
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
RNA polimerasi legata al DNA per la trascrizione in vitro programmabile e il calcolo molecolare
In questo articolo
Riepilogo
Descriviamo l'ingegneria di una nuova T7 RNA polimerasi legata al DNA per regolare le reazioni di trascrizione in vitro. Discutiamo i passaggi per la sintesi e la caratterizzazione proteica, convalidiamo la regolazione trascrizionale proof-of-concept e discutiamo le sue applicazioni nel calcolo molecolare, nella diagnostica e nell'elaborazione delle informazioni molecolari.
Abstract
La nanotecnologia del DNA consente l'autoassemblaggio programmabile degli acidi nucleici in forme e dinamiche prescritte dall'utente per diverse applicazioni. Questo lavoro dimostra che i concetti della nanotecnologia del DNA possono essere utilizzati per programmare l'attività enzimatica della T7 RNA polimerasi derivata dai fagi (RNAP) e costruire reti di regolazione genica sintetica scalabili. In primo luogo, un RNAP T7 legato all'oligonucleotide viene ingegnerizzato tramite l'espressione di un RNAP con tag SNAP N-terminale e il successivo accoppiamento chimico del tag SNAP con un oligonucleotide modificato con benzilguanina (BG). Successivamente, lo spostamento del filamento di acido nucleico viene utilizzato per programmare la trascrizione della polimerasi su richiesta. Inoltre, gli assemblaggi di acidi nucleici ausiliari possono essere utilizzati come "fattori di trascrizione artificiale" per regolare le interazioni tra l'RNAP T7 programmato dal DNA con i suoi modelli di DNA. Questo meccanismo di regolazione della trascrizione in vitro può implementare una varietà di comportamenti circuitali come la logica digitale, il feedback, la cascata e il multiplexing. La componibilità di questa architettura di regolazione genica facilita l'astrazione, la standardizzazione e il ridimensionamento del design. Queste caratteristiche consentiranno la prototipazione rapida di dispositivi genetici in vitro per applicazioni quali il biorilevamento, il rilevamento di malattie e l'archiviazione dei dati.
Introduzione
Il calcolo del DNA utilizza un insieme di oligonucleotidi progettati come mezzo per il calcolo. Questi oligonucleotidi sono programmati con sequenze per assemblarsi dinamicamente secondo la logica specificata dall'utente e rispondere a specifici input di acido nucleico. Negli studi proof-of-concept, l'output del calcolo consiste tipicamente in un insieme di oligonucleotidi marcati fluorescentemente che possono essere rilevati tramite elettroforesi su gel o lettori di piastre di fluorescenza. Negli ultimi 30 anni sono stati dimostrati circuiti computazionali del DNA sempre più complessi, come varie cascate di logica digitale, reti di reazioni chimiche e reti neurali1,2,3. Per aiutare con la preparazione di questi circuiti del DNA, sono stati utilizzati modelli matematici per prevedere la funzionalità dei circuiti genici sintetici4,5e sono stati sviluppati strumenti computazionali per la progettazione di sequenze di DNA ortogonali6,7,8,9,10 . Rispetto ai computer a base di silicio, i vantaggi dei computer a DNA includono la loro capacità di interfacciarsi direttamente con le biomolecole, operare in soluzione in assenza di un alimentatore, nonché la loro compattezza e stabilità complessive. Con l'avvento del sequenziamento di nuova generazione, il costo della sintesi dei computer del DNA è diminuito negli ultimi due decenni a un ritmo più veloce della Legge11di Moore. Le applicazioni di tali computer basati sul DNA stanno ora iniziando ad emergere, come per la diagnosi delle malattie12,13, per alimentare la biofisica molecolare14e come piattaforme di archiviazione dei dati15.

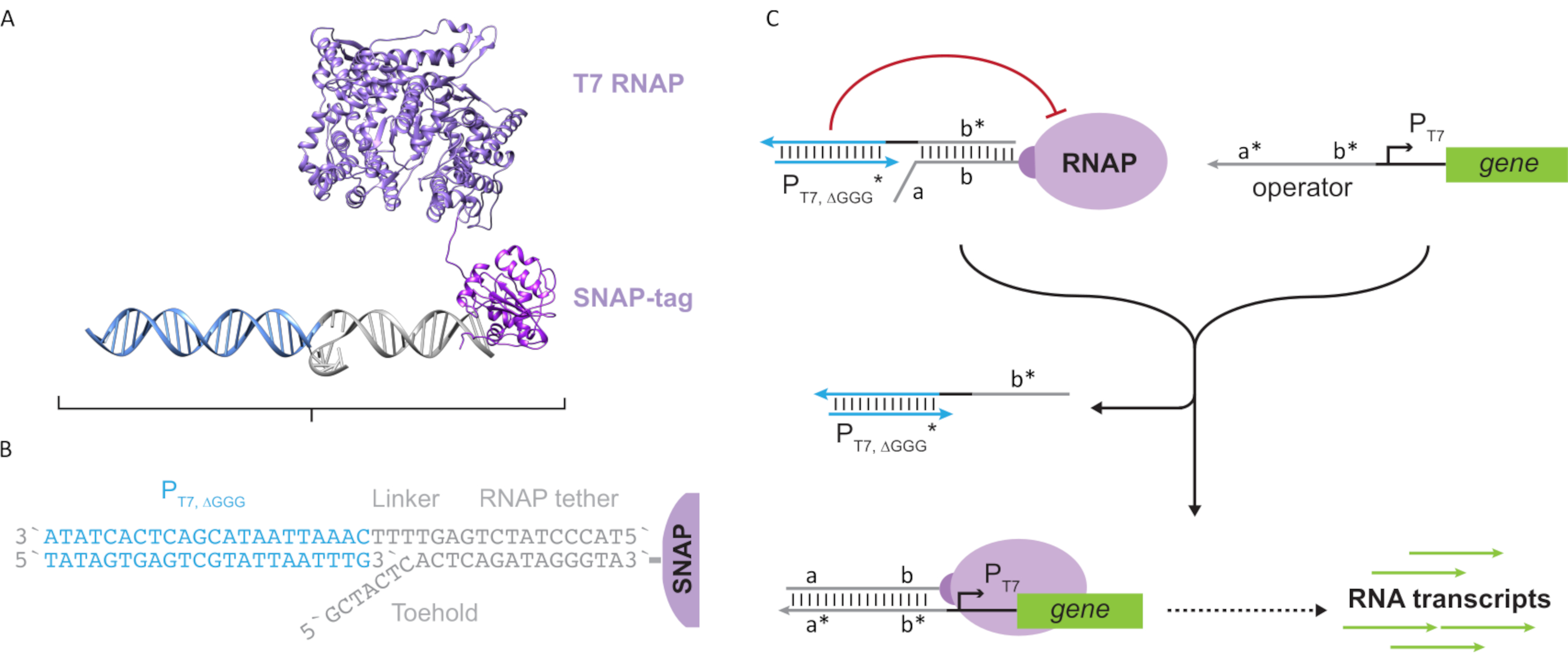
Figura 1: Meccanismo di spostamento del filamento di DNA mediato dalla morsa. Il punto d'appoggio, δ, è una sequenza libera e non legata su un duplex parziale. Quando un dominio complementare (δ*) viene introdotto su un secondo filamento, il dominio δ libero funge da punto d'appoggio per l'ibridazione, consentendo al resto del filamento (ɑ*) di spostare lentamente il suo concorrente attraverso una reazione reversibile di zipping/decompressione nota come migrazione del filamento. All'aumentare della lunghezza della δ, il ΔG per la reazione in avanti diminuisce e lo spostamento avviene più facilmente. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Ad oggi, la maggior parte dei computer del DNA utilizza un motivo ben consolidato nel campo della nanotecnologia dinamica del DNA noto come spostamento del filamento di DNA mediato dalla soglia (TMDSD, Figura 1)16. Questo motivo è costituito da un duplex di DNA parzialmente a doppio filamento (dsDNA) che mostra brevi sporgenze "toehold" (cioè da 7 a 10 nucleotidi (nt)). I filamenti di "input" dell'acido nucleico possono interagire con i duplex parziali attraverso il punto d'appoggio. Ciò porta allo spostamento di uno dei fili dal duplex parziale, e questo filamento liberato può quindi servire come input per i duplex parziali a valle. Pertanto, TMDSD consente la cascata del segnale e l'elaborazione delle informazioni. In linea di principio, i motivi TMDSD ortogonali possono operare indipendentemente in soluzione, consentendo l'elaborazione parallela delle informazioni. Ci sono state una serie di variazioni sulla reazione TMDSD, come lo scambio di filamenti di DNA mediato da trattenute (TMDSE)17,i punti di appoggio "senza perdite" con domini a doppia lunghezza18,i punti di riferimento non corrispondenti alla sequenza19e lo spostamento del filamento mediato da "handhold"20. Questi principi di progettazione innovativi consentono un'energetica e una dinamica TMDSD più finemente sintonizzate per migliorare le prestazioni di calcolo del DNA.
I circuiti genici sintetici, come i circuiti genici trascrizionali, sono anche in grado dicalcolare 21,22,23. Questi circuiti sono regolati da fattori di trascrizione proteica, che attivano o reprimono la trascrizione di un gene legandosi a specifici elementi regolatori del DNA. Rispetto ai circuiti basati sul DNA, i circuiti trascrizionali presentano diversi vantaggi. In primo luogo, la trascrizione enzimatica ha un tasso di turnover molto più elevato rispetto ai circuiti catalitici del DNA esistenti, generando così più copie di output per singola copia di input e fornendo un mezzo più efficiente di amplificazione del segnale. Inoltre, i circuiti trascrizionali possono produrre diverse molecole funzionali, come aptameri o RNA messaggero (mRNA) che codifica per proteine terapeutiche, come output di calcolo, che possono essere sfruttati per diverse applicazioni. Tuttavia, una delle principali limitazioni degli attuali circuiti trascrizionali è la loro mancanza di scalabilità. Questo perché esiste un insieme molto limitato di fattori di trascrizione ortogonali basati su proteine e la progettazione de novo di nuovi fattori di trascrizione proteica rimane tecnicamente impegnativa e dispendiosa in termini di tempo.

Figura 2: Astrazione e meccanismo del complesso della polimerasi "tether" e "cage". (A e B) Un tether oligonucleotide è enzimaticamente etichettato in una polimerasi T7 attraverso la reazione SNAP-tag. Una gabbia costituita da un promotore T7 "finto" con una sporgenza del complemento di legame consente di ibridarsi al legame e bloccare l'attività trascrizionale. (C) Quando l'operatore (a*b*) è presente, si lega al punto d'appoggio sul legame oligonucleotidico (ab) e sposta la regione b* della gabbia, permettendo la trascrizione. Questa figura è stata modificata da Chou e Shih27. Abbreviazioni: RNAP = RNA polimerasi. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Questo documento introduce un nuovo elemento costitutivo per il calcolo molecolare che combina le funzionalità dei circuiti trascrizionali con la scalabilità dei circuiti basati sul DNA. Questo blocco di costruzione è un T7 RNAP attaccato covalentemente con un legame di DNA a singolo filamento (Figura 2A). Per sintetizzare questo RNAP T7 legato al DNA, la polimerasi è stata fusa in un snap-tag24 N-terminale ed espressa in modo ricombinante in Escherichia coli. Lo SNAP-tag è stato quindi reagito con un oligonucleotide funzionalizzato con il substrato BG. Il tether oligonucleotidico consente il posizionamento di ospiti molecolari in prossimità della polimerasi tramite ibridazione del DNA. Uno di questi ospiti era un bloccante trascrizionale competitivo denominato "gabbia", che consiste in un "finto" duplex di DNA promotore T7 senza gene a valle (Figura 2B). Quando è legata all'RNAP tramite il suo legame oligonucleotidico, la gabbia blocca l'attività della polimerasi superando altri modelli di DNA per il legame RNAP, rendendo l'RNAP in uno stato "OFF" (Figura 2C).
Per attivare la polimerasi in uno stato "ON", sono stati progettati modelli di DNA T7 con domini "operatore" a singolo filamento a monte del promotore T7 del gene. Il dominio operatore (cioè il dominio a*b* Figura 2C) può essere progettato per spostare la gabbia dall'RNAP tramite TMDSD e posizionare l'RNAP prossimale al promotore T7 del gene, avviando così la trascrizione. In alternativa, sono stati progettati anche modelli di DNA in cui la sequenza dell'operatore era complementare ai filamenti ausiliari di acido nucleico che sono indicati come "fattori di trascrizione artificiale" (cioè filamenti TFA e TFB nella Figura 3A). Quando entrambi i filamenti vengono introdotti nella reazione, si assemblano nel sito dell'operatore, creando un nuovo dominio pseudo-contiguo a*b*. Questo dominio può quindi spostare la gabbia tramite TMDSD per avviare la trascrizione (Figura 3B). Questi fili possono essere forniti sia esogenamente che prodotti.

Figura 3: Programmazione selettiva dell'attività della polimerasi attraverso un attivatore a commutazione a tre componenti. (A) Quando i fattori di trascrizione (TFA e TFB) sono presenti, si legano al dominio operatore a monte del promotore, formando una pseudo sequenza a singolo filamento (a*b*) in grado di spostare la gabbia attraverso lo spostamento del DNA mediato dalla presa. (B) Questo dominio a*b* può spostare la gabbia tramite TMDSD per avviare la trascrizione. Questa figura è stata modificata da Chou e Shih27. Abbreviazioni: TF = fattore di trascrizione; RNAP = RNA polimerasi; TMDSD = spostamento del filamento di DNA mediato dalla punta. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
L'uso di fattori di trascrizione basati su acidi nucleici per la regolazione trascrizionale in vitro consente l'implementazione scalabile di sofisticati comportamenti circuitali come la logica digitale, il feedback e la cascata del segnale. Ad esempio, si possono costruire cascate di gate logici progettando sequenze di acidi nucleici in modo tale che i trascritti di un gene a monte attivino un gene a valle. Un'applicazione che sfrutta il cascading e il multiplexing resi capaci da questa tecnologia proposta è lo sviluppo di circuiti di calcolo molecolare più sofisticati per la diagnostica portatile e l'elaborazione di dati molecolari. Inoltre, l'integrazione delle capacità di calcolo molecolare e di sintesi dell'RNA de novo può consentire nuove applicazioni. Ad esempio, un circuito molecolare può essere progettato per rilevare uno o una combinazione di RNA definiti dall'utente come input e output RNA terapeutici o mRNA che codificano peptidi funzionali o proteine per applicazioni mediche point-of-care.
Protocollo
1. Preparazione del buffer
NOTA: la preparazione del tampone di purificazione delle proteine può avvenire in qualsiasi giorno; qui, è stato fatto prima di iniziare gli esperimenti.
- Preparare un tampone di lisi/equilibrazione contenente 50 mM di tris (idrossimetil)amminometano (Tris), 300 mM di cloruro di sodio (NaCl), 5% di glicerolo e 5 mM di β-mercaptoetanolo (BME), pH 8. Aggiungere 1,5 mL di 1M Tris, 1,8 mL di 5M NaCl, 1,5 mL di glicerolo, 25,2 mL di acqua deionizzata (ddH2O) in un tubo centrifugo da 50 mL e aggiungere 10,5 μL di 14,2 M BME appena prima dell'uso.
NOTA: Tris può causare tossicità acuta; quindi, evitare di respirare la sua polvere ed evitare il contatto con la pelle e gli occhi. Il BME è tossico e deve essere utilizzato solo in una cappa aspirante. È importante aggiungere BME per ultimo, appena prima della risospensione e della lisi cellulare. Vedere la Tabella 1 per la formula del tampone di lisi. - Preparare un tampone di lavaggio (pH 8) contenente 50 mM Tris, 800 mM NaCl, 5% glicerolo, 5 mM BME e 20 mM imidazolo. Aggiungere 1,5 mL di 1 M Tris, 4,8 mL di 5 M NaCl, 1,5 mL di glicerolo e 22,2 mL di ddH2O in un tubo centrifugo da 50 mL. Appena prima dell'uso, aggiungere 7 μL di 14,2 M BME e 200 μL di 2 M imidazolo a 20 mL della soluzione di cui sopra.
NOTA: Per prevenire la tossicità acuta dovuta all'imidazolo, utilizzare dispositivi di protezione individuale. È importante aggiungere BME e imidazolo per ultimi, appena prima di lavare la proteina dalla colonna. Vedere la Tabella 2 per la formula del tampone di lavaggio. - Preparare un tampone di eluizione (pH8) contenente 50 mM Tris, 800 mM NaCl, 5% glicerolo, 5 mM BME e 200 mM imidazolo. Aggiungere 0,5 mL di 1 M Tris, 1,6 mL di 5 M NaCl, 0,5 mL di glicerolo e 6,4 mL di ddH2O a un tubo centrifugo da 15 mL. Appena prima dell'uso, aggiungere 3,5 μL di 14,2 M BME e 1 mL di 2 M di imidazolo a 10 mL della soluzione di cui sopra.
NOTA: È importante aggiungere BME e imidazolo per ultimi, appena prima di eluire la proteina fuori dalla colonna. Vedere la Tabella 3 per la formula del tampone di eluizione. - Preparare 2x tampone di stoccaggio (da miscelare 1:1 con glicerolo) contenente 100 mM Tris, 200 mM NaCl, 40 mM BME e 2 mM di acido etilendiammidetraacetico (EDTA), 0,2% di un tensioattivo non ionico (vedere la Tabella dei materiali). Preparare 50 mL del tampone di stoccaggio aggiungendo 5 mL di 1 M Tris, 2 mL di 5 M NaCl, 42,56 mL di ddH2O, 200 μL di 0,5 M EDTA, 100 μL del tensioattivo non ionico in un tubo centrifuga da 50 mL. Miscelare fino a quando la soluzione è omogenea, filtrare il tampone di stoccaggio attraverso un filtro a siringa da 0,2 μm e aggiungere 140,8 μL di BME alla soluzione di cui sopra prima dell'uso.
NOTA: Per evitare tossicità acuta dovuta all'EDTA, evitare di respirare la polvere ed evitare il contatto con la pelle e gli occhi. È importante aggiungere BME per ultimo e mescolare l'intero buffer di stoccaggio 1: 1 con glicerolo, appena prima di conservare la proteina purificata. Vedere la Tabella 4 per la formula del buffer di archiviazione.
2. Crescita della cultura durante la notte: Giorno 1
- Preparare 1.000x kanamicina brodo sciogliendo 500 mg di kanamicina in 10 ml di ddH2O.
NOTA: Utilizzare dispositivi di protezione individuale per prevenire la tossicità acuta dovuta alla kanamicina. - Aggiungere 20 μL del brodo di kanamicina 1.000x a 20 ml di brodo di lisogenesi. Usando una punta di pipetta sterile, colpire un brodo di glicerolo BL21 E. coli trasformato e quindi inoculare la coltura introducendo la punta nel brodo del mezzo di crescita.

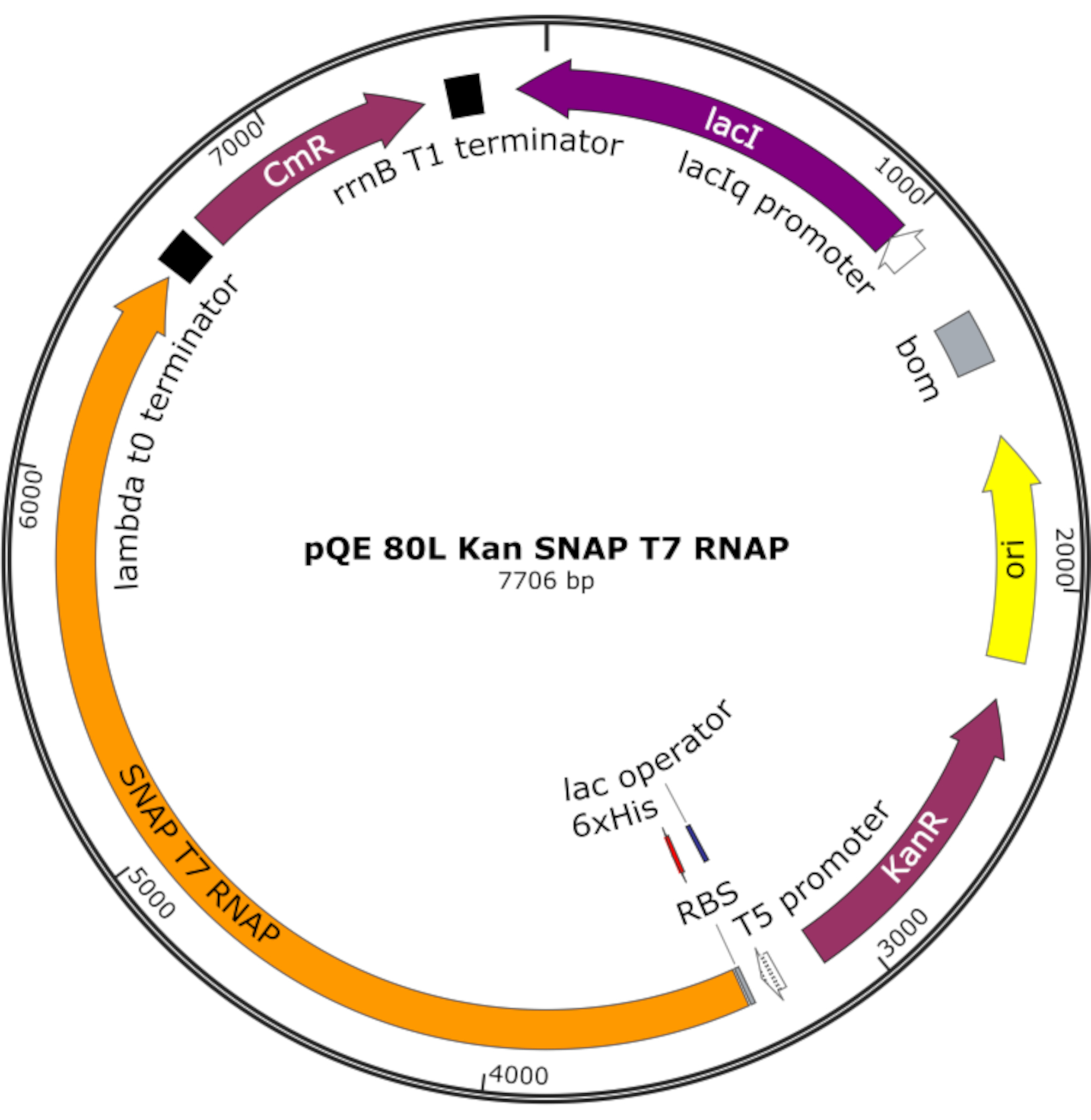
Figura 4: Mappa plasmidica per SNAP T7 RNAP. Il plasmide codifica un T7 RNAP contenente un tag istidina N-terminale (6x His) e un dominio SNAP-tag (SNAP T7 RNAP) sotto un repressore lac (lacI) su una dorsale pQE-80L. Altre caratteristiche includono i geni di resistenza alla kanamicina (KanR) e resistenza al cloramfenicolo (CmR). Abbreviazione: RNAP = RNA polimerasi. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: Il plasmide codifica un RNAP T7 contenente un tag istidina N-terminale e un dominio SNAP-tag (SNAP T7 RNAP), nonché un gene di resistenza alla kanamicina sotto una dorsale pQE-80L (Figura 4)25.
- Ancora una volta, aggiungere 20 μL del brodo di kanamicina 1.000x in un pallone di coltura separato contenente 20 ml di brodo di lisogenesi e incubarlo come controllo.
- Incubare i due campioni (dai passaggi 2.2 e 2.3) durante la notte per 12-18 ore a 37 °C, ruotando a 10 × g.
3. Crescita e induzione cellulare: Giorno 2
- Inoculare 400 mL di brodo di lisogenia contenente 400 μL di stock di kanamicina con 4 mL di coltura di crescita notturna dalla fase 2.4. Incubare i palloni di coltura a 37 °C, ruotando a 10 × g.
- Una volta che la coltura ha raggiunto una densità ottica (OD) a 600 nm di ~0,5, estrarre 1 mL di campione dal pallone di crescita come controllo. Conservare il campione di controllo a 4 °C.
- Indurre le cellule con isopropil β-D-1-tiogalattopiranoside (IPTG) aggiungendo 40 μL di 1M IPTG per 100 mL di coltura per raggiungere una concentrazione finale di 0,4 mM IPTG. Incubare il campione per 3 ore a 37 °C, ruotando a 10 × g,quindi ruotare la coltura indotta a 8.000 × g per 10 minuti per pellettizzare le cellule. Rimuovere il surnatante e conservare il pellet a -20 °C fino a nuovo utilizzo.
NOTA: Per evitare tossicità acuta dovuta a IPTG, evitare di respirare la polvere ed evitare il contatto con la pelle e gli occhi. Se necessario, puoi mettere in pausa l'esperimento qui e continuare il giorno successivo.
4. Lisi cellulare, purificazione delle proteine: Giorno 3
- Risospesciare il pellet cellulare immagazzinato con 10 ml di tampone di lisi sul ghiaccio e ruotare delicatamente per garantire che l'intero pellet venga risospeso. Quindi, pipettare 1 mL di campione in dieci tubi da 1,5 mL che vengono tenuti sul ghiaccio.
- Sonicare ogni campione con un'impostazione di ampiezza di "1", pulsato per 2 s con un ciclo di lavoro del 50% per un periodo di 30 s. Prima e dopo ogni campione, pulire la punta di sonicazione con etanolo al 70% e ddH2O. Tenere tutti i campioni sul ghiaccio durante e dopo la sonicazione.
NOTA: Tenere il 70% di etanolo lontano da fonti di calore e fiamme libere. - Equilibrare una colonna di spin di purificazione dell'acido nitrilotriacetico (Ni-NTA) caricato con nichel a una temperatura di esercizio di 4 °C. Posizionare/conservare la colonna a 4 °C e tenerla sul ghiaccio durante l'uso.
- Centrifugare i dieci campioni da 1 mL a 15.000 × g per 20 min a 4 °C. Pipettare con cura il surnatante contenente l'RNAP ricombinante senza disturbare il pellet. Se necessario, utilizzare un buffer di bilanciamento aggiuntivo per regolare il volume totale a ≥ 6 ml.
- Rimuovere delicatamente la linguetta inferiore dalla colonna di rotazione Ni-NTA per consentire il flusso attraverso la colonna. Posizionare la colonna in un tubo di centrifuga e tenerla sul ghiaccio.
NOTA: utilizzare un tubo centrifuga da 50 mL con le colonne di rotazione Ni-NTA da 3 mL. - Centrifugare la colonna a 700 × g e 4 °C per 2 minuti per rimuovere il buffer di stoccaggio. Equilibrare la colonna aggiungendo 6 mL di buffer di equilibrio alla colonna. Lasciare che il tampone entri completamente nel letto di resina.
- Rimuovere il tampone di equilibratura dalla colonna mediante centrifugazione a 700 × g e 4 °C per 2 minuti. Prima di aggiungere l'estratto di cella preparato alla colonna, posizionare un tappo inferiore sulla colonna per evitare di perdere qualsiasi prodotto. Quindi, aggiungere l'estratto della cella alla colonna e mescolare su un miscelatore shaker orbitale per 30 minuti a 4 °C.
- Rimuovere il tappo inferiore dalla colonna e posizionare la colonna in un tubo centrifugo da 50 mL etichettato flusso attraverso. Centrifugare la colonna a 700 × g per 2 minuti per raccogliere il flusso attraverso.
- Aggiungere 6 mL di tampone di lavaggio alla colonna per lavare la resina. Centrifugare la colonna a 700 × g per 2 min per raccogliere la frazione in un nuovo tubo centrifuga etichettato lavaggio 1. Ripetere questo passaggio altre due volte per un totale di 3 frazioni separate e raccogliere le frazioni in tubi centrifughi separati (lavare 2 e lavare 3).
- Aggiungere 3 ml di tampone di eluizione per eluire le proteine His-tagged dalla resina. Centrifugare la colonna a 700 × g per 2 minuti per raccogliere la frazione in un nuovo tubo di centrifuga etichettato eluato 1. Ripetere questo passaggio altre due volte per un totale di 3 frazioni separate e raccogliere le frazioni in tubi centrifughi separati (eluato 2 ed eluato 3).
- Unire gli eluati ed eseguire la desalinizzazione per rimuovere i sali dalla soluzione proteica.
- Pipetta 15 mL di polisorbato 0,05 % p/v 20 su un'unità filtrante centrifuga da 100 kDa. Centrifugare a 4.000 × g per 40 minuti ed eliminare il flusso attraverso.
- Utilizzare il filtro rivestito per concentrare gli eluati 1, 2 e 3 (9 mL di eluato proteico totale + 6 mL di tampone di stoccaggio) a ~1.500 μL. Centrifugare il filtro a 3.220 × g per 20 minuti e lavare delicatamente la membrana con pipetta per evitare precipitazioni.
- Diluire il campione a 15 ml con buffer di stoccaggio. Eseguire uno scambio di buffer utilizzando il buffer di archiviazione 1:1.000 ripetendo il passaggio 4.11.2 altre due volte.
- Quantificare la proteina purificata misurando l'assorbanza della frazione a 280 nm. Spegnere lo spettrofotometro con buffer di memorizzazione (2x buffer di archiviazione a 4 °C). Mescolare delicatamente il campione degli eluati combinati e misurarne l'assorbanza.
NOTA: eseguire tre letture separate a diluizioni 1x, 10x e 50x del campione proteico per calcolare la media e quantificare la proteina. Diluire i campioni nel buffer di conservazione. - Regolare i campioni proteici a 100 μM utilizzando 2x buffer di stoccaggio. Diluire il campione aggiustato 1:1 in volume con glicerolo al 100%. Conservare la soluzione proteica risultante a -80 °C.
5. Elettroforesi su gel di sodio dodecilsolfato-poliacrilammide (SDS-PAGE) del prodotto proteico: Giorno 3
- Eseguire un gel SDS-PAGE per l'analisi delle proteine. Mescolare 9 μL del campione con 3 μL di 4x colorante a carico proteico di litio dodecil solfato (LDS). Riscaldare i campioni a 95 °C per 10 min.
- Caricare i campioni su una configurazione del gel Bis-Tris SDS-PAGE al 4-12%. Caricare la scala proteica in ben 1, quindi con campioni (da sinistra a destra): flow-through, lavare 1, lavare 2, lavare 3, eluizione 1, eluizione 2, eluizione 3 e eluizione totale desalata.
NOTA: la tabella 5 contiene una tabella di caricamento del campione per il gel SDS-PAGE. - Eseguire i campioni di gel caricati in tampone di acido etansolfonico (MES) 2-(N-morfolino) per 35 minuti a 200 V. Risciacquare il gel in un vassoio pulito tre volte per 10 minuti ciascuno utilizzando 200 ml di ddH2O, con una delicata agitazione per rimuovere qualsiasi SDS dalla matrice del gel.
NOTA: Indossare dispositivi di protezione individuale per evitare tossicità acuta dovuta al MES. - Macchiare il gel con 20 ml di blu Coomassie e incubare il gel durante la notte a temperatura ambiente con una leggera agitazione. Demacchia il gel due volte per 1 ora ciascuna con 200 mL di ddH2O con una delicata agitazione su uno shaker orbitale.
NOTA: Lavare il gel per un periodo più lungo o sostituire frequentemente l'acqua migliorerà la sensibilità. Inoltre, posizionare un tessuto piegato per pulire delicatamente nel contenitore per assorbire il colorante in eccesso accelererà il processo di decolorazione.
6. Verifica funzionale di SNAP T7 RNAP tramite trascrizione in vitro
NOTA: Questo protocollo utilizza il modello di DNA, che codifica per l'aptamero fluorescente Broccoli RNA e consente l'uso della fluorescenza per monitorare la cinetica della trascrizione su un lettore di piastre di fluorescenza.
- Impostare tre reazioni di trascrizione in vitro (IVT) per confrontare l'attività di SNAP T7 RNAP con RNAP T7 wild-type (WT) T7 da una fonte commerciale e un controllo solo tampone. Regolare il volume di ogni reazione a 20 μL.
- Preparare la reazione SNAP T7 RNAP IVT mescolando 2 μL di tampone di trascrizione 10x, 0,4 μL di miscela di ribonucleoside trifosfato (rNTP) da 25 mM, 5 μL di modello di DNA da 500 nM, 2 μL da 500 nM SNAP T7 RNAP e 10,6 μL di ddH2O.
- Preparare la reazione WT RNAP IVT mescolando 2 μL di tampone di trascrizione 10x, 0,4 μL di miscela rNTP da 25 mM, 5 μL di modello di DNA 500 nM, 2 μL di WT T7 RNAP e 10,6 μL di ddH2O.
- Preparare la reazione IVT solo tampone mescolando 2 μL di tampone di trascrizione 10x, 0,4 μL di miscela rNTP da 25 mM, 5 μL di modello di DNA 500 nM e 12,6 μL di ddH2O.
NOTA: Aggiungere l'RNAP per ultimo, mantenendo i campioni sul ghiaccio fino alla sua introduzione. La Tabella 6, la Tabella 7e la Tabella 8 contengono le formule di reazione IVT.
- Monitorare la cinetica di trascrizione su un lettore di piastre a fluorescenza per 2 ore a intervalli di 2 minuti a 37 °C utilizzando una lunghezza d'onda di eccitazione di 470 nm e una lunghezza d'onda di emissione di 512 nm.
7. Preparazione di oligonucleotidi modificati con BG: Giorno 1
- Sciogliere l'oligonucleotide con modificazione 3'-ammina in ddH2O ad una concentrazione finale di 1 mM. Etichetta questo S1.
- Mescolare 25 μL di bicarbonato di sodio 1 M (NaHCO3), 284 μL di solfossido di dimetile (DMSO) al 100%, 125 μL di S1 (stock di oligonucleotidi) e 66 μL di 50 mM di estere BG-N-idrossisuccinimide (NHS) diluito con DMSO, regolare il volume a 500 μL e incubare durante la notte a temperatura ambiente a 100 × g.
NOTA: Tenere il DMSO lontano da calore e fiamma in quanto è un liquido combustibile. La Tabella 9 contiene la formula di reazione per la coniugazione BG all'oligonucleotide.
- Mescolare 25 μL di bicarbonato di sodio 1 M (NaHCO3), 284 μL di solfossido di dimetile (DMSO) al 100%, 125 μL di S1 (stock di oligonucleotidi) e 66 μL di 50 mM di estere BG-N-idrossisuccinimide (NHS) diluito con DMSO, regolare il volume a 500 μL e incubare durante la notte a temperatura ambiente a 100 × g.
8. Precipitazione di etanolo/acetone del coniugato BG-oligonucleotide: Giorno 2
- Centrifugare il prodotto della fase 7.1.1. a 13.000 × g per 5 min. Trasferire con cautela il surnatante in un tubo fresco ed eliminare qualsiasi BG precipitato. Dividere la reazione in due aliquote uguali da 250 μL per evitare il trabocco ed eseguire i passaggi seguenti su entrambe le aliquote.
- Aggiungere 1/10 del volume di 3 M acetato di sodio (25 μL), seguito da 2,5 volte il volume in etanolo al 100% (625 μL). Incubare a -80 °C per 1 ora.
NOTA: Utilizzare dispositivi di protezione individuale quando si maneggia sia l'acetato di sodio (può causare irritazione agli occhi, alla pelle, al tratto digestivo e respiratorio) che l'etanolo (estremamente infiammabile, provoca irritazione al contatto). Se necessario, metti in pausa l'esperimento qui e continua il giorno successivo. - Posizionare i tubi nella centrifuga e contrassegnare il bordo esterno. Centrifugare i tubi a 17.000 × g per 30 min a 4 °C.
NOTA: Il pellet di oligonucleotidi apparirà sul bordo marcato del tubo. - Senza disturbare il pellet, scartare il surnatante. Rabboccare con 750 μL di etanolo refrigerato al 70% e girare a 17.000 × g per 10 minuti a 4 °C.
- Senza disturbare il pellet, scartare il surnatante. Rabboccare con 750 μL di acetone al 100% e ruotare a 17.000 × g per 10 minuti a 4 °C.
NOTA: Utilizzare dispositivi di protezione individuale quando si maneggia l'acetone in quanto è estremamente infiammabile e provoca irritazione al contatto. - Con il coperchio del tubo aperto, asciugare all'aria per 5 minuti per rimuovere l'acetone in eccesso attraverso l'evaporazione. Ri-dissolvere l'oligonucleotide in 250 μL di 1x tampone Tris-EDTA (TE) per produrre una soluzione di BG-oligonucleotide ~850 μM.
- Ripetere i passaggi da 8,2 a 8,6 e ri-dissolvere in 70 μL di 1x te buffer. Etichetta questo S2.
9. Pulizia BG-oligonucleotide tramite cromatografia con filtrazione su gel
- Sospendere la matrice invertendo vigorosamente le colonne più volte; rimuovere il cappuccio superiore e staccare la punta inferiore della colonna. Posizionare la colonna in un tubo centrifuga da 1,5 mL e centrifugare il tubo a 1.000 × g per 1 minuto a temperatura ambiente. Scartare il tampone e il tubo di raccolta eluiti.
NOTA: è importante prevenire la formazione di vuoto. Utilizzare immediatamente le colonne preparate. - Posizionare le colonne imballate in tubi centrifugati puliti da 1,5 ml. Aggiungere 300 μL di 1x TE buffer al centro del letto della colonna e centrifugare a 1.000 × g per 2 minuti per sostituire la soluzione tampone. Ancora una volta, scartare il tampone e il tubo di raccolta eluiti.
- Posizionare le colonne a scambio tampone in tubi centrifuga puliti da 1,5 ml. Applicare fino a 75 μL di campione al centro del letto. Girare a 1.000 × g per 4 min.
NOTA: Non disturbare il letto o toccare i lati della colonna; il punto più alto del fluido gel dovrebbe puntare verso il rotore esterno. - Raccogliere l'eluato dal tubo di raccolta, in quanto contiene l'acido nucleico purificato. Per quantificare il campione, misurarne l'assorbanza a 260 nm; etichettare questo S3.
NOTA: prendere nota della lunghezza del percorso utilizzata nella misurazione e calcolare la concentrazione utilizzando la legge di Beer-Lambert.
10. Analisi PAGE denaturante del coniugato BG-oligonucleotide
- Lanciare un gel 18% Tris-borato-EDTA (TBE)-Urea PAGE. Sciogliere 4,8 g di UREA, 4,5 mL di acrilammide al 40% (19:1) e 1 mL di 10x TBE in 2,8 mL di ddH2O; aggiungere 5 μL di tetrametiletilendiammina (TEMED) e mescolare accuratamente. Ripetere con 100 μL di persolfato di ammonio al 10% (APS). Versare la soluzione in una cassetta di gel vuota e consentire la polimerizzazione per 40 minuti.
NOTA: Utilizzare dispositivi di protezione individuale appropriati quando si maneggia urea (provoca irritazione agli occhi e alla pelle), acrilammide (tossica e cancerogena) e TEMED (tossico, infiammabile, corrosivo). La Tabella 10 contiene la formula di reazione per un gel di poliacrilammide TBE-UREA al 18%. - Microonde 500 mL di tampone TBE (0,5x) per 2 min e 30 s o fino a ~70 °C e versare in un apparecchio gel. Preparare il colorante di carico di formammide (denaturazione) contenente il 95% di formammide + 1 mM di EDTA e blu bromofenolo. Mescolare il colorante di carico con ciascun campione e caricare la miscela sul gel di poliacrilammide.
NOTA: Utilizzare dispositivi di protezione individuale appropriati quando si maneggia la formammide in quanto cancerogena. La tabella 11 contiene una tabella di carico del gel campione. - Eseguire il gel a 270 V per 35 minuti o fino a quando il fronte del colorante migra verso la fine. Mettere il gel in una scatola di gel e macchiare con colorante alla cianina per acidi nucleici per 15 minuti a temperatura ambiente prima dell'imaging.
NOTA: Utilizzare dispositivi di protezione individuale appropriati quando si maneggia il colorante alla cianina in quanto è combustibile.
11. Coniugazione di oligonucleotidi a SNAP T7 RNAP e analisi PAGE
- Preparare i reagenti per l'accoppiamento su scala analitica di BG-oligonucleotide a SNAP T7 RNAP: effettuare 9 diluizioni di oligo DNA a singolo filamento (ssDNA) con ddH2O per creare rapporti oligo:RNAP che vanno da 5:1 a 1:5. Diluire il brodo proteico a 50 μM.
NOTA: i rapporti di esempio sono disponibili nella Tabella 12; questi rapporti sono calcolati utilizzando una concentrazione di RNAP di 50 μM. - Per ogni diluizione di oligo ssDNA, effettuare 10 μL della miscela di reazione contenente 2 μL di tampone SNAP, 4 μL di BG-oligonucleotide e 4 μL di SNAP T7 RNAP.
NOTA: la tabella 13 contiene le formule di reazione per la reazione di etichettatura SNAP-tag.- Preparare altri due campioni di controllo: 1) un controllo RNAP sostituendo BG-oligonucleotide con ddH2O; 2) un controllo del DNA sostituendo SNAP T7 RNAP con ddH2O (per la più bassa concentrazione oligonucleotidica di SNAP T7 RNAP). Incubare tutti i campioni a temperatura ambiente per 1 ora e mantenere il ghiaccio fino a quando necessario.
- Impostare undici reazioni da 10 μL aggiungendo 2 μL di ciascun campione a 4 μL di tampone SNAP e 2 μL di colorante a carico proteico e riscaldare a 70 °C per 10 min. Caricare 2 μL di ciascun campione sul gel proteico Bis-Tris al 4-12% ed eseguire l'elettroforesi su gel a 200 V per 35 min.
NOTA: la tabella 14 contiene le formule di reazione per i campioni di carico del gel.- Lavare la SDS tramite 3x scambio d'acqua su uno shaker, ogni lavaggio dura 10 minuti ciascuno. Stain con colorante cianonico per acidi nucleici per 15 minuti prima dell'imaging. Macchiare nuovamente il gel utilizzando 20 ml di macchia blu Coomassie per 1 ora. Smacchiare con ddH2O per 1 ora (o durante la notte) prima dell'imaging.
NOTA: Nel gel, una delle reazioni produrrà la polimerasi più legata insieme alla minima quantità di BG-oligonucleotide libero in eccesso; questo è il rapporto ottimale.
- Lavare la SDS tramite 3x scambio d'acqua su uno shaker, ogni lavaggio dura 10 minuti ciascuno. Stain con colorante cianonico per acidi nucleici per 15 minuti prima dell'imaging. Macchiare nuovamente il gel utilizzando 20 ml di macchia blu Coomassie per 1 ora. Smacchiare con ddH2O per 1 ora (o durante la notte) prima dell'imaging.
- Preparare i reagenti per l'accoppiamento della scala preparativa BG-oligonucleotide a SNAP T7 RNAP. Eseguire la reazione di accoppiamento con il rapporto ottimale trovato nella scala analitica.
NOTA: Ridurre al minimo l'esposizione delle proteine a temperatura ambiente posizionando la proteina sul ghiaccio quando non è in uso.
12. Purificazione di SNAP-T7 legato a oligonucleotidi mediante colonne a scambio ionico
- Seguire le istruzioni del produttore per la configurazione del tubo se si discosta dalle istruzioni elencate qui. Preparare un tampone di purificazione con pH superiore al punto isoelettrico della proteina.
NOTA: Per la proteina di esempio in questo protocollo, è stato utilizzato un tampone di purificazione di tampone fosfato di sodio 10 mM (pH 7).- Preparare 1.000 μL di tampone di eluizione contenente concentrazioni finali di 50 mM Tris e 0,5 M NaCl. Miscelare 50 μL di 1 M Tris, 100 μL di 5 M NaCl e 850 μL di ddH2O.
NOTA: la tabella 15 contiene la formula di reazione per il tampone di eluizione.
- Preparare 1.000 μL di tampone di eluizione contenente concentrazioni finali di 50 mM Tris e 0,5 M NaCl. Miscelare 50 μL di 1 M Tris, 100 μL di 5 M NaCl e 850 μL di ddH2O.
- Posizionare una colonna in un tubo di centrifuga da 2 ml e lavare con tampone di purificazione a 2.000 × g per 15 minuti o fino a quando tutto il tampone è stato eluito. Scartare il tampone eluito.
- Diluire ogni campione con tampone di purificazione a un rapporto tampone di purificazione 3:1:1 e caricare il campione nella colonna 400 μL alla volta. Ruotare a 2.000 × g per 10 minuti o fino a quando tutto il buffer non è stato eluito. Raccogliete il flusso passante ed etichettatelo come flow-through.
- Aggiungere 400 μL di tampone di purificazione al centro della colonna. Ruotare a 2.000 × g per 15 minuti o fino a quando tutto il buffer non è stato eluito. Raccogliere il flow-through ed etichettarlo come wash 1. Ripeti altre due volte per lavare 2 e lavare 3.
- Aggiungere 50 μL di tampone di eluizione al centro della colonna. Girare a 2.000 × g per 5 minuti o fino a quando tutto il buffer è stato eluito. Raccogliere il flusso passante ed etichettarlo come eluate 1. Ripeti altre due volte per l'eluato 2 e l'eluito 3.
- Pool eluisce 1, 2 e 3 (etichettare questo eluato totale), lasciando una piccola frazione di ciascun eluato per il gel e misurare l'assorbanza a 260 nm (A260) e 280 nm (A280). Dopo la misurazione, aggiungere glicerolo in rapporto 1:1 e conservare a -20 °C fino a nuovo utilizzo.
- Utilizzare un'unità filtrante centrifuga (0,5 mL; 30 kDa) per sostituire il buffer con 2x buffer buffer (~1:100) (etichettare questo prodotto). Misurare di nuovo A260/280. Aggiungere glicerolo in rapporto 1:1 e conservare a -20 °C fino a nuovo utilizzo.
- Caricare ogni eluato: flow-through, lavare 1-3, eluato totale e prodotto in un gel Bis-Tris SDS-PAGE al 4-12%, insieme a una scala proteica. Funzionare a 200 V per 35 minuti o fino a quando il fronte del colorante migra verso la fine.
13. Dimostrazione del controllo su richiesta dell'attività dell'RNA polimerasi legata
- Preparare 5x tampone di ricottura contenente 25 mM Tris, 5 mM EDTA e 25 mM cloruro di magnesio (MgCl2). Mescolare 2,4 μL di ciascun modello (1 μM) con 5 μL di tampone di ricottura e 14,2 μL di ddH2O per formare 25 μL di gabbia dsDNA da 1 μM. Incubare questa soluzione a 75 °C per 2 min. Allo stesso modo, ricottura i filamenti di senso e antisenso del promotore e del modello di DNA dell'aptamero verde malachite. Preparare una soluzione da 1 mM di ossalato verde malachite.
NOTA: la tabella 16 contiene la formula di reazione per il tampone di ricottura 5x, la tabella 17 contiene la formula di reazione per la ricottura di due modelli di ssDNA. - Incubare lo SNAP T7 RNAP legato con la gabbia dsDNA in un rapporto molare 1:5 a temperatura ambiente per 15 minuti fino a una concentrazione finale di 500 nM RNAP. Tenere sul ghiaccio fino a quando necessario.
- Preriscaldare il lettore di piastre a 37 °C. Impostare tre reazioni IVT da 25 μL sul ghiaccio
- Impostare una reazione contenente lo SNAP T7RNAP in gabbia con fattori di trascrizione dell'acido nucleico. Miscelare 2,5 μL di 10x tampone IVT, 1 μL di miscela rNTP da 25 mM, 1 μL di 1 mM di verde malachite, 2,5 μL della miscela RNAP-gabbia, 2,5 μL ciascuno di filamenti oligonucleotidici A e B del fattore di trascrizione A e B da 1 μL di 1 mM modello di aptamero verde malachite in 10 μL di ddH2O.
- Impostare una reazione contenente lo SNAP T7RNAP in gabbia senza fattori di trascrizione dell'acido nucleico. Miscelare 2,5 μL di 10x tampone IVT, 1 μL di miscela rNTP da 25 mM, 1 μL di 1 mM di verde malachite, 2,5 μL della miscela RNAP-gabbia e 3 μL di modello aptamero verde malachite da 1 mM in 15 μL di ddH2O.
- Impostare una reazione contenente solo buffer. Miscelare 2,5 μL di tampone IVT 10x, 1 μL di miscela rNTP da 25 mM, 1 μL di 1 mM di verde malachite e 3 μL di modello aptamero verde malachite da 1 mM in 17,5 μL di ddH2O.
NOTA: La tabella 18 contiene un riferimento generale per le reazioni di trascrizione in vitro.
- Trasferire ogni reazione su una piastra a 384 pozzetti. Monitorare la trascrizione dell'aptamero verde malachite su un lettore di piastre di fluorescenza per 2 ore a 37 °C e con eccitazione a 610 nm ed emissione di 655 nm. Una volta terminato, tenere la piastra su ghiaccio fino a quando non è necessario.
- Microonde 0,5x TBE buffer per 2 min 30 s o fino a ~70 °C. Eseguire i prodotti a RNA di ciascun pozzo in un gel di poliacrilammide TBE-Urea al 12% denaturante nel tampone TBE riscaldato 0,5x a 280 V per 20 minuti o fino a quando il fronte del colorante raggiunge la fine. Macchiare il gel con colorante di cianina colorazione di acido nucleico per 10 minuti su uno shaker orbitale prima dell'imaging.
NOTA: La Tabella 19 contiene la formula di reazione per un gel TBE-Urea PAGE denaturante al 12%.
Risultati

Figura 5: Analisi SDS-PAGE dell'espressione di SNAP T7 RNAP e test di trascrizione in vitro. (A) Analisi di purificazione della proteina SNAP T7 RNAP, peso molecolare SNAP T7 RNAP: 119.4kDa. FT = flusso dalla colonna, W1 = frazioni di eluizione del tampone di lavaggio contenente impurità, E1-3 = frazioni di eluizione contenenti prodotto puri...
Discussione
Questo studio dimostra un approccio ispirato alla nanotecnologia del DNA per controllare l'attività della T7 RNA polimerasi accoppiando covalentemente un RNAP T7 ricombinante N-terminale con tag SNAP con un oligonucleotide funzionalizzato BG, che è stato successivamente utilizzato per programmare reazioni TMDSD. In base alla progettazione, lo SNAP-tag è stato posizionato al N-terminus della polimerasi, poiché il C-terminus del wild-type T7 RNAP è sepolto all'interno del nucleo della struttura proteica e stabilisce i...
Divulgazioni
Non ci sono interessi finanziari concorrenti da dichiarare da parte di nessuno degli autori.
Riconoscimenti
L.Y.T.C riconosce il generoso sostegno del New Frontiers in Research Fund-Exploration (NFRF-E), del Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant e della Medicine by Design Initiative dell'Università di Toronto, che riceve finanziamenti dal Canada First Research Excellence Fund (CFREF).
Materiali
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
Riferimenti
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -. J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981 (2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324 (2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -. B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607 (2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C., Doudna, J. A. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -. B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati