dimensions | MD6-02 | Grease for sealing 24-well plates |
| Hirschmann 22 x 22 mm glaser cover slides | Hirschmann | 8000104 | Cover slides for sealing 24-well sitting drop plates |
| Crystal pins | PSI | Manufactured inhouse | Thin-film supports for micro-crystals. |
| 1-1.3 mm SiLibeads Type S | Faust | 6239547 | Glass beads for making mico-seed stocks |
| Macbook Pro | Apple | Macbook Pro | Computer for performing data analysis |
| CCP4 software suite | CCP4 | Diffraction pattern data processing software | |
| Excel | Microsoft | Microsoft Office | Plotting tool for phase diagram |
| Hausser Scientific Bright-Line counting chamber | Thermo Fischer Scientific | 02-671-51B | Tool to calculate crystal concentration |
| PACT Premier | Molecular Dimensions | MD1-29-ECO | Sparse-matrix crystallization screen |
| Rock Imager | Formulatrix | Rock Imager | Temperature controlled crystal plate storage and imager |
| Rock MakerWeb | Formulatrix | Rock MakerWeb | Crystal plate creation and image storage stoftware |
| Formulator | Formulatrix | Formulator | 96-well crystal screen creation liquid handling robot |
| Leica MZ16 Microscope | Leica | Leica MZ16 | Light microscope |
| LAS V4.6 | Leica | LAS V4.6 | Software for Leica microscopes |
| Spectra/Por 3.5 kDa dialysis tubing | Spectrumlabs | Spectra/Por 3 Dialysis Membrane | 3.5 kDa dialysis membrane |
| Dialysis tubing closures | Spectrumlabs | Spectra/Por 3 Duniversal Closures | Clips to seal the dialysis tubing ends |
| Amicon 10 kDa centrifugal concentrator | Merck-Millipore | Amicon Ultra-15 10 kDa centrifugal concentrator | 10 kDa centrifugal filter |
| 5810 R swing bucket centrifuge | Eppendorf | 5810 R Centrifuge | Swing bucket centrifuge |
Riferimenti
- DePonte, D. P., et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. Journal of Physics D: Applied Physics. 41 (19), 195505 (2008).
- Hunter, M. S., et al. Fixed-target protein serial microcrystallography with an x-ray free electron laser. Scientific Reports. 4 (1), 6026 (2014).
- Weierstall, U., et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nature Communications. 5 (1), 1-6 (2014).
- Roessler, C. G. G., et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure. 24 (4), 631-640 (2016).
- Sherrell, D. A., et al. A modular and compact portable mini-endstation for high-precision, high-speed fixed target serial crystallography at FEL and synchrotron sources. Journal of Synchrotron Radiation. 22 (6), 1372-1378 (2015).
- Roedig, P., et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Scientific Reports. 5 (1), 1-11 (2015).
- Botha, S., et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Crystallographica Section D Biological Crystallography. 71 (2), 387-397 (2015).
- Weinert, T., et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nature Communications. 8 (1), 542 (2017).
- Tenboer, J., et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science. 346 (6214), 1242-1246 (2014).
- Nango, E., et al. A three-dimensionalmovie of structural changes in bacteriorhodopsin. Science. 354 (6319), 1552-1557 (2016).
- Suga, M., et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature. 543 (7643), 131-135 (2017).
- Mehrabi, P., et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nature Methods. 16 (10), 979-982 (2019).
- Halle, B. Biomolecular cryocrystallography: structural changes during flash-cooling. Proceedings of the National Academy of Sciences of the United States of America. 101 (14), 4793-4798 (2004).
- Fraser, J. S., et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 462 (7273), 669-673 (2009).
- Fenwick, R. B., van den Bedem, H., Fraser, J. S., Wright, P. E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proceedings of the National Academy of Sciences of the United States of America. 111 (4), 445-454 (2014).
- Keedy, D. A., et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife. 4, (2015).
- Thomaston, J. L., et al. XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction. Proceedings of the National Academy of Sciences of the United States of America. 114 (51), 13357-13362 (2017).
- Haas, D. J., Rossmann, M. G. Crystallographic studies on lactate dehydrogenase at -75 °C. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry. 26 (7), 998-1004 (1970).
- Hope, H. Cryocrystallography of biological macromolecules: a generally applicable method. Acta Crystallographica Section B Structural Science. 44 (1), 22-26 (1988).
- Wu, W., et al. Batch crystallization of rhodopsin for structural dynamics using an X-ray free-electron laser. Acta Crystallographica Section:F Structural Biology Communications. 71 (7), 856-860 (2015).
- Ishchenko, A., Cherezov, V., Liu, W. Preparation and delivery of protein microcrystals in lipidic cubic phase for serial femtosecond crystallography. Journal of Visualized Experiments. (115), e54463 (2016).
- Andersson, R., et al. Well-based crystallization of lipidic cubic phase microcrystals for serial X-ray crystallography experiments. Acta Crystallographica Section D: Structural Biology. 75 (10), 937-946 (2019).
- Luft, J. R., et al. The detection and subsequent volume optimization of biological nanocrystals. Structural Dynamics. 2 (4), 041710 (2015).
- Lee, D. B., et al. Supersaturation-controlled microcrystallization and visualization analysis for serial femtosecond crystallography. Scientific Reports. 8 (1), 1-10 (2018).
- Kupitz, C., et al. Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system. Philosophical transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1647), 20130316 (2014).
- Beale, J. H., et al. Successful sample preparation for serial crystallography experiments. Journal of Applied Crystallography. 52, 1385-1396 (2019).
- Rayment, I. Small-scale batch crystallization of proteins revisited: An underutilized way to grow large protein crystals. Structure. 10 (2), 147-151 (2002).
- García-Ruiz, J. M. Nucleation of protein crystals. Journal of Structural Biology. 142 (1), 22-31 (2003).
- McPherson, A., Kuznetsov, Y. G. Mechanisms, kinetics, impurities and defects: Consequences in macromolecular crystallization. Acta Crystallographica Section F:Structural Biology Communications. 70 (4), 384-403 (2014).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta crystallographica. Section F, Structural biology communications. 71, 247-260 (2015).
- Luft, J. R., DeTitta, G. T. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallographica Section D Biological Crystallography. 55 (5), 988-993 (1999).
- Ireton, G. C., Stoddard, B. L. Microseed matrix screening to improve crystals of yeast cytosine deaminase. Acta crystallographica. Section D, Biological crystallography. 60 (3), 601-605 (2004).
- Forsythe, E. L., Maxwell, D. L., Pusey, M. Vapor diffusion, nucleation rates and the reservoir to crystallization volume ratio. Acta Crystallographica Section D Biological Crystallography. 58 (10), 1601-1605 (2002).
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L., Blow, D. M. IUCr An automated system for micro-batch protein crystallization and screening. Journal of Applied Crystallography. 23 (4), 297-302 (1990).
- Chayen, N. E., Shaw Stewart, P. D., Blow, D. M. Microbatch crystallization under oil - a new technique allowing many small-volume crystallization trials. Journal of Crystal Growth. 122 (1-4), 176-180 (1992).
- Chayen, N. E. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallographica Section D: Biological Crystallography. 54 (1), 8-15 (1998).
- D'Arcy, A., Mac Sweeney, A., Stihle, M., Haber, A. The advantages of using a modified microbatch method for rapid screening of protein crystallization conditions. Acta Crystallographica - Section D Biological Crystallography. 59 (2), 396-399 (2003).
- Darmanin, C., et al. Protein crystal screening and characterization for serial femtosecond nanocrystallography. Scientific Reports. 6, 25345 (2016).
- Gati, C., et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2247-2252 (2017).
- Cheng, R. K. Y. Towards an optimal sample delivery method for serial crystallography at XFEL. Crystals. 10 (3), 215 (2020).
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Advances in Condensed Matter Physics. 2013, 1-10 (2013).
- Oberthuer, D., et al. Double-flow focused liquid injector for efficient serial femtosecond crystallography. Scientific Reports. 7 (1), 44628 (2017).
- McPherson, A., Gavira, J. A. Introduction to protein crystallization. Acta crystallographica. Section F, Structural biology communications. 70, 2-20 (2014).
- Yaoi, M., et al. Effect of stirring method on protein crystallization. Japanese Journal of Applied Physics, Part 2: Letters. 43 (10), 1318 (2004).
- Castro, F., Ferreira, A., Teixeira, J. J., Rocha, F. Influence of Mixing Intensity on Lysozyme Crystallization in a Meso Oscillatory Flow Reactor. Crystal Growth & Design. 18 (10), 5940-5946 (2018).
- Moews, P. C., Bunn, C. W. An X-ray crystallographic study of the rennin-like enzyme of Endothia parasitica. Journal of Molecular Biology. 54 (2), 395-397 (1970).
- Newman, J., et al. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta crystallographica. Section D, Biological. 61, 1426-1431 (2005).
- Ebrahim, A., et al. Resolving polymorphs and radiation-driven effects in microcrystals using fixed-target serial synchrotron crystallography. Acta Crystallographica Section D Structural Biology. 75, 151-159 (2019).
- Davy, B., et al. Reducing sample consumption for serial crystallography using acoustic drop ejection. Journal of Synchrotron Radiation. 26 (5), (2019).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati