
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Imaging in vivo del tessuto cerebrale completamente attivo in larve di zebrafish svegli e giovani mediante rimozione del cranio e della pelle
In questo articolo
Riepilogo
Qui presentiamo un metodo per immaginare il cervello embrionale del pesce zebra in vivo fino agli stadi larvale e giovanile. Questa procedura microinvasiva, adattata da approcci elettrofisiologici, fornisce l'accesso ai dettagli cellulari e subcellulari del neurone maturo e può essere combinata con studi optogenetici e neurofarmacologici per caratterizzare la funzione cerebrale e l'intervento farmacologico.
Abstract
Comprendere i cambiamenti effimeri che si verificano durante lo sviluppo e la maturazione del cervello richiede un'imaging dettagliato ad alta risoluzione nello spazio e nel tempo a risoluzione cellulare e subcellulare. I progressi nelle tecnologie molecolari e di imaging ci hanno permesso di ottenere numerose informazioni dettagliate sui meccanismi cellulari e molecolari dello sviluppo del cervello nell'embrione trasparente di zebrafish. Recentemente, i processi di raffinamento della connettività neuronale che si verificano in fasi larvali successive diverse settimane dopo la fecondazione, che sono ad esempio il controllo del comportamento sociale, il processo decisionale o il comportamento guidato dalla motivazione, si sono spostati al centro della ricerca. In queste fasi, la pigmentazione della pelle del pesce zebra interferisce con la penetrazione della luce nel tessuto cerebrale e le soluzioni per gli stadi embrionali, ad esempio l'inibizione farmacologica della pigmentazione, non sono più fattibili.
Pertanto, viene fornita una soluzione chirurgica minimamente invasiva per l'accesso al microscopio al cervello del pesce zebra sveglio che deriva da approcci elettrofisiologici. Nei teleostei, la pelle e la cartilagine cranica morbida possono essere accuratamente rimosse micro-peeling di questi strati, esponendo i neuroni sottostanti e i tratti assonali senza danni. Ciò consente di registrare la morfologia neuronale, comprese le strutture sinaptiche e il loro contenuto molecolare, e l'osservazione di cambiamenti fisiologici come i transitori di Ca2+ o gli eventi di trasporto intracellulare. Inoltre, è possibile interrogare questi processi mediante inibizione farmacologica o manipolazione optogenetica. Questo approccio di esposizione cerebrale fornisce informazioni sui cambiamenti strutturali e fisiologici nei neuroni, nonché sulla correlazione e l'interdipendenza di questi eventi nel tessuto cerebrale vivo nell'intervallo di minuti o ore. La tecnica è adatta per l'imaging cerebrale in vivo di larve di zebrafish fino a 30 giorni dopo la fecondazione, l'ultimo stadio di sviluppo testato finora. Fornisce quindi l'accesso a questioni importanti come il raffinamento e il ridimensionamento sinaptico, il trasporto assonale e dendritico, il targeting sinaptico del carico citoscheletrico o l'espressione locale dipendente dall'attività. Pertanto, è possibile prevedere un ampio utilizzo di questo approccio di montaggio e imaging.
Introduzione
Negli ultimi decenni, il pesce zebra (Danio rerio) si è evoluto come uno degli organismi modello di vertebrati più popolari per gli studi sullo sviluppo embrionale e larvale. La grande fecondità delle femmine di zebrafish unita al rapido sviluppo ex utero dell'embrione e alla sua trasparenza durante le prime fasi di sviluppo embrionale sono solo alcuni dei fattori chiave che rendono il pesce zebra un potente organismo modello per affrontare le domande sullo sviluppo1. I progressi nelle tecnologie genetiche molecolari combinati con studi di imaging in vivo ad alta risoluzione hanno permesso di affrontare i meccanismi biologici cellulari alla base dei processi di sviluppo2. In particolare, nel campo della differenziazione neuronale, della fisiologia, della connettività e della funzione, zebrafish ha fatto luce sull'interazione tra dinamica molecolare, funzioni cerebrali e comportamento organismico con dettagli senza precedenti.
Tuttavia, la maggior parte di questi studi sono limitati agli stadi embrionali e larvali precoci durante la prima settimana di sviluppo, poiché la trasparenza del tessuto del sistema nervoso viene progressivamente persa. In queste fasi, il tessuto cerebrale è impedito dall'accesso con approcci microscopici ad alta risoluzione che vengono schermati dalla differenziazione del cranio e dalla pigmentazione3.
Pertanto, le questioni chiave della differenziazione neuronale, della maturazione e della plasticità come il perfezionamento della connettività neuronale o il ridimensionamento sinaptico sono difficili da studiare. Questi processi cellulari sono importanti per definire i meccanismi cellulari che guidano, ad esempio, il comportamento sociale, il processo decisionale o il comportamento basato sulla motivazione, aree a cui la ricerca sui pesci zebra su larve di diverse settimane ha recentemente contribuito con risultati chiave basati su studi comportamentali4.
Gli approcci farmacologici per inibire la pigmentazione nelle larve di zebrafish per diverse settimane sono a malapena fattibili o possono persino causare effetti dannosi5,6,7,8. I ceppi mutanti doppi o tripli con specifici difetti di pigmentazione, come casper9 o crystal10, sono diventati strumenti tremendamente preziosi, ma sono laboriosi nell'allevamento, forniscono pochi figli e rappresentano il pericolo di accumulare malformazioni genetiche a causa di un'eccessiva consanguineità.
Qui, viene fornita una procedura minimamente invasiva come alternativa applicabile a qualsiasi ceppo di zebrafish. Questa procedura è stata adattata da studi elettrofisiologici per registrare l'attività neuronale nelle larve di zebrafish viventi e sveglie. Nei teleostei, la pelle e la cartilagine morbida del cranio possono essere accuratamente rimosse micro-peeling di questi strati, perché non sono strettamente intrecciati con la vascolarizzazione cerebrale. Ciò consente di esporre il tessuto cerebrale contenente neuroni e tratti assonali senza danni e di registrare la morfologia neuronale, comprese le strutture sinaptiche e il loro contenuto molecolare, che a sua volta include l'osservazione di cambiamenti fisiologici come transitori di Ca2+ o eventi di trasporto intracellulare per un massimo di diverse ore. Inoltre, al di là delle caratterizzazioni descrittive, l'accesso diretto al tessuto cerebrale consente l'interrogazione delle funzioni neuronali mature mediante la somministrazione di sostanze neurofarmacologiche e approcci optogenetici. Pertanto, le vere relazioni funzionali alla struttura possono essere rivelate nel cervello del pesce zebra giovanile utilizzando questa strategia di esposizione cerebrale.
Protocollo
Tutto il lavoro sugli animali qui descritto è conforme alle normative legali (Direttiva UE 2010/63). La manutenzione e la manipolazione del pesce sono state approvate dalle autorità locali e dal rappresentante per il benessere degli animali della Technische Universität Braunschweig.
1. Preparazione di liquido cerebrospinale artificiale (ACSF), acarosio a bassa fusione e aghi di vetro affilati
- Preparare l'ACSF sciogliendo le sostanze chimiche elencate alle seguenti concentrazioni in acqua distillata. 134 mM NaCl (58,44 g/mol), 2,9 mM KCl (74,55 g/mol), 2,1 mM CaCl2 (110,99 g/mol), 1,2 mM MgCl2 6x H2O (203,3 g/mol), 10 mM HEPES (238,31 g/mol) e 10 mM d-Glucosio (180,16 g/mol).
NOTA: Per MgCl2,CaCl2e KCl, le soluzioni stock da 1 M vengono preparate in acqua sterile dissalcata e conservate a 4 °C per la successiva preparazione di ACSF fresco. Glucosio, HEPES e NaCl sono disciolti come composti solidi nella soluzione acsf fresca. Per sciogliere le sostanze chimiche, seguire le istruzioni del produttore. - Regolare il pH dell'ACSF a 7,8 con 10 M NaOH. La preparazione di ACSF richiede una misurazione precisa delle sostanze chimiche e una regolazione fine del pH in quanto sostituisce il liquido cerebrospinale e mantiene le condizioni fisiologiche richieste affinché i neuroni siano pienamente funzionali, altrimenti potrebbe causare malfunzionamenti cerebrali e morte neuronale.
- Conservare l'ACSF appena preparato a 4 °C per un massimo di 4 settimane. Per le condizioni di lavoro, aliquotare il volume richiesto di ACSF per il giorno/esperimento e pre-riscaldamento a 25-28 °C (e facoltativamente ossigenarlo, fase 2.5)
NOTA: ASCF appena preparato va bene per 1 giorno. Se si prevede di utilizzarlo per diversi giorni, ACSF deve essere filtrato sterile. - Per l'anestesia successiva delle larve, preparare una soluzione stock di 50 mM di d-Tubocurarina in acqua distillata e conservare la soluzione a -20 °C come aliquote da 100 μL nel congelatore fino a quando necessario.
- Per incorporare il pesce, preparare il 2,5% di agarose a bassa fusione (LM) sciogliendo 1,25 g di LM-agarose(Tabella dei materiali)in 50 ml di ACSF e far bollire fino a quando l'agarose è completamente dissolta.
NOTA: In alternativa, possono essere utilizzate concentrazioni più alte o più basse di LM-agarose a seconda dell'allestimento sperimentale. Tuttavia, se l'agarose è troppo morbido, non sarà in grado di tenere il pesce in posizione quando apre il cranio. - Conservare l'agarose a 37 °C bagno d'acqua, per evitare la solidificazione e perché questa temperatura non danneggerà le larve durante l'incorporamento. Dopo che l'acarosio bollito è stato raffreddato a 37 °C nel bagno d'acqua, aggiungere la quantità necessaria di d-tubocurarina all'agarose aliquota necessaria affinché il giorno raggiunga una concentrazione di lavoro di 10 μM. Per un uso futuro, conservare l'acarosio residuo a 4 °C per evitare contaminazioni.
- Preparare aghi di vetro affilati e sottili da capillari di vetro (Figura supplementare 1) utilizzando un estrattore di micropipette con le seguenti impostazioni.
- Estrattore I, capillare tipo 1: Calore 1: 65,8; Calore 2: 55.1; Trazione in 2 fasi
Estrattore II, capillare tipo 2: Calore = 700; Fil = 4; Vel = 55; Del = 130; Pul = 55; Trazione in 1 passaggio.
NOTA: Le unità sono specifiche per ogni estrattore e capillare di vetro qui utilizzato, rispettivamente (vedi Tabella dei materiali). Altri capillari e tiranti possono anche essere utilizzati per preparare gli aghi di vetro. Ma gli aghi di vetro non dovrebbero essere troppo sottili in quanto potrebbero rompersi quando entrano in contatto con il cranio. Capillare: lunghezza: 100 mm (4 pollici); OD: 1,5 mm; ID: 0,84 mm; filamento: Sì
- Estrattore I, capillare tipo 1: Calore 1: 65,8; Calore 2: 55.1; Trazione in 2 fasi
2. Anestesia delle larve e preparati per l'incorporamento
- Quando inizi l'esperimento per la giornata, trasferisci gli animali necessari con una pipetta di plastica Pasteur in una capsula di Petri di 90 mm di diametro, che viene riempita con Danieau (per le larve che sono ancora conservate in una capsula di Petri con Danieau) o acqua dall'impianto ittico (per le larve che sono più vecchie di 7 dpf e sono tenute nella struttura ittica).
- Quando si pipettaggio di pesci più vecchi di 2 settimane, assicurarsi che l'apertura della pipetta sia abbastanza grande da evitare di ferire il pesce durante il trasferimento. Non usare una rete perché danneggerà fisicamente soprattutto le larve più giovani.
- Aggiungere naupli rotifera o artemia adatti alle dimensioni delle larve conservate nella capsula di Petri, per garantire il libero accesso al cibo e il massimo stato di salute delle larve e per ridurre lo stress.
- Per l'incorporamento, trasferire le larve selezionate su una capsula di Petri di 35 mm di diametro riempita con ACSF. Aggiungere il volume necessario di d-Tubocurarina per raggiungere una concentrazione di lavoro/dose efficace di 10 μM e attendere qualche minuto fino a quando le larve sono completamente immobilizzate11.
NOTA: Quando il pesce invecchia o se è necessaria un'anestesia completa più veloce (meno di 5 min), è possibile aumentare la concentrazione di d-Tubocurarina (LD50 per i topi è 0,13 mg/kg per via endovenosa12). È anche possibile utilizzare un anestetico diverso, come α-bungarotossina (concentrazione di lavoro: 1 mg / mL), che ha lo stesso effetto di curare e mantiene anche il cervello completamente attivo13. Se un cervello completamente attivo non è necessario per il soggetto di interesse, la tricaina in una dose non letale (0,02%) è anche un'opzione per anestetizzare completamente le larve. Tuttavia, la tricaina blocca i canali del sodio, compromettendo così l'attività cerebrale14. - Preparare la camera di montaggio prendendo il coperchio della capsula di Petri da 35 mm di diametro, capovolgere il coperchio e posizionare un coperchio quadrato di vetro (24 x 24 mm) sul fondo del coperchio. Vedere la Figura 1 (parte superiore) per una descrizione schematica di questi passaggi. La superficie più liscia del vetro impedisce lo scivolamento via del blocco di agarosio, che contiene le larve durante la procedura di apertura del cranio.
- Aliquotare la quantità di ACSF necessaria per il giorno in un flaconcino appropriato (ad esempio, tubo da 50 ml, becher, flacone Schott, ecc.) e ossigenarlo con carbogeno (5% CO2, 95% O2). Se l'imaging solo morfologia (ad esempio, modelli di fluorescenza) ACSF è ancora necessario per garantire l'integrità del cervello e che le cellule non sono influenzate negativamente dagli effetti di osmolarità, ma l'ossigenazione dell'ACSF non è necessaria. Questo passaggio deve essere eseguito solo quando è necessaria la piena attività cerebrale per l'imaging.
NOTA: Per una saturazione ottimale dell'ossigeno del mezzo, aggiungere una pietra d'aria all'estremità del tubo di carbogeno. Per garantire un livello di ossigeno sufficientemente elevato, è necessario scambiare l'ACSF nelle camere di imaging con ACSF appena ossigenato ogni 20-60 minuti, a seconda del numero e dell'età delle larve incorporate nella stessa camera di imaging (ad esempio, per una singola larva incorporata è sufficiente uno scambio ACSF ogni ora. Per sei larve di età superiore a 14 dpf incorporate in parallelo, è necessario scambiare ACSF ogni 20 minuti) quindi pianificare la quantità necessaria di ACSF saturo di ossigeno secondo l'esperimento pianificato.
3. Incorporamento delle larve
- Trasferire le larve completamente anestetizzate con una pipetta Pasteur di plastica nella camera di montaggio preparata (al punto 2.4). Quindi, rimuovere con attenzione il mezzo in eccesso per evitare la diluizione dell'LM-agarose. Tutti i seguenti passaggi devono essere eseguiti al microscopio stereo con ingrandimento sufficiente.
NOTA: l'inclinazione della camera di montaggio può aiutare a rimuovere completamente il mezzo. - Procedere immediatamente al passaggio successivo, aggiungendo una goccia di LM-agarose sufficientemente grande sopra le larve (circa 1 ml, a seconda delle dimensioni delle larve) per proteggere gli animali dall'essiccazione e ridurre lo stress inutile.
- Orientare le larve in posizione prima che l'agarose si solidifichi. Assicurarsi che la parte dorsale delle larve sia diretta verso l'alto. Inoltre, assicurati di incorporare le larve il più vicino possibile alla superficie dell'agarose.
NOTA: A seconda delle dimensioni e del numero di larve pianificate per l'incorporazione allo stesso tempo, è possibile regolare la concentrazione di agarose. Ad esempio, per 1-3 larve che hanno 30 dpf, si raccomanda una concentrazione dell'1,8% -2% di LM-agarose. Per 1-4 larve che hanno 7 dpf, è più pratico usare il 2,5% di LM-agarose, mentre, per 5-8 larve, il 2% è più adatto. Se è necessario un cervello completamente attivo, si consiglia di incorporare solo tre pesci contemporaneamente per ridurre il tempo necessario per far funzionare le larve. In generale, si raccomanda di utilizzare concentrazioni più basse (1,8% -2%) più le larve invecchiano o più larve sono pianificate per essere incorporate allo stesso tempo. - Se le immagini verranno registrate utilizzando un microscopio invertito, tagliare il blocco di agarose contenente le larve in una piccola forma cuboide. Questo è importante per trasferire le larve nella camera di imaging in seguito. Se si utilizza un microscopio verticale, tale rifilatura non è necessaria, perché la camera di montaggio può essere utilizzata anche come camera di imaging. Nella Figura 1 (parte superiore), è possibile trovare una descrizione schematica di questi passaggi.
4. Esporre il cervello
NOTA: Tutti i seguenti passaggi devono essere eseguiti con la massima cura per non ferire inutilmente le larve. Se è necessario un cervello completamente attivo per l'esperimento, tieni presente che con ogni secondo che passa, mentre il pesce è ancora completamente montato in agarose e ha un cranio aperto senza ACSF ossigenato, il cervello soffrirà di una mancanza di ossigeno e si asciugherà anche. Gli effetti della carenza di ossigeno diventeranno ancora più drammatici, più vecchie sono le larve incorporate. Pertanto, è importante eseguire l'intervento chirurgico non solo nel più breve tempo possibile, ma anche con la massima precisione per non evocare danni cerebrali meccanici con l'ago. Una volta addestrati, i passaggi 4.2-4.4 non dovrebbero richiedere più di 30 s per pesce.
- Iniziare l'intervento chirurgico non appena l'agarose si è solidificato. In primo luogo, tagliare via tutta l'agarose in eccesso sopra la regione del cervello di interesse per ottenere libero accesso alla testa e uno spazio di lavoro libero. Se la parte dorsale della testa sporge già dall'agarose, salta questo passaggio.
- A seconda della regione di interesse, scegli un punto per iniziare con l'intervento chirurgico. Prendi l'ago di vetro e fai una piccola incisione attraverso la pelle ma senza penetrare troppo in profondità nel tessuto. Questo sarà il punto di partenza per staccare la pelle sovrapposta.
NOTA: per risultati ottimali, non iniziare mai direttamente al di sopra della regione di interesse per ridurre il rischio di danneggiare strutture importanti. Se necessario, è possibile anche iniziare posteriormente al cervello posteriore e da lì lavorare in avanti fino a quando l'area indesiderata della pelle viene staccata. - Continuare con tagli molto piccoli lungo la parte della pelle con l'obiettivo di rimuovere spostando a malapena l'ago appena sotto la superficie. Il più delle volte non è necessario muoversi completamente intorno al cervello e ritagliare un pezzo di pelle e cranio simile a un cerchio, ma piuttosto fare solo due incisioni lungo la testa e poi spingere la pelle lontano verso l'uno o l'altro lato. La Figura 2 mostra una rappresentazione schematica della strategia di taglio ottimale per ottenere libero accesso al cervelletto.
NOTA: Questa micro-chirurgia è una procedura delicata e molto probabilmente avrà bisogno di un po 'di allenamento per rimuovere perfettamente la pelle senza danneggiare il cervello sottostante. Si raccomanda inoltre di scoprire la strategia di taglio ottimale per la regione del cervello di interesse e attenersi ad essa per il periodo dell'esperimento. - Immediatamente dopo aver rimosso la pelle da tutte le larve incorporate, procedere versando ACSF (ossigenato) sull'agarosio per inondare via particelle di pelle indesiderate e sangue e per mantenere il cervello completamente attivo e proteggerlo dall'essiccazione.
NOTA: Se è necessario un cervello sano per l'esperimento, si consiglia di scegliere un massimo di tre pesci alla volta. - Se si utilizza un microscopio verticale, iniziare direttamente con l'imaging.
- Quando si utilizza un microscopio invertito, far scorrere una piccola spatola sotto il blocco cuboide di agarose (passaggio 3.4).
- Aggiungere una piccola goccia di LM-agarose sul fondo della camera di imaging (ad esempio, piatto con fondo di vetro) e capovolgere immediatamente il blocco di agarose contenente le larve con la spatola per 180 ° e spingerlo delicatamente sul fondo della camera di imaging, mentre la goccia di agarose liquido funge da colla.
- Quando l'agarose si è solidificato, riempire la camera di imaging con ACSF (ossigenato), quindi iniziare l'imaging. Vedere la Figura 1 (parte inferiore) per una descrizione schematica.
- Quando è richiesta la piena attività cerebrale per l'esperimento, assicurarsi sempre che l'ACSF nella camera di imaging abbia un livello di ossigeno sufficientemente alto. Per garantire ciò, sostituire attentamente il mezzo con ACSF appena ossigenato quando possibile ogni 20-60 minuti (a seconda del numero e delle dimensioni del pesce, delle dimensioni e della superficie della camera di imaging e della durata dell'imaging).

Figura 1: Procedura schematica per la preparazione di zebrafish a cranio aperto per l'imaging in vivo in modo graduale. Le istruzioni di lavoro per i diversi passaggi possono essere trovate nella grafica stessa. Grafica disegnata da Florian Hetsch e adattata da Paul Schramm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Rappresentazione schematica dettagliata della microchirurgia eseguita per rimuovere pezzi di pelle e cranio sopra la regione cerebrale di interesse. Il cerchio rosso indica il punto in cui deve essere effettuato il primo taglio. La linea tratteggiata di rosso delinea il percorso ottimale da tagliare insieme all'ago per ottenere libero accesso al cervelletto senza danneggiarlo. La freccia verde segna la direzione in cui la pelle eccessiva e i pezzi del cranio possono essere facilmente spinti via. Assicurati di non penetrare mai nel tessuto cerebrale durante l'intera procedura. Dopo aver staccato con successo la pelle, la regione cerebrale di interesse (qui, il cervelletto) sarà liberamente accessibile per qualsiasi tipo di imaging in vivo ad alta risoluzione. Fare clic qui per visualizzare una versione più grande di questa figura.
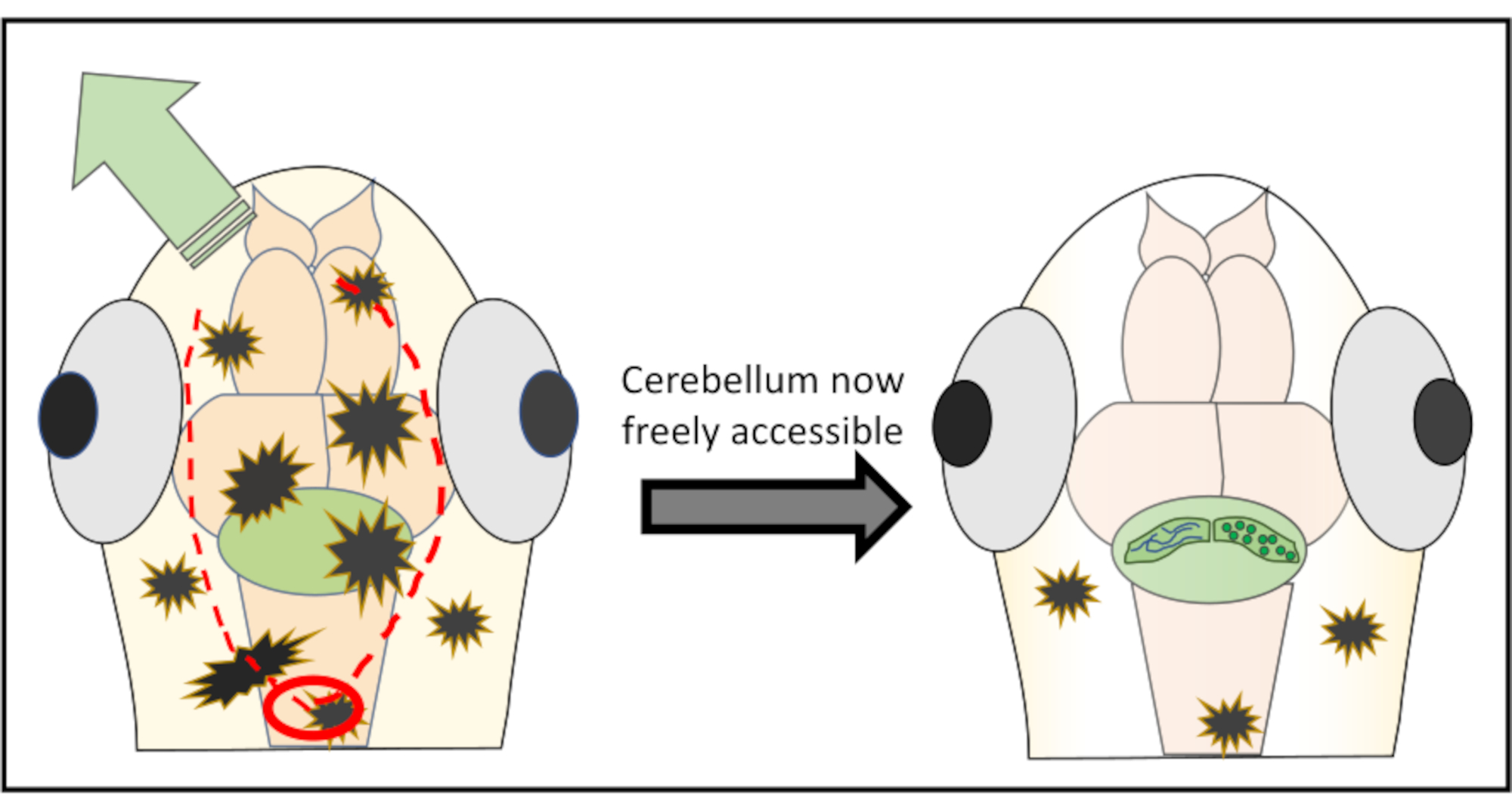{kind=link}
Risultati
Figura 3A,C mostrano una larva di 14 dpf della linea transgenica Tg[-7.5Ca8:GFP]bz12[15] con il cranio ancora intatto. Le cellule del pigmento nella pelle sovrapposta sono distribuite su tutta la testa e interferiscono con il segnale di fluorescenza nella regione di interesse (qui, cervelletto). Con la larva in questa condizione, non è possibile ottenere immagini ad alta risoluzione del cervello....
Discussione
Il metodo presentato fornisce un approccio alternativo all'isolamento cerebrale o al trattamento delle larve di zebrafish con farmaci che inibiscono la pigmentazione per la registrazione di immagini ad alta risoluzione di neuroni nel loro ambiente in vivo. La qualità delle immagini registrate con questo metodo è paragonabile alle immagini provenienti da cervelli espiantati, ma in condizioni naturali.
Inoltre, si evita una perdita di intensità di fluorescenza, perché non è necessa...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Ringraziamo in particolare Timo Fritsch per l'eccellente cura degli animali e Hermann Döring, Mohamed Elsaey, Sol Pose-Méndez, Jakob von Trotha, Komali Valishetti e Barbara Winter per il loro utile supporto. Siamo anche grati a tutti gli altri membri del laboratorio Köster per il loro feedback. Il progetto è stato finanziato in parte dal progetto della Fondazione tedesca per la ricerca (DFG, KO1949/7-2) 241961032 (a RWK) e dal Bundesministerium für Bildung und Forschung (BMBF; È riconosciuto il progetto ERA-Net NEURON II CIPRESS 01EW1520 to JCM).
Materiali
| Name | Company | Catalog Number | Comments |
| Calcium chloride | Roth | A119.1 | |
| Confocal Laser scanning microscope | Leica | TCS SP8 | |
| d-Glucose | Sigma | G8270-1KG | |
| d-Tubocurare | Sigma-Aldrich | T2379-100MG | |
| Glass Capillary type 1 | WPI | 1B150F-4 | |
| Glass Capillary type 2 | Harvard Apparatus | GC100F-10 | |
| Glass Coverslip | deltalab | D102424 | |
| HEPES | Roth | 9105.4 | |
| Hoechst 33342 | Invitrogen (Thermo Fischer) | H3570 | |
| Imaging chamber | Ibidi | 81156 | |
| Potassium chloride | Normapur | 26764298 | |
| LM-Agarose | Condalab | 8050.55 | |
| Magnesium chloride (Hexahydrate) | Roth | A537.4 | |
| Microscope Camera | Leica | DFC9000 GTC | |
| Needle-Puller type 1 | NARISHIGE | Model PC-10 | |
| Needle-Puller type 2 | Sutter Instruments | Model P-2000 | |
| Pasteur-Pipettes 3ml | A.Hartenstein | 20170718 | |
| Sodium chloride | Roth | P029.2 | |
| Sodium hydroxide | Normapur | 28244262 | |
| Tricain | Sigma-Aldrich | E10521-50G | |
| Waterbath | Phoenix Instrument | WB-12 | |
| 35 mm petri dish | Sarstedt | 833900 | |
| 90 mm petri dish | Sarstedt | 821473001 |
Riferimenti
- Embryology. Zebrafish Development Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Zebrafish_Development (2020)
- Sassen, W. A., Köster, R. W. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. Dove Medical Press. 2015 (5), 151-163 (2015).
- Singh, A. P., Nüsslein-Volhard, C. Zebrafish stripes as a model for vertebrate colour pattern formation. Current Biology. 25 (2), 81-92 (2015).
- Kalueff, A. V., et al. Time to recognize zebrafish 'affective' behavior. Brill: Behaviour. 149 (10-12), 1019-1036 (2012).
- Karlsson, J., von Hofsten, J., Olsson, P. -. E. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology. 3, 522-527 (2001).
- Bohnsack, B. L., Gallina, D., Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PloS One. 6, 22991 (2011).
- Elsalini, O. A., Rohr, K. B. Phenylthiourea disrupts thyroid function in developing zebrafish. Development Genes and Evolution. 212, 593-598 (2003).
- Baumann, L., Ros, A., Rehberger, K., Neuhauss, S. C. F., Segner, H. Thyroid disruption in zebrafish (Danio rerio) larvae: Different molecular response patterns lead to impaired eye development and visual functions. Aquatic Toxicology. 172, 44-55 (2016).
- White, R., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2, 183-189 (2008).
- Antinucci, P., Hindges, R. A crystal-clear zebrafish for in vivo imaging. Scientific Reports. 6, 29490 (2016).
- Burr, S. A., Leung, Y. L. Curare (d-Tubocurarine). Encyclopedia of Toxicology (3rd Edition). , 1088-1089 (2014).
- Gesler, H. M., Hoppe, J. 3,6-bis(3-diethylaminopropoxy) pyridazine bismethiodide, a long-acting neuromuscular blocking agent. The Journal of Pharmacology and Experimental Therapeutics. 118 (4), 395-406 (1956).
- Furman, B. . Alpha Bungarotxin. Reference Module in Biomedical Sciences. , (2018).
- Attili, S., Hughes, S. M. Anaesthetic tricaine acts preferentially on neural voltage-gated sodium channels and fails to block directly evoked muscle contraction. PLoS One. 9 (8), 103751 (2014).
- Namikawa, K., et al. Modeling neurodegenerative spinocerebellar ataxia type 13 in zebrafish using a Purkinje neuron specific tunable coexpression system. Journal of Neuroscience. 39 (20), 3948-3969 (2019).
- Hennig, M. Theoretical models of synaptic short term plasticity. Frontiers in Computational Neuroscience. 7 (45), (2013).
- Wang, Y., et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 137, 3119-3128 (2010).
- Hobro, A., Smith, N. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Knogler, L. D., Kist, A. M., Portugues, R. Motor context dominates output from purkinje cell functional regions during reflexive visuomotor behaviours. eLife. 8, 42138 (2019).
- Hsieh, J., Ulrich, B., Issa, F. A., Wan, J., Papazian, D. M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Frontier in Neural Circuits. 8 (147), (2014).
- Scalise, K., Shimizu, T., Hibi, M., Sawtell, N. B. Responses of cerebellar Purkinje cells during fictive optomotor behavior in larval zebrafish. Journal of Neurophysiology. 116 (5), 2067-2080 (2016).
- Harmon, T. C., Magaram, U., McLean, D. L., Raman, I. M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife. 6, 22537 (2017).
- Zehendner, C. M., et al. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8 (12), 82823 (2013).
- Dhabhar, F. S. The short-term stress response - mother nature's mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Frontiers of Neuroendocrinology. 49, 175-192 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati