
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Ablazioni di volume profonde e controllate spazialmente utilizzando un microscopio a due fotoni nella Zebrafish Gastrula
In questo articolo
Riepilogo
Lo sviluppo embrionale richiede una coordinazione su larga scala del movimento cellulare. L'ablazione laser mediata dall'eccitazione a due fotoni consente l'ablazione tridimensionale controllata spazialmente di grandi gruppi di cellule profonde. Inoltre, questa tecnica può sondare la reazione delle cellule che migrano collettivamente in vivo alle perturbazioni nel loro ambiente meccanico.
Abstract
La morfogenesi coinvolge molti movimenti cellulari per organizzare le cellule in tessuti e organi. Per un corretto sviluppo, tutti questi movimenti devono essere strettamente coordinati e l'accumulo di prove suggerisce che ciò si ottiene, almeno in parte, attraverso interazioni meccaniche. Testare questo nell'embrione richiede perturbazioni fisiche dirette. Le ablazioni laser sono un'opzione sempre più utilizzata che consente di alleviare i vincoli meccanici o di isolare fisicamente due popolazioni cellulari l'una dall'altra. Tuttavia, molte ablazioni vengono eseguite con un laser ultravioletto (UV), che offre una risoluzione assiale limitata e una penetrazione tissutale. Qui viene descritto un metodo per ablare volumi profondi, significativi e spazialmente ben definiti utilizzando un microscopio a due fotoni. Le ablazioni sono dimostrate in una linea transgenica di zebrafish che esprime la proteina fluorescente verde nel mesendoderma assiale e utilizzata per recidere il mesendoderma assiale senza influenzare l'ectoderma sovrastante o la cellula del tuorlo sottostante. Il comportamento cellulare viene monitorato mediante imaging dal vivo prima e dopo l'ablazione. Il protocollo di ablazione può essere utilizzato in diverse fasi dello sviluppo, su qualsiasi tipo di cellula o tessuto, su scale che vanno da pochi micron a più di cento micron.
Introduzione
Le interazioni cellula-cellula svolgono un ruolo vitale nello sviluppo. Le cellule forniscono segnali che i loro vicini diretti, o le cellule più lontane, possono percepire, influenzando così il loro destino e / o comportamento. Molti di questi segnali sono di natura chimica. Ad esempio, negli eventi di induzione ben caratterizzati, un gruppo cellulare produce molecole diffusibili che influenzano il destino di un'altra popolazione cellulare1. Altri segnali, tuttavia, sono meccanici; le cellule esercitano forze e vincoli sui loro vicini, che i vicini percepiscono e a cui rispondono2.
Un modo per studiare l'importanza di queste interazioni cellula-cellula in vivo è quello di eliminare alcune cellule e osservare lo sviluppo successivo. Sfortunatamente, le tecniche disponibili per rimuovere o distruggere le cellule sono limitate. Le cellule possono essere rimosse chirurgicamente3,4, utilizzando aghi o piccoli fili, ma tali trattamenti sono invasivi, non molto precisi e di solito eseguiti sotto uno stereomicroscopio, impedendo l'imaging immediato al microscopio. Inoltre, prendere di mira le cellule profonde implica perforare un buco nei tessuti sovrastanti, creando perturbazioni indesiderate. I fotosensibilizzatori geneticamente codificati, come KillerRed, sono stati utilizzati per indurre la morte cellulare tramite l'illuminazione della luce5. I fotosensibilizzatori sono cromofori che generano specie reattive dell'ossigeno dopo l'irradiazione della luce. Il loro principale limite è che richiedono lunghe illuminazioni di luce (circa 15 minuti), che possono essere difficili da ottenere se le cellule sono in movimento, e che inducono la morte cellulare attraverso l'apoptosi, che non è immediata.
Infine, le ablazioni laser sono state sviluppate e ampiamente utilizzate negli ultimi 15 anni6,7,8,9,10,11,12. Un raggio laser è focalizzato sulla cellula / tessuto mirato. Induce la sua ablazione attraverso il riscaldamento, la fotoablazione o l'ablazione indotta dal plasma; il processo coinvolto dipende dalla densità di potenza e dal tempo di esposizione13. La maggior parte dei protocolli di ablazione utilizza laser UV per la loro alta energia. Tuttavia, la luce UV viene assorbita e dispersa dai tessuti biologici. Pertanto, il targeting di cellule profonde richiede un'elevata potenza laser, che quindi induce danni nei tessuti più superficiali e fuori piano. Ciò limita l'uso dei laser UV alle strutture superficiali e spiega la loro risoluzione assiale relativamente bassa. L'ottica non lineare (la cosiddetta microscopia a due fotoni) utilizza proprietà non lineari della luce per eccitare un fluoroforo con due fotoni di circa mezza energia nel dominio infrarosso. Se applicato alle ablazioni, questo ha tre vantaggi principali. In primo luogo, la luce infrarossa è meno dispersa e meno assorbita della luce UV dai tessuti biologici14, consentendo di raggiungere strutture più profonde senza aumentare la potenza laser richiesta. In secondo luogo, l'uso di un laser pulsato a femtosecondi fornisce densità di potenza molto elevate, creando un'ablazione attraverso l'induzione del plasma, che, contrariamente al riscaldamento, non si diffonde spazialmente15. In terzo luogo, la densità di potenza che induce la formazione di plasma viene raggiunta solo nel punto focale. Grazie a queste proprietà, le ablazioni laser a due fotoni possono essere utilizzate per colpire con precisione le cellule profonde senza influenzare l'ambiente tissutale circostante.
Le migrazioni collettive sono un ottimo esempio di processi di sviluppo in cui le interazioni cellula-cellula sono fondamentali. Le migrazioni collettive sono definite come migrazioni cellulari in cui le cellule vicine influenzano il comportamento di una cellula16. La natura di queste interazioni (chimiche o meccaniche) e il modo in cui influenzano la migrazione cellulare possono variare notevolmente e spesso non sono del tutto comprese. La capacità di rimuovere le cellule e osservare come questo influisce sugli altri è fondamentale per svelare ulteriormente questi processi collettivi. Alcuni anni fa, abbiamo stabilito, utilizzando approcci chirurgici, che la migrazione del polster durante la gastrulazione del pesce zebra è una migrazione collettiva17. Il polster è un gruppo di cellule che costituisce le prime cellule internalizzanti sul lato dorsale dell'embrione18. Queste cellule, etichettate in verde nella linea transgenica Tg(gsc:GFP), si trovano in profondità nell'embrione, al di sotto di diversi strati di cellule epiblastiche. Durante la gastrulazione, questo gruppo guida l'estensione del mesoderma assiale, migrando dall'organizzatore embrionale al polo animale19,20,21,22,23 (Figura 1A). Abbiamo stabilito che le cellule richiedono il contatto con i loro vicini per orientare la loro migrazione nella direzione del polo animale. Tuttavia, una migliore comprensione delle basi cellulari e molecolari di questa migrazione collettiva comporta la rimozione di alcune cellule per vedere come questo influenza quelle rimanenti. Abbiamo quindi sviluppato ablazioni di volumi grandi e profondi utilizzando una configurazione di microscopia a due fotoni. Qui, dimostriamo l'uso di questo protocollo per recidere il polster nel suo mezzo e osservare le conseguenze sulla migrazione cellulare tracciando i nuclei etichettati con Histone2B-mCherry.
Protocollo
Tutto il lavoro sugli animali è stato approvato dal Comitato Etico N 59 e dal Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche con il numero di file APAFIS#15859-2018051710341011v3. Alcuni dei passaggi descritti di seguito sono specifici per le nostre apparecchiature e software, ma potrebbero essere facilmente adattati a diverse apparecchiature.
1. Preparazione dell'iniezione
- Preparare 75 ml di soluzione di agarosio all'1% in Embryo Medium (EM).
- Posizionare lo stampo di iniezione in una capsula di Petri da 90 mm e versare circa 50 ml di agarosio, sufficienti per far galleggiare lo stampo. Lasciare che l'agarosio si solidifichi e rimuovere lo stampo di iniezione.
- Preparare un piatto rivestito di agarosio versando 1 ml di agarosio in una capsula di Petri da 30 mm.
- Preparare 4 μL di 30 ng/μL di soluzione di mRNA di istone2B-mCherry diluendo la soluzione madre in acqua priva di RNasi e conservarla sul ghiaccio.
NOTA: Fare attenzione a indossare i guanti durante la manipolazione dell'mRNA per evitare la degradazione mediata dalla RNasi. - Estrarre un ago per iniezione da un capillare usando l'estrattore di micropipette.
2. Preparazione degli embrioni
- Una volta che i pesci hanno deposto le uova, raccogliere, risciacquare e raccogliere in una capsula di Petri da 90 mm in EM. Collocare gli embrioni in un'incubatrice a 28,5 °C.
- Attendere 20 minuti affinché la prima cella diventi visibile.
- Trasferire 30 embrioni nella piastra di iniezione riempita con EM. Spremere gli embrioni nei solchi usando una pinza leggermente smussata e orientarli con il palo animale verso l'alto.
- Utilizzando una punta del microloader, riempire un ago per iniezione con 2 μL di soluzione di mRNA. Inserire l'ago nel supporto capillare posto in un micropolatore collegato con tubo in politetrafluoroetilene (PTFE) a un iniettore d'aria.
- Sotto lo stereomicroscopio, rompere con attenzione la punta dell'ago.
- Iniettare la soluzione di mRNA negli embrioni allo stadio a 1 cellula inserendo l'ago nella cellula.
NOTA: il volume iniettato è circa un terzo del volume della cella. - Riposizionare gli embrioni iniettati nell'incubatrice a 28,5 °C.
3. Preparazione del microscopio a due fotoni
NOTA: in questo protocollo vengono utilizzati due laser. Uno è usato per visualizzare GFP (a 920 nm) ed eseguire ablazioni (a 820 nm). Sarà indicato come il laser verde / ablazione. L'altro è usato a 1160 nm per l'immagine mCherry. Sarà indicato come il laser rosso.
- Impostare il laser verde/ablazione su 820 nm (lunghezza d'onda di ablazione) e il laser rosso su 1160 nm (eccitazione mCherry).
- Utilizzando specchi mobili sul percorso ottico, allineare i raggi laser verdi/ablazione e rossi sia all'ingresso che all'uscita della testa di scansione.
NOTA: questo aumenta la messa a fuoco del raggio laser e riduce al minimo il volume focale per l'eccitazione e l'ablazione. - Misurare la potenza massima del laser verde/ablazione a 820 nm sotto l'obiettivo. Per fare ciò, posizionare il misuratore di potenza sotto l'obiettivo, chiudere la camera nera, impostare la potenza del laser verde / ablazione al 100% e aprire le persiane. Calcola la percentuale di potenza laser necessaria per raggiungere i 300 mW.
- Riportare il laser verde/ablazione a 920 nm (eccitazione GFP) e impostare la potenza del laser al 7%. Impostare la potenza del laser rosso su 15%.
- Attivare i rivelatori epi-PhotoMultiplier Tubes (PMT) per linee verdi e rosse; impostare la sensibilità PMT della linea verde e rossa su 65.
- Impostare il campo visivo su 400 x 400 μm, la risoluzione dell'immagine su 512 x 512 pixel e la frequenza di scansione su 800 Hz.
- Selezionare la modalità 3D Timelapse Imaging . Quindi, creare una cartella e attivare il salvataggio automatico per i dati dopo ogni acquisizione.
- Assemblare la camera di riscaldamento e impostarla a 28 °C. Attendere almeno 10 minuti affinché la camera e l'obiettivo si riscaldino.
4. Montare l'embrione
- Sotto uno stereomicroscopio a fluorescenza, identificare gli embrioni al 70% epiboly che esprimono GFP.
NOTA: selezionare embrioni con un segnale luminoso nel mesoderma assiale e senza fluorescenza di fondo per una migliore qualità di imaging. - Trasferire da tre a quattro embrioni selezionati nel piatto rivestito di agarosio (fase 1.3) utilizzando una pipetta pasteur di plastica e decoerniarli accuratamente con una pinza fine.
NOTA: Gli embrioni decoionati sono molto delicati e scoppiano a contatto con aria o plastica. - Versare 1 mL di agarosio allo 0,2% in 1x penicillina-streptomicina EM in una piccola fiala di vetro. Posizionare il flaconcino in un riscaldatore a blocchi a secco preriscaldato a 42 °C.
NOTA: I seguenti passaggi devono essere eseguiti rapidamente per consentire l'orientamento dell'embrione prima che si depositi l'agarosio. - Trasferire un embrione decorionato nel flaconcino di vetro di agarosio allo 0,2% utilizzando una pipetta di vetro lucidata a fuoco. Fare attenzione a non aggiungere troppo EM nell'agarosio per evitare di diluirlo. Scartare l'EM rimanente dalla pipetta e aspirare l'embrione indietro insieme a abbastanza agarosio per coprire il vetrino del piatto inferiore di vetro prima che l'embrione cada dalla pipetta.
- Soffiare l'agarosio e l'embrione sul vetrino del piatto. Fare attenzione a non lasciare che l'embrione tocchi l'aria o il lato di plastica del piatto. Quindi, riempire la camera attorno al vetrino con agarosio.
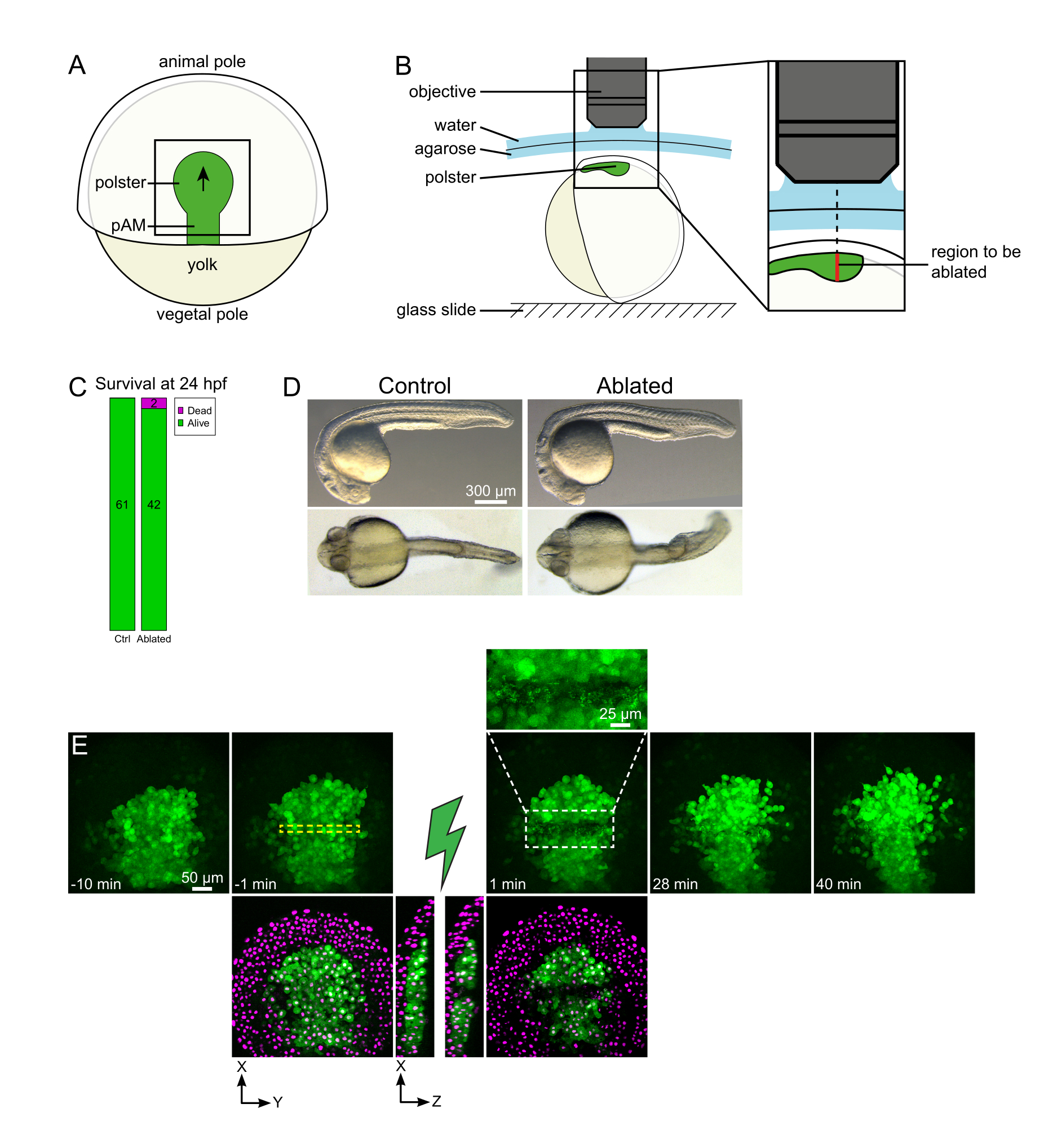
- Utilizzare una ciglia per orientare l'embrione in modo che la regione target sia in alto (Figura 1B).
NOTA: Quando si orientano gli embrioni, fare attenzione a toccare solo il blastoderma, non il tuorlo molto fragile. L'agarosio si stabilirà in circa 1 minuto, a seconda della temperatura ambiente. - Attendere ~ 5 minuti affinché l'agarosio si fissi completamente, quindi aggiungere alcune gocce di penicillina-streptomicina EM.
5. Localizzazione dell'embrione e imaging pre-ablazione
- Posizionare il piatto con fondo di vetro sotto l'obiettivo nella camera riscaldata. Immergere l'obiettivo in penicillina-streptomicina EM e chiudere la camera riscaldata.
- Spostare il dispositivo di scorrimento per impostare il percorso della luce sugli oculari. Quindi, usando oculari, lampade fluorescenti e controllo del palcoscenico, trova un embrione e imposta l'attenzione sulla superficie dell'embrione.
- Spegnere la lampada a fluorescenza, impostare il percorso della luce verso i PMT e chiudere la camera nera.
NOTA: Fare attenzione a spegnere tutte le sorgenti luminose nella camera nera in quanto potrebbe danneggiare i PMT. - Avvia l'imaging dal vivo e individua il mesoderma assiale. Regola le potenze laser verde/ablazione e rosso per avere un buon segnale (cioè tra 1.000 e 20.000 fotoni per pixel per le aree di espressione GFP). Usa il canale rosso per spostare lo stadio verso l'alto dell'embrione e imposta questa posizione come Z = 0.
- Scegli un time-step di 1 min e uno Z-step di 2 μm. Un corso Z di 110 μm è sufficiente per comprendere l'intero polster e viene acquisito in meno di 1 minuto con queste impostazioni. Impostare la prima fetta 15 μm sopra il mesoderma assiale (nell'ectoderma più superficiale).
NOTA: il polster si muove lungo una linea curva in modo che la fetta inferiore dello Z-stack debba essere impostata 30 μm più in profondità rispetto alla posizione più profonda del polster per adattarsi al suo movimento durante l'imaging time-lapse (Figura 1E). - Registra 10-15 minuti di filmato pre-ablazione.

Figura 1: Esito positivo delle ablazioni laser. (A) Schema di un embrione gastrulante al 70% epiboly in vista dorsale; pAM: mesoderma assiale posteriore; freccia nera segna la direzione della migrazione polster; il quadrato nero indica un tipico campo visivo per le ablazioni nel polster. (B) Schema di montaggio dell'embrione per la separazione del polster. Vista laterale. L'embrione è montato in modo tale che il piano del polster sia perpendicolare all'asse ottico. (C) sopravvivenza e (D) morfologia degli embrioni di controllo e ablati a 24 ore dopo la fecondazione. La barra della scala è di 300 μm. (E) Sequenza temporale dall'ablazione laser nel polster di un embrione Tg(gsc:GFP) che esprime Histone2B-mCherry. Le viste con solo il canale verde sono proiezioni massime. Il primo piano mostra l'area ablata contenente detriti cellulari. Le viste con canali verde e rosso (visualizzati come magenta) sono sezioni XY e XZ prima e dopo l'ablazione (il fulmine verde rappresenta l'ablazione). Le fette XZ mostrano che i tessuti sovrastanti (nuclei magenta senza espressione GFP) non sono stati influenzati dall'ablazione delle strutture sottostanti. La casella tratteggiata gialla corrisponde al ROI selezionato per il trattamento di ablazione laser. La barra della scala è di 50 μm nelle viste grandi e 25 μm nel primo piano. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
6. Posizione del bersaglio e ablazione laser
- Individua il contorno polster sull'imaging dal vivo e, utilizzando lo strumento EOM (Electro-Optic Modulator Region of Interest), disegna un rettangolo di 20 pixel (15 μm) che copre la larghezza del polster. Posizionare questo rettangolo al centro del polster (Figura 1E).
- Si noti la posizione assiale dei piani più alto e più basso contenenti celle polster. Le ablazioni saranno eseguite ogni 10 μm tra questi due piani. Fai attenzione che il ROI non si sovrapponga alla cellula del tuorlo su nessuno di questi piani.
- Posizionate lo stage nella posizione Z più bassa dell'intervallo. Le ablazioni devono essere eseguite dal basso verso l'alto poiché i detriti assorbono la luce.
- Impostare la lunghezza d'onda del laser verde/ablazione su 820 nm e impostare la percentuale di potenza per ottenere una potenza di uscita di 300 mW (passaggio 3.3).
- Impostare la frequenza di imaging su 200 Hz.
- Impostare l'EOM di imaging laser verde/ablazione su 0 e selezionare la modalità ROI-Treat .
- Accendere l'EOM e impostare il trattamento in modo che inizi immediatamente (dopo 0 fotogrammi).
- Impostare la modalità di imaging su Timelapse e disattivare il salvataggio automatico.
- Impostare Time Step sulla modalità Veloce.
- Impostate Numero di fotogrammi di trattamento e Numero di fotogrammi sul valore corrispondente alla profondità di destinazione (Tabella 1).
| Profondità (μm) | Telai per trattamenti |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Tabella 1: Numero suggerito di fotogrammi di trattamento laser in funzione della profondità cellulare mirata nell'embrione (0 è la superficie dell'embrione).
- Avviare l'imaging. L'acquisizione è nera poiché l'otturatore del PMT si chiude durante il trattamento EOM.
- Spostarsi verso l'alto dello stage fino alla successiva posizione Z dell'elenco (passaggio 6.2).
- Ripetere i passaggi da 6.10 a 6.12 fino a raggiungere la parte superiore del polster.
7. Verifica post-ablazione e imaging
- Impostare il laser verde/ablazione su 920 nm e potenza del 5%. Impostare l'EOM di imaging laser verde/ablazione su 100 e selezionare la modalità Fullfield .
- Impostare frequenza di imaging su 800 Hz. Disattivare EOM.
- Passa attraverso l'intero stack in modalità live per verificare se ogni aereo è stato ablato. In caso contrario, tornare al passaggio 6.2.
NOTA: l'ablazione a volte induce uno spostamento verticale dei tessuti vicini in modo che lo Z-stack potrebbe dover essere ridefinito. - Impostare la modalità di imaging su Timelapse 3D e riattivare il salvataggio automatico. Registra 40-60 minuti di filmato post-ablazione.
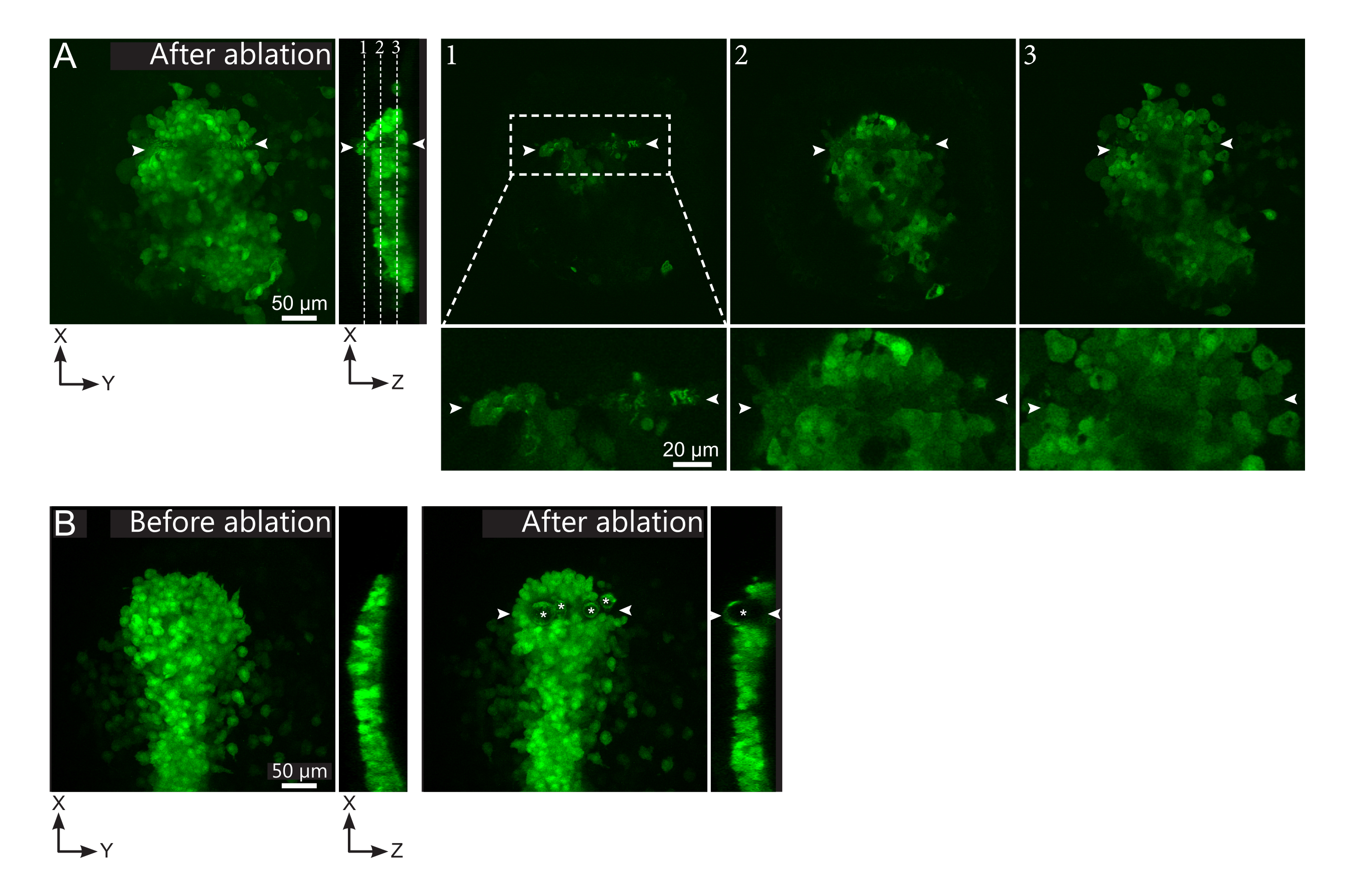
- Controlla, nel filmato post-ablazione, se le cellule bersaglio sono state effettivamente ablate. Il recupero della fluorescenza, o le cellule mirate che occupano spazio e impediscono alle cellule follower di muoversi, indicano che le cellule bersaglio sono state solo fotolette e non ablate (Figura 1E e Figura 2A).

Figura 2: Risultati negativi delle ablazioni laser. (A) Esempi tipici di potenziali guasti nell'ablazione laser. Le viste XY di grandi dimensioni sono proiezioni massime, la vista XZ è una sezione ricostruita. L'area trattata al laser si trova tra le due punte di freccia bianche. Tre piani focali sono evidenziati nella sezione ricostruita e visualizzati a destra. Corrispondono a tre diversi tipi di guasti. Il piano 1 mostra che le cellule sopra il polster sono state ablate. Ciò può essere identificato dalla presenza di detriti autofluorescenti su questo piano focale (vedi primo piano) sopra il polster (vedi posizione del piano 1 sulla sezione ricostruita). Ciò probabilmente deriva da una definizione errata della regione da ablare. Il piano 2 mostra le cellule che sono state sbiancate ma non ablate. Possono essere identificati come il segnale di bassa fluorescenza rivela ancora contorni cellulari intatti (vedi primo piano). Il piano 3 mostra cellule intatte, che sono state appena sbiancate dal trattamento laser. Ciò potrebbe derivare da una definizione errata della regione da ablare o da un cattivo trattamento. Nelle situazioni raffigurate nei piani 2 e 3, è possibile riapplicare il trattamento di ablazione alle cellule bersaglio non ablate. La barra della scala è di 50 μm in ampie viste e 20 μm in primi piani. (B) Un tipico esempio di bolle (contrassegnate da asterischi bianchi) formate da cavitazione a causa di un trattamento laser troppo intenso. Tali bolle non sono limitate a un piano Z, a volte anche che copre l'intera altezza del polster, deformando i tessuti vicini. La barra della scala è di 50 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
8. Analisi dei dati
- Apri la serie time-lapse con il software di analisi delle immagini e imposta la dimensione corretta dei pixel.
- Nella funzione Spot , impostare la Dimensione oggetto su 10 μm, poiché questa è la dimensione media del nucleo durante la gastrulazione. Quindi, eseguire la funzione Spot per rilevare e tracciare i nuclei.
NOTA: il rilevamento può essere leggermente migliorato considerando la risoluzione assiale inferiore, adattando una forma ellissoidale lunga 12 μm lungo l'asse Z. - Utilizzare i filtri per rimuovere i falsi positivi. Nella linea Tg(Gsc:GFP), le cellule dell'asse embrionale e alcune cellule endodermiche sono etichettate in verde. Quindi, il filtraggio sull'intensità del verde consente una rapida selezione di queste celle (Figura 3A).
- Impostare la distanza massima tra punti consecutivi su un valore compatibile con la velocità delle celle.
NOTA: prestare attenzione a considerare l'intervallo di tempo tra due fotogrammi. Le celle polster migrano a 2,8 ± 0,8 μm/min. Quindi, consentire 4 μm di spostamento massimo per un intervallo di tempo di 1 minuto rimuove la maggior parte delle tracce artefatte. - Consentire spazi vuoti su uno o due punti temporali fornisce tracce continue più lunghe, ma può introdurre errori di tracciamento. Se un nucleo non viene rilevato correttamente in un punto una tantum, prendere in considerazione la possibilità di eseguire nuovamente il rilevamento spot con parametri/filtri diversi.
- Controllare visivamente le tracce e, se necessario, correggerle.
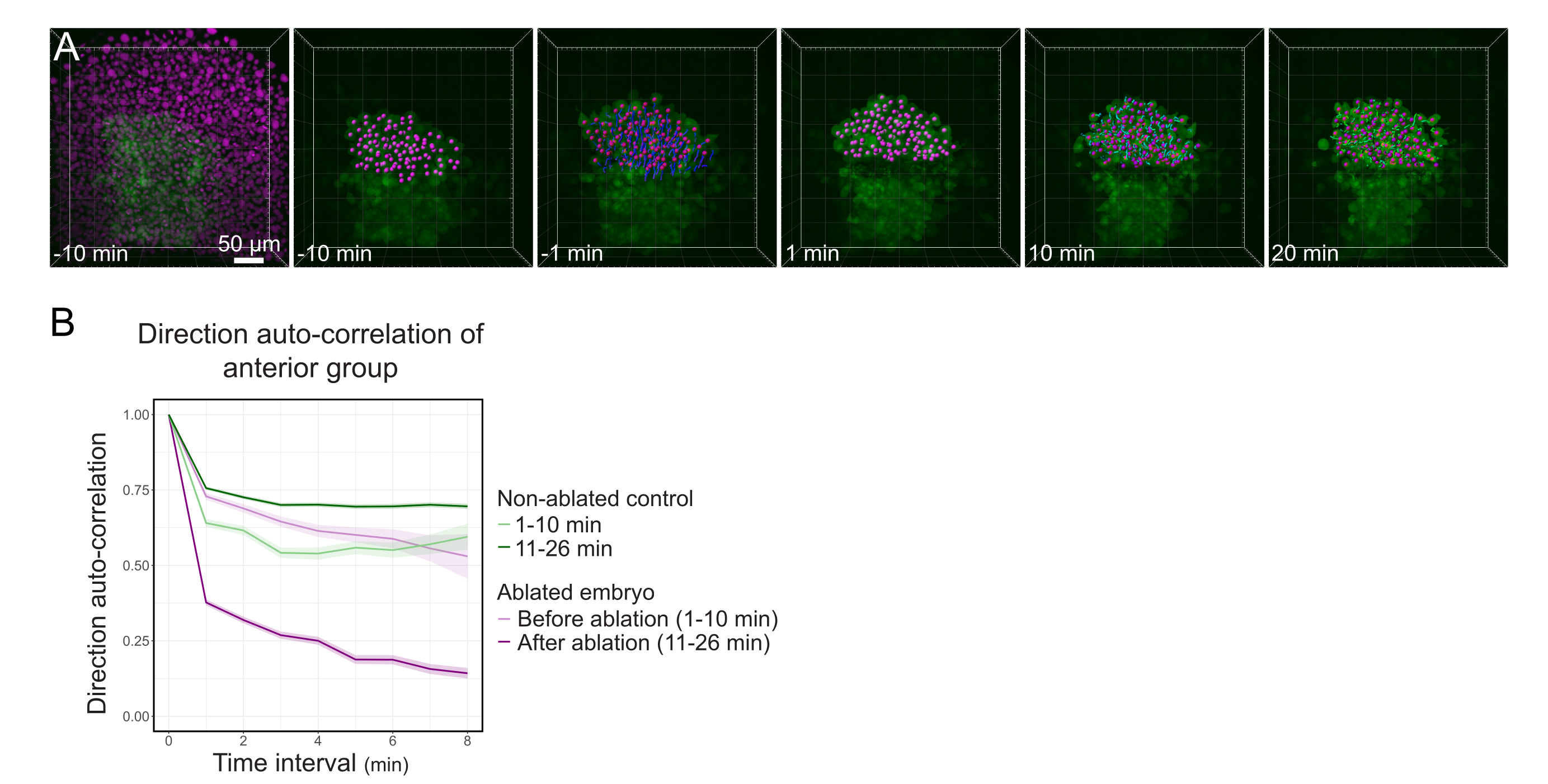
- Esportare i risultati come file .xlsx. Elaborare il file utilizzando routine di fogli di calcolo pubblicate24 (Figura 3B) e routine personalizzate su software di analisi dei dati (disponibile su richiesta).

Figura 3: L'isolamento della metà anteriore del polster influisce sulla direzionalità cellulare. (A) Ricostruzioni 3D di un embrione Tg(gsc:GFP) che esprime Histone2B-mCherry (visualizzato in magenta), prima e dopo un'ablazione laser che taglia il polster nel suo mezzo. I nuclei appartenenti alla metà anteriore del polster sono contrassegnati con un punto magenta e tracciati nel tempo prima e dopo l'ablazione (vedi Film S1). La barra della scala è 50 μm. (B) Come misura della persistenza della migrazione, l'auto-correlazione di direzione delle cellule appartenenti alla parte anteriore del polster prima e dopo l'ablazione. Le cellule mostrano un movimento continuo prima dell'ablazione, che diminuisce drasticamente dopo l'ablazione, indicando la perdita della migrazione orientata collettivamente. L'auto-correlazione della direzione è stata misurata anche sulle cellule che formano la metà anteriore del polster di un embrione non ablato, come controllo. Gli inviluppi del grafico indicano un errore standard. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Risultati
Per recidere il polster nel suo mezzo, un embrione Tg (gsc: GFP), iniettato con mRNA histone2B-mCherry è stato montato allo stadio epibolico al 70%, come descritto nel passaggio 4. Il polster è stato identificato dall'espressione GFP e l'embrione è stato montato in modo che il piano del polster sia perpendicolare all'asse ottico (Figura 1B). Inclinare l'embrione lontano da questa posizione complicherà la procedura. La luce dovrà passare attraverso più tessuti per raggiungere i...
Discussione
Qui, descriviamo un protocollo che utilizza ottiche non lineari per eseguire ablazioni di volume profonde e spazialmente ben definite. Il passo più critico del protocollo è trovare condizioni di trattamento che forniscano energia sufficiente per consentire ablazioni, ma non troppa energia, per evitare detriti o cavitazioni eccessivi. La quantità di energia erogata nel sito target dipende principalmente da: (1) la potenza di uscita del laser, (2) la qualità dell'allineamento laser, (3) la natura del tessuto attraverso...
Divulgazioni
Gli autori non dichiarano interessi concorrenti.
Riconoscimenti
Ringraziamo Emilie Menant per la cura dei pesci, la Polytechnique Bioimaging Facility, in particolare Pierre Mahou, per l'assistenza con l'imaging dal vivo sulle loro apparecchiature in parte supportate da Région Ile-de-France (interDIM) e Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging). Questo lavoro è stato sostenuto dalle sovvenzioni ANR 15-CE13-0016-1, 18-CE13-0024, 20-CE13-0016 e dal programma di ricerca e innovazione Horizon 2020 dell'Unione europea nell'ambito della convenzione di sovvenzione Marie Skłodowska-Curie n. 840201, del Ministère de l'Enseignement Supérieur et de la Recherche e del Centre National de la Recherche Scientifique.
Materiali
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
Riferimenti
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati