
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Metodi in vitro bottom-up per saggiare l'organizzazione ultrastrutturale, il rimodellamento della membrana e il comportamento di sensibilità alla curvatura delle settine
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Le settine sono proteine citoscheletriche. Interagiscono con le membrane lipidiche e possono percepire ma anche generare curvatura della membrana su scala micron. Descriviamo in questo protocollo metodologie bottom-up in vitro per l'analisi delle deformazioni della membrana, del legame della settina sensibile alla curvatura e dell'ultrastruttura del filamento di settino.
Abstract
Il rimodellamento della membrana avviene costantemente a livello della membrana plasmatica e all'interno degli organelli cellulari. Per analizzare completamente il ruolo dell'ambiente (condizioni ioniche, composizioni proteiche e lipidiche, curvatura della membrana) e dei diversi partner associati a specifici processi di rimodellamento della membrana, intraprendiamo approcci bottom-up in vitro . Negli ultimi anni, c'è stato un vivo interesse nel rivelare il ruolo delle proteine della settina associate alle principali malattie. Le settine sono proteine citoscheletriche essenziali e ubiquitarie che interagiscono con la membrana plasmatica. Sono implicati nella divisione cellulare, nella motilità cellulare, nella neuro-morfogenesi e nella spermiogenesi, tra le altre funzioni. È quindi importante capire come le settine interagiscono e si organizzano a livello delle membrane per indurre successivamente deformazioni di membrana e come possono essere sensibili a specifiche curvature di membrana. Questo articolo mira a decifrare l'interazione tra l'ultra-struttura delle settine a livello molecolare e il rimodellamento della membrana che avviene su scala micron. A tal fine, il lievito in erba e i complessi di septina dei mammiferi sono stati espressi e purificati in modo ricombinante. Una combinazione di saggi in vitro è stata quindi utilizzata per analizzare l'autoassemblaggio delle settine sulla membrana. Sono stati utilizzati doppi strati lipidici supportati (SLB), vescicole unilamellari giganti (GUV), grandi vescicole unilamellari (LUV) e substrati ondulati per studiare l'interazione tra auto-assemblaggio della settina, rimodellamento della membrana e curvatura della membrana.
Introduzione
Le settine sono proteine che formano filamenti citoscheletrici che interagiscono con le membrane lipidiche. Le settine sono onnipresenti negli eucarioti ed essenziali per numerose funzioni cellulari. Sono stati identificati come i principali regolatori della divisione cellulare nei lieviti in erba e nei mammiferi 1,2. Sono coinvolti negli eventi di rimodellamento della membrana, nella ciliogenesi3 e nella spermiogenesi4. All'interno delle cellule di mammifero, le settine possono anche interagire con actina e microtubuli 5,6,7 in un legante di Rho GTPasi (BORG)-dipendentemodo 8. In vari tessuti (neuroni9, ciglia3, spermatozoi10), le settine sono state identificate come regolatori delle barriere di diffusione per componenti legati alla membrana11. Le settine hanno anche dimostrato di regolare il blebbing della membrana e la formazione di protrusioni12. Le septine, essendo proteine multi-tasking, sono implicate nell'emergere di varie malattie prevalenti13. La loro errata regolazione è associata all'emergere di tumori14 e malattie neurodegenerative15.
A seconda dell'organismo, diverse subunità della settina (due in Caenorhabditis elegans a 13 nell'uomo) si assemblano per formare complessi la cui organizzazione varia in modo tessuto-dipendente16. Il blocco di base della settina raccoglie da due a quattro subunità, presenti in due copie e auto-assemblati in modo palindromico simile a un'asta. Nel lievito in erba, le settine sono ottameriche17,18. In situ, le settine sono spesso localizzate in siti con curvatura micrometrica; Si trovano nei siti di costrizione della divisione, alla base delle ciglia e dei dendriti e nell'anello degli spermatozoi19,20. Alla membrana, il ruolo delle settine sembra essere duplice: sono implicate nel rimodellamento del doppio strato lipidico e nel mantenimento dell'integrità della membrana21. Quindi, studiare le proprietà biofisiche delle proteine e/o delle subunità che formano filamenti di septina sulla membrana è cruciale per comprendere il loro ruolo. Per analizzare le proprietà specifiche delle settine in un ambiente ben controllato, sono appropriati approcci in vitro bottom-up. Finora, solo pochi gruppi hanno descritto le proprietà biofisiche delle settine in vitro20,22,23. Quindi, rispetto ad altri filamenti citoscheletrici, le attuali conoscenze sul comportamento delle settine in vitro rimangono limitate.
Questo protocollo descrive come l'organizzazione dei filamenti di settina, il rimodellamento della membrana e la sensibilità alla curvatura possono essere analizzati19. A tal fine, è stata utilizzata una combinazione di metodi di microscopia ottica ed elettronica (microscopia a fluorescenza, microscopia crioelettronica [cryo-EM] e microscopia elettronica a scansione [SEM]). Il rimodellamento della membrana delle vescicole unilamellari giganti (GUV) di dimensioni micrometriche viene visualizzato utilizzando la microscopia ottica a fluorescenza. L'analisi della disposizione e dell'ultrastruttura dei filamenti di settina legati alle vescicole lipidiche viene eseguita utilizzando la crio-EM. L'analisi della sensibilità alla curvatura della settina viene effettuata utilizzando SEM, studiando il comportamento dei filamenti di settina legati a doppi strati lipidici solidi, depositati su substrati ondulati di curvature variabili, che consente l'analisi della sensibilità alla curvatura sia per curvature positive che negative. Rispetto alla precedente analisi20,24, qui proponiamo di utilizzare una combinazione di metodi per analizzare a fondo come le settine possono auto-assemblarsi, deformare sinergicamente la membrana ed essere sensibili alla curvatura. Si ritiene che questo protocollo sia utile e adattabile a qualsiasi proteina filamentosa che mostri un'affinità per le membrane.
Protocollo
1. Determinazione del rimodellamento della membrana mediante vescicole unilamellari giganti (GUV)
NOTA: In questa sezione, i GUV sono generati per imitare le deformazioni della membrana eventualmente indotte dalle settine in un contesto cellulare. Infatti, nelle cellule, le settine si trovano frequentemente in siti con curvature micrometriche. I GUV hanno dimensioni che vanno da pochi a decine di micrometri e possono essere deformati. Sono quindi appropriati per dosare qualsiasi deformazione indotta dalla settina su scala micrometrica. I lipidi fluorescenti, così come le settine marcate con fluorescenza (utilizzando la proteina fluorescente verde [GFP]), vengono utilizzati per seguire il comportamento di lipidi e proteine tramite microscopia a fluorescenza.
- Preparazione di tamponi e soluzioni
- Preparare il tampone di crescita GUV (50 mM NaCl, 50 mM saccarosio e 10 mM Tris [pH = 7,8]) e il tampone di osservazione (75 mM NaCl e 10 mM Tris [pH = 7,8]).
- Misurare (utilizzando un osmometro commerciale) e regolare l'osmolarità dei tamponi di osservazione e di crescita (170 mOsmol· L-1, in teoria) aggiungendo piccole quantità di NaCl fino a quando le loro rispettive osmolarità sono uguali. Filtrare i buffer utilizzando un filtro da 0,2 μm. Aliquot il tampone di crescita e conservarlo a -20 °C per un ulteriore utilizzo. Conservare il buffer di osservazione a 4 °C.
NOTA: la differenza di osmolarità tra i due buffer non deve superare il 5%. - Preparare una soluzione di β-caseina da 5 mg·mL-1 nel tampone di osservazione. Assicurare la completa dissoluzione (dopo alcune ore a 4 °C con agitazione magnetica). Filtrare la soluzione con un filtro da 0,2 μm, aliquota e conservare a -20 °C.
- Preparare le miscele lipidiche ad una concentrazione lipidica totale di 3 mg·mL-1 in cloroformio. Utilizzare una composizione (mol%) di 56,8% Uovo L-α-fosfatidilcolina (EggPC), 15% colesterolo, 10% 1,2-dioleoil-sn-glicero-3-fosfoetanolammina (DOPE), 10% 1,2-dioleoil-sn-glicero-3-fosfo-L-serina (DOPS), 8% cervello L-α-fosfatidilinositolo-4,5-bisfosfato (PI(4,5)P 2) e 0,2% Bodipy-TR-Ceramide per migliorare le interazioni proteina-lipidi e favorire l'incorporazione di PI(4,5)P225.
NOTA: Maneggiare il cloroformio sotto una cappa aspirante utilizzando guanti in nitrile e occhiali di sicurezza. Pipettare le soluzioni di cloroformio con siringhe di vetro ed evitare la plastica poiché il cloroformio scioglie la plastica. Prima e dopo l'uso, sciacquare le siringhe pipettando il cloroformio 5x-10x. Utilizzare siringhe separate per pipettare lipidi fluorescenti specifici per prevenire la contaminazione incrociata. I lipidi possono essere conservati in cloroformio a -20 °C in una fiala di vetro ambrato ricoperta di Teflon. I flaconcini devono essere riempiti con argon prima della chiusura e sigillati con parafilm per evitare qualsiasi ossidazione lipidica. - Elettroformazione di GUV utilizzando una configurazione di fili di platino
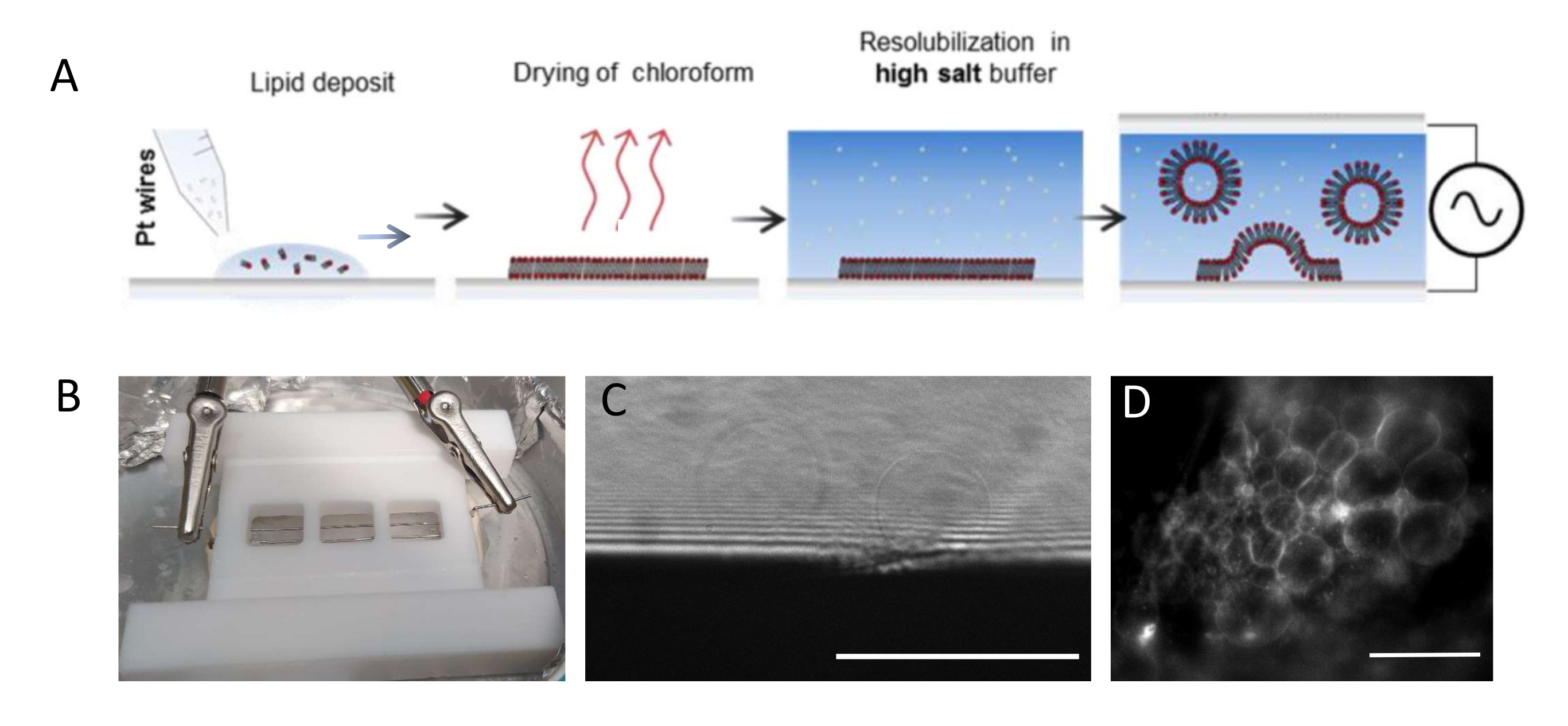
NOTA: La figura 1 presenta lo schema delle fasi sperimentali e un'immagine della camera.- Pulire accuratamente la camera e i fili di platino come segue per rimuovere eventuali residui lipidici.
- Immergere i fili e la camera nell'acetone e sonicare per 10 minuti. Pulirli accuratamente con un fazzoletto di carta usando acetone.
- Assemblare la camera inserendo i fili, immergere nuovamente nell'acetone e sonicare per 10 minuti. Pulire ancora una volta con acetone, assicurandosi che i fili siano completamente puliti. Immergere la camera in etanolo, sonicare per 10 minuti e pulire con etanolo.
- Infine, immergere la camera in acqua deionizzata, sonicare per 10 minuti e asciugare con un flusso di azoto o aria.
NOTA: La camera in teflon (Figura 1B) è stata realizzata su misura nell'officina interna. Ospita tre scomparti che possono essere sigillati su entrambi i lati utilizzando vetrine di copertura. I fili di platino possono essere inseriti nella camera attraverso fori di 1,3 mm di diametro. - Dopo aver pulito la camera, depositare 3-4 gocce per compartimento (ogni goccia è di circa 0,1 μL)) della miscela lipidica 3 mg·mL-1 su ciascun filo di platino. Ruotare i fili di 180° e depositare 3-4 goccioline lipidiche per compartimento sul lato opposto di ciascun filo di platino. Assicurarsi che le gocce non si contattino l'una con l'altra. Sono necessari circa 5 μL della miscela lipidica per camera intera.
- Posizionare la camera di crescita in una camera a vuoto per 30 minuti per rimuovere qualsiasi traccia di cloroformio.
NOTA: il vuoto profondo (0,1 mbar) è il migliore. Una volta essiccati, i lipidi sono vulnerabili all'ossidazione e, pertanto, non dovrebbero essere lasciati all'aria per più di pochi minuti. - Depositare il grasso ad alto vuoto sul fondo della camera (il lato più vicino ai fili) lungo la periferia dei tre scomparti utilizzando una siringa e premere un coprislip pulito (22 mm x 40 mm) contro il grasso per garantire una perfetta tenuta. Sigillare entrambe le estremità della camera (cioè nei siti di entrata/uscita dei fili) utilizzando pasta sigillante (piastre di cera). Allo stesso modo, applicare grasso sottovuoto sull'altro lato della camera.
- Riempire gli scomparti con tampone di crescita (~1 mL per camera) usando una pipetta. Non mescolare la soluzione troppo rapidamente o fortemente per evitare qualsiasi distacco del film lipidico dai fili. Sigillare ermeticamente la parte superiore della camera utilizzando un coprivetro da 22 mm x 40 mm premendolo contro il grasso. Per evitare la formazione di bolle d'aria, premere delicatamente il coperchio di vetro dal centro verso i bordi.
- Collocare la camera in un frigorifero a 4 °C e collegare i fili a un generatore di funzioni d'onda (funzione sinusoidale a 500 Hz). Impostare una tensione effettiva di 350 mV per un periodo di crescita più breve (cioè 6 h) o 250 mV per un periodo di crescita più lungo (cioè 12-16 h), come già presentato e ottimizzato in uno studio di Beber et al.25.
- Rimuovere i coperchi, rimuovere il sigillante e il grasso e rimuovere i fili. Lavare e strofinare la camera con un fazzoletto di carta usando alternativamente acqua ed etanolo (≥70%).
NOTA: La tensione e la scala temporale ottimali per la crescita del GUV dipendono da molti parametri, dalla concentrazione di sale tampone alla geometria della camera (cioè la distanza tra i fili e le dimensioni della camera). Utilizzare la stessa camera ogni volta che l'esperimento viene ripetuto per garantire la riproducibilità. I fili sono in prossimità del fondo della camera in modo che i lipidi possano essere ripresi utilizzando la microscopia fluorescente. Immagine dei lipidi in ogni fase (vedi Figura 1C,D) per assicurarsi che il processo di elettroformazione abbia successo.
- Incubazione di septini con le vescicole
- Raccogliere i GUV dai fili utilizzando punte per pipette pretagliate (apertura ~1 mm) che vengono portate in prossimità dei fili. Quindi, pipettare la soluzione lungo tutto il filo. Questa procedura impedisce la generazione di forti flussi lamellari che potrebbero interrompere i GUV. Dopo questa fase, non è più necessario tagliare le punte delle pipette. Infatti, i flussi lamellari non danneggiano i GUV nella soluzione.
NOTA: A causa della presenza di PI(4,5)P 2 nella miscela lipidica, i GUV che sono stati raccolti devono essere conservati per non più di2-3 ore prima dell'esperimento. Infatti, PI(4,5)P2 si solubilizza rapidamente e le settine non si legano più alle membrane poche ore dopo la loro formazione. Tuttavia, una volta che le settine sono legate alla membrana, rimangono legate per alcuni giorni. - Diluire la soluzione madre di settine in Tris 10 mM (pH 8) esclusivamente per raggiungere un'osmolarità pari a quella del tampone di crescita; Se necessario, diluire ulteriormente la soluzione di septina nel tampone di osservazione. Aggiungere il volume previsto di GUV raccolti (50-100 μL per un volume totale di 200 μL). Eseguire l'incubazione direttamente nella camera di osservazione dopo la passivazione con β-caseina (vedi sotto). Sono necessari 20-30 minuti di attesa per raggiungere l'equilibrio.
NOTA: L'espressione e la purificazione dei complessi ottamerici della settina (lieviti umani o in erba) sono ampiamente descritti in altri articoli17. In breve, le settine sono state espresse in Escherichia coli, purificate in laboratorio utilizzando fasi di cromatografia per affinità, esclusione dimensionale e scambio ionico e conservate a -80 °C in una soluzione acquosa di 50 mM Tris-HCl (pH 8), 300 mM KCl e 5 mM MgCl2 a ~1 mg·mL-1 (3 μM) di concentrazione. Un'alta concentrazione di sale viene utilizzata per evitare l'aggregazione della settina. I complessi di septina non devono essere concentrati attraverso un dispositivo di centrifugazione del filtro, che induce l'aggregazione e quindi riduce la resa proteica.
- Raccogliere i GUV dai fili utilizzando punte per pipette pretagliate (apertura ~1 mm) che vengono portate in prossimità dei fili. Quindi, pipettare la soluzione lungo tutto il filo. Questa procedura impedisce la generazione di forti flussi lamellari che potrebbero interrompere i GUV. Dopo questa fase, non è più necessario tagliare le punte delle pipette. Infatti, i flussi lamellari non danneggiano i GUV nella soluzione.
- Imaging con microscopio a disco confocale e/o rotante
- Per evitare che i GUV si attacchino alla superficie e/o esplodano, passivare la camera di osservazione incubandola con una soluzione di β-caseina da 5 mg·mL-1 per 30 minuti.
- Rimuovere la soluzione di β-caseina e trasferire la soluzione di septina-GUV (punto 1.4.2.) nella camera di osservazione utilizzando una pipetta. Lasciare sedimentare i GUV sul fondo della camera per 10-15 minuti.
NOTA: La discrepanza di composizione tra l'interno del GUV e il buffer esterno crea sia una mancata corrispondenza della densità che dell'indice di rifrazione. A causa della mancata corrispondenza dell'indice di rifrazione, i GUV sono visibili con la microscopia ottica a luce di trasmissione. - Utilizzando la microscopia confocale, visualizzare il segnale fluorescente dei lipidi per verificare la qualità dei GUV e lo stato di lamellarità della membrana. Valutare la densità delle settine legate ai GUV registrando il segnale fluorescente delle settine dopo aver eseguito una corretta calibrazione25. Eseguire acquisizioni Z-stack a intervalli spaziali di 0,4 μm per analizzare e visualizzare le deformazioni 3D delle vescicole indotte dall'interazione tra le settine e la membrana.
NOTA: sono stati utilizzati obiettivi ad immersione in olio con ingrandimenti di 60 o 100 volte. Sono stati utilizzati microscopi a disco confocale o rotante standard (Table of Materials) con dimensioni dei pixel rispettivamente di 250 nm e 110 nm. È necessario adattare le condizioni di imaging a un determinato apparecchio. Nessun agente sbiancante anti-foto specifico è stato aggiunto alla soluzione.

Figura 1: Elettroformazione dei GUV . (A) Rappresentazione schematica del processo di elettroformazione mediante fili di platino. (B) Immagine del dispositivo fatto in casa in teflon assemblato con fili di platino utilizzati per generare GUV mediante elettroformazione. I fili hanno un diametro di 0,5 mm e una distanza di 3 mm. (C) GUV (oggetti sferici) osservati al microscopio ottico a trasmissione durante il processo di crescita. La zona opaca nella parte inferiore dell'immagine è il filo di platino. (D) GUV (oggetti fluorescenti rotondi) osservati al microscopio fluorescente durante la crescita sul filo di platino. Barre della scala = 100 μm. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
2. Analisi dell'organizzazione ultrastrutturale dei filamenti di septina mediante crio-microscopia elettronica
NOTA: le vescicole non sono adatte per l'imaging con metodi di microscopia elettronica standard. Infatti, i campioni vengono essiccati utilizzando metodi standard di colorazione negativa. Dopo la disidratazione, è probabile che le vescicole subiscano deformazioni aspecifiche, spesso con conseguenti sporgenze lipidiche. La microscopia crioelettronica è quindi una strategia molto migliore per osservare specifiche deformazioni delle vescicole. Utilizzando la crio-EM, i campioni sono incorporati all'interno di un sottile strato (~ 100-200 nm) di ghiaccio vetrificato, che conserva i campioni vicino allo stato nativo. I GUV sono, tuttavia, troppo grandi (diverse decine di micrometri) per essere incorporati nel ghiaccio sottile e quindi ripresi dalla microscopia elettronica a trasmissione. Quindi, vengono generate grandi vescicole unilamellari (LUV), i cui diametri vanno da ~ 50-500 nm, per determinare come le settine possono deformare le vescicole e come si dispongono sulle vescicole.
- Generazione di grandi vescicole unilamellari (LUV)
- Preparare 50 μg di miscela lipidica solubilizzata in cloroformio con una composizione molare di 57% EggPC, 15% colesterolo, 10% DOPE, 10% DOPS e 8% cervello PI(4,5)P2), che è ottimizzato per migliorare l'interazione della settina con la membrana in una fiala di vetro.
- Asciugare la soluzione sotto il flusso di argon per generare un film lipidico essiccato nel flaconcino. Mettere il flaconcino sotto vuoto per 30 minuti per asciugare completamente il lipide.
- Risolubilizzare il film lipidico in 50 μL di soluzione acquosa (50 mM Tris-HCl [pH 8]; 50 mM KCl; 2 mM MgCl2) per ottenere una concentrazione finale di 1 mg·mL-1, vortice per 10 s, e trasferire la soluzione in un tubo.
NOTA: i LUV devono essere utilizzati immediatamente per l'incubazione con septine. In caso contrario, l'interazione proteina-lipide sarà debole a causa della solubilizzazione di PI(4,5)P2. Questo processo di risolubilizzazione grezzo genera una popolazione eterogenea di vescicole con diametri che vanno da 50 nm a 500 nm. Quindi, un'intera gamma di diametri e quindi curvature vengono analizzati simultaneamente.
- Incubare le settine con i lipidi solubilizzati a concentrazioni finali di lipidi e settina di 0,1 mg·mL-1 (circa 300 nM) e 20 nM, rispettivamente, in un tampone ad alto contenuto di sale (50 mM Tris-HCl [pH 8], 300 mM KCl, 2 mM MgCl2). Incubare il campione per 1 ora a temperatura ambiente.
- Congelamento a tuffo per vetrificare il campione
- Griglie di carbonio holey a scarica di bagliore (300 mesh) sul lato carbonio per 30 s a 5 mA utilizzando apparecchiature di generazione al plasma e inserire la griglia all'interno di una macchina di congelamento a tuffo in un ambiente umido.
- Adsorbire 4 μL del campione (Fase 2.2.) sul lato del carbonio scaricato a bagliore della griglia. Immediatamente prima dell'adsorbimento del campione, aggiungere perle d'oro a 5-10 nm nella soluzione, nel caso in cui i campioni vengano utilizzati per generare serie inclinate mediante criotomografia.
NOTA: La densità delle perle d'oro deve essere vagliata e regolata empiricamente e dipende dal fornitore. La densità ottimizzata è di 10-15 perle d'oro nel campo visivo. - Asciugare i campioni dal lato nudo per aspirare la goccia del campione sul lato opposto.
NOTA: Il tempo di aspirazione è in genere di 4 s e la posizione della carta da filtro e la forza di estrazione vengono regolate empiricamente testando e vagliando posizioni alternative della carta da filtro per ottimizzare la qualità del ghiaccio (spessore) e la densità del materiale. - Trasferire le griglie in un microscopio o conservarle in un contenitore di azoto liquido.
NOTA: L'uso di griglie di carbonio holey è essenziale per accogliere la polidispersità nelle dimensioni delle vescicole. La macchiatura del campione dal lato opposto favorisce un maggiore adsorbimento del materiale biologico sulla griglia.
- Imaging al microscopio crioelettronico
- Inserire le griglie in un microscopio elettronico (EM) attrezzato per l'osservazione crio-EM. Scherma l'intera griglia generando una mappa dell'intero campione a basso ingrandimento (tipicamente a 120x ingrandimento) per selezionare le aree che mostrano ghiaccio migliore (cioè sottile e ben vetrificato).
- Per la raccolta dati 2D cryo-EM, raccogliere immagini con una dimensione in pixel di circa 2 Å per pixel per verificare la qualità del campione. Assicurarsi che sia i filamenti di settina che il doppio strato lipidico siano visibili.
- Per la raccolta dei dati della tomografia crioelettronica, selezionare le aree di interesse che mostrano vescicole deformate. Assicurarsi che siano presenti abbastanza perle d'oro (almeno 10) nel campo visivo.
- In base al software utilizzato per la raccolta dei dati in serie inclinata, selezionare le posizioni di messa a fuoco e di tracciamento in modo che siano abbastanza lontane dall'area di interesse. Raccogli le serie inclinate variando l'angolo di inclinazione da -60° a +60°, raccogliendo un'immagine ogni 2°-3° gradi.
NOTA: La dose totale deve essere di circa 100 elettroni/Å2. La dimensione dei pixel varia da 1,3 Å a 2,1 Å, a seconda del microscopio utilizzato per la raccolta dei dati. Idealmente, uno schema simmetrico angolare per la raccolta dei dati è preferito come segue: 0°, -3°, +3°, -6°, +6°, -9°, +9° [...] -60°, +60°. Tuttavia, i goniometri dei microscopi ad ingresso laterale non forniscono sufficiente stabilità meccanica per ottenere tali schemi simmetrici. In alternativa, l'acquisizione può essere avviata da 0° a 34°, seguita da una seconda sequenza angolare da -2° a -60°, e terminata da una sequenza finale da 36° a 60°. Lo scopo è quello di raccogliere le prime immagini (con i danni da radiazioni più bassi) agli angoli più bassi. Inoltre, per visualizzare l'ultrastruttura delle vescicole e dei filamenti di settina legati, può essere utilizzato un microscopio crio-EM standard (filamento di esaboruro di lantanio (LaB6) e ingresso laterale del campione). Tuttavia, se si mira a perseguire un'ulteriore elaborazione delle immagini (media sub-tomogramma, per esempio), è meglio utilizzare microscopi a emissione di campo (FEG) di ultima generazione dotati di rivelatori diretti. In questo protocollo, limitiamo la nostra descrizione all'acquisizione di ricostruzioni 3D e tralasciamo la media dei sottotomogrammi.
- Ricostruzione 3D da criotomografia e segmentazione
- Utilizza la suite software IMOD (Table of Materials) per l'allineamento delle immagini in serie inclinate e la ricostruzione 3D26,27. All'interno di IMOD, eseguire l'allineamento della serie di inclinazione in base al posizionamento dei fiduciali (perle d'oro). Inoltre, se necessario, eseguire la determinazione e la correzione della funzione di trasferimento del contrasto (CTF) all'interno dell'IMOD26. Infine, ottieni la ricostruzione 3D con IMOD, seguendo rigorosamente ogni passaggio.
- Segmentare manualmente i doppi strati lipidici e i filamenti di settina utilizzando 3Dmod27 dalla suite software IMOD, per la visualizzazione.
3. Analisi della sensibilità alla curvatura della settina mediante SEM
NOTA: Per capire come le settine possono essere sensibili alle curvature micrometriche, è stato utilizzato un approccio in vitro per incubare complessi di filamenti di settina con doppi strati lipidici supportati da solidi depositati su modelli ondulati ondulati su scala micrometrica.
- Progettazione di una replica ondulata di NOA (adesivo ottico Norland) da modelli ondulati di polidimetilsilossano (PDMS)
- Utilizzare modelli ondulati PDMS di ampiezza di 250 nm e periodicità laterale di 2 μm o altre dimensioni per saggiare le curvature adatte alla proteina di interesse.
NOTA: I modelli ondulati PDMS sono progettati e generati come descritto in Nania et al.28,29. - In un ambiente di camera bianca, depositare 5 μL di NOA liquido su un coprivetrino circolare di 1 cm di diametro e posizionare la dima PDMS sulla goccia. Trattare con luce UV (320 nm) per 5 minuti per fotopolimerizzare il NOA liquido in un sottile film polimerico. Quindi, staccare delicatamente il modello PDMS dal vetrino con il NOA appena polimerizzato.
NOTA: NOA è una colla otticamente trasparente comune adatta per l'imaging al microscopio ottico. Inoltre, NOA è resistente ai processi di fissazione chimica e colorazione effettuati prima dell'imaging SEM. Entrambe le resine NOA 71 e NOA 81 possono essere utilizzate con risultati simili. Il modello PDMS iniziale potrebbe essere utilizzato più volte per produrre repliche NOA. Le repliche NOA ottenute possono essere conservate in una scatola a temperatura ambiente per mesi.
- Utilizzare modelli ondulati PDMS di ampiezza di 250 nm e periodicità laterale di 2 μm o altre dimensioni per saggiare le curvature adatte alla proteina di interesse.
- Generazione di un doppio strato lipidico supportato e incubazione proteica
- Trattare i film NOA utilizzando un detergente al plasma ad aria per 5 minuti per rendere la superficie idrofila.
- Preparare una soluzione di piccole vescicole unilamellari (SUV) con una composizione molare del 57% di EggPC, del 15% di colesterolo, del 10% di DOPE, del 10% di DOPS e dell'8% di PI(4,5)P 2 cerebrale ad una concentrazione lipidica totale di 1 mg·mL-1. Preparare i SUV risospendendo un film lipidico essiccato nel tampone di osservazione, come descritto al punto 2.1.3. Sonicare delicatamente la soluzione usando un sonicatore da bagno per 5-10 minuti fino a quando la soluzione è trasparente.
NOTA: La soluzione dei SUV può essere conservata congelata per diverse settimane a -20 °C. - Inserire i vetrini che supportano i modelli NOA all'interno dei pozzetti delle scatole di coltura cellulare. Depositare 100 μL di soluzione SUV da 1 mg·mL-1 sui modelli NOA appena scaricati (fase 3.2.1.) e incubare per 30 minuti a temperatura ambiente. Questo passaggio induce la fusione dei SUV con la superficie del modello NOA per generare un doppio strato lipidico supportato.
- Risciacquare accuratamente i vetrini 6 volte con il tampone septina (50 mM Tris-HCl [pH 8], 50 mM KCl, 2 mM MgCl2) per rimuovere il SUV non fuso. Dopo ogni risciacquo, non lasciare mai asciugare completamente il campione.
- Diluire la soluzione madre di septina ottamerica in tampone ad alto contenuto di sale della settina (50 mM Tris-HCl [pH 8], 300 mM KCl, 2 mM MgCl 2) utilizzando tampone di settina (50 mM Tris-HCl [pH 8], 50 mM KCl, 2 mM MgCl2) a concentrazioni finali comprese tra 10 nM e 100 nM e volumi di 1 mL. Incubare la soluzione proteica sui vetrini per ~ 1 h a temperatura ambiente.
NOTA: il volume è abbastanza grande in modo che l'evaporazione non sia un problema.
- Preparazione del campione per l'analisi SEM
NOTA: Sono stati sviluppati vari protocolli al microscopio elettronico per analizzare le strutture proteiche e il loro assemblaggio. L'attuale protocollo preserva l'organizzazione dei filamenti di settino, utilizzando un protocollo di fissazione derivato da Svitkina et al.30 che è facile da implementare. Inoltre, questo protocollo ottimizza le osservazioni SEM ad alta risoluzione.- Preparazione di reagenti e soluzioni madre
NOTA: Questo protocollo richiede diversi reagenti e soluzioni che possono essere preparati in anticipo o appena prima dell'incubazione. Seguire le istruzioni fornite per evitare artefatti o mancanza di reattività chimica.- Cacodilato di sodio 0,2 M soluzione madre: Per preparare questa soluzione a doppia resistenza, sciogliere 2,14 g di polvere di cacodilato di sodio in ~ 40 ml di acqua distillata sotto agitazione magnetica. Dopo completa dissoluzione, regolare il pH a 7,4 aggiungendo delicatamente 0,1 M HCl (~1 mL per 50 mL di soluzione) e portare il volume finale con acqua distillata. Questa soluzione può essere conservata per 24-48 h a 4°C.
- 2% glutaraldeide (GA) in cacodilato di sodio 0,1 M (soluzione fissativa): preparare questa soluzione subito prima dell'uso diluendo GA di grado EM altamente puro con una soluzione di cacodilato di sodio 0,2 M (vedere sopra). Utilizzare una soluzione stock GA commerciale al 25%-50% grazie alla sua capacità di stoccaggio ottimizzata a 4 °C. Per preparare 10 ml di soluzione fissativa, diluire 0,8 mL di soluzione GA commerciale al 25% con 4,2 mL di acqua distillata e 5 mL di cacodilato di sodio 0,2 M.
- Tetrossido di osmio all'1% (OsO 4) in cacodrilato di sodio 0,1 M: preparare questa seconda soluzione fissativa subito prima dell'uso diluendo la soluzione madre commerciale di OsO4 al4% con cacodilato di sodio 0,2 M (vedi sopra). Per 4 mL di soluzione fissativa OsO 4, diluire 1 mL di soluzione commerciale di OsO 4 al4% con 1 mL di acqua distillata e 2 mL di cacodilato di sodio 0,2 M.
NOTA: Utilizzare la soluzione stock commerciale al 4% di OsO4 nei bulbi di vetro pieghevoli a causa delle loro proprietà di conservazione e manipolazione. Si noti che OsO4 è altamente reattivo. Assicurati che il suo colore sia leggermente giallo e non scuro. - 1% di acido tannico (TA) in acqua: preparare questa soluzione subito prima dell'uso. Preparare la soluzione di TA per ottenere una concentrazione finale dell'1% di TA in acqua distillata a temperatura ambiente. Sciogliere 10 mg in 1 mL di acqua distillata e vortice per alcuni minuti. La soluzione di TA non può essere conservata e deve essere filtrata con un filtro da 0,2 μm prima dell'uso.
- Soluzione di acetato di uranile (UA) all'1% in acqua Preparare la soluzione di UA per ottenere una concentrazione finale dell'1% di UA in acqua distillata. Sciogliere 10 mg in 1 mL di acqua distillata e vortice per almeno 30 minuti a 1 ora mediante vortice o agitazione a temperatura ambiente. Questa soluzione può essere conservata per 1 mese a 4 °C ma può facilmente precipitare e deve quindi essere filtrata con un filtro da 0,2 μm prima dell'uso.
- Serie graduata di soluzioni di etanolo Preparare soluzioni di etanolo al 50%, 70%, 95% e 100% in acqua. Preparare il bagno finale da bottiglie appena aperte di etanolo al 100% o da etanolo al 100% disidratato per almeno 24 ore con setacci molecolari (diametro nominale dei pori = 4 Å) per rimuovere dai campioni tutte le tracce di acqua che potrebbero interferire con l'essiccazione e il rivestimento. Fare attenzione a non agitare questa soluzione in quanto potrebbe causare la risospensione delle particelle di silicato.
NOTA: Prestare attenzione quando si maneggiano i reagenti chimici utilizzati in questo protocollo. La glutaraldeide, il tetrossido di osmio, il cacodilato di sodio e l'acetato di uranile sono tutti altamente tossici, essendo radioattivo anche l'acetato di uranile. Tutte le manipolazioni dei reagenti e dei loro rifiuti devono essere effettuate utilizzando protezioni individuali (guanti, camici da laboratorio, occhiali di sicurezza) e collettive (cappe aspiranti e schermi in plexiglass), secondo le procedure specifiche del laboratorio.
- Fissazione del campione
- Lavare i campioni (cioè vetrini NOA con doppi strati lipidici supportati fusi e proteine incubate) con PBS. Sostituire il PBS con la soluzione fissativa GA preriscaldata a 37 °C e lasciare procedere la reazione per 15 minuti. I campioni possono quindi essere conservati a 4 °C.
- Rimuovere la soluzione fissativa e lavare i campioni fissi 3 volte con cacodilato di sodio 0,1 M (5 minuti per lavaggio) agitando delicatamente.
- Incubare i campioni nella soluzione fissativa OsO4 per 10 minuti con la massima protezione dalla luce per consentire il fissaggio delle strutture membranose e aumentare la conducibilità elettrica dei campioni. Rimuovere la soluzione fissativa e lavare i campioni 3 volte in acqua distillata (5 minuti per lavaggio) agitando delicatamente.
- Incubare i campioni lavati nella soluzione fissativa TA filtrata per un massimo di 10 minuti. Rimuovere la soluzione di TA e lavare i campioni 3 volte in acqua distillata (5 minuti per lavaggio) agitando delicatamente.
- Incubare i campioni lavati in una soluzione fissativa UA appena filtrata per 10 minuti con la massima protezione dalla luce. Rimuovere la soluzione UA e lavare i campioni 3 volte in acqua distillata (5 minuti per lavaggio) agitando delicatamente.
- Disidratazione del campione e essiccazione dei punti critici
NOTA: l'essiccazione all'aria non è consentita per evitare di danneggiare i campioni attraverso tensioni superficiali indesiderate quando si passa dallo stato liquido a quello gassoso. In EM sono stati stabiliti due metodi: il metodo fisico per raggiungere lo stato supercritico di CO2 e bypassare il suo punto critico (31 °C, 74 bar), o il metodo chimico di evaporazione dell'esametildisilazano (HMDS), un agente essiccante con ridotta tensione superficiale. CO2 e HMDS sono scarsamente miscibili con l'acqua. Di conseguenza, tutte le tracce di acqua devono essere sostituite con un solvente di transizione (etanolo) per evitare danni successivi durante i processi di essiccazione.- Incubare i campioni per 2-3 minuti in ciascuna soluzione di etanolo, a partire dal 50% al 100% (anidro) bagno di etanolo.
NOTA: Poiché l'evaporazione dell'etanolo tra i bagni è rapida, anche la manipolazione del campione deve essere rapida per evitare qualsiasi essiccazione all'aria. - Trasferire i vetrini all'interno dell'essiccatore a punti critici preriempito con etanolo e seguire le istruzioni del produttore.
NOTA: In questo protocollo è stato utilizzato un apparecchio automatico, ma è possibile utilizzare qualsiasi sistema. I protocolli possono differire per ogni apparato, ma devono essere ottimizzati per rimuovere completamente l'etanolo (25 bagni in questo protocollo) e ridurre le velocità per i diversi scambi di solventi (ingressi di CO 2 e uscite di etanolo / CO2) e la depressurizzazione finale (quasi 1 ora in questo protocollo). - Al termine dell'essiccazione del punto critico, conservare immediatamente i campioni in un essiccatore fino al montaggio e al rivestimento. Poiché i campioni essiccati sono altamente igroscopici, rivestirli (vedi sotto) il prima possibile.
NOTA: In alternativa, HMDS può offrire un metodo più economico e veloce. Sebbene l'HDMS non sia mai stato testato sui nostri campioni, questo approccio ha prodotto buoni risultati per l'osservazione delle proteine sulla faccia interna delle membrane cellulari31,32.
- Incubare i campioni per 2-3 minuti in ciascuna soluzione di etanolo, a partire dal 50% al 100% (anidro) bagno di etanolo.
- Montaggio e rivestimento del campione.
NOTA: Poiché i campioni biologici presentano scarse proprietà di conducibilità elettrica, devono essere rivestiti con un film metallico conduttivo prima dell'osservazione SEM. Viene quindi utilizzato lo sputtering plasma-magnetron.- Allegare la copertina agli stub che verranno utilizzati per future osservazioni SEM. Utilizzare la vernice argentata a causa della sua migliore conduttività elettrica, rispetto ai dischi di carbonio. Aggiungi una striscia di vernice argentata sulla faccia superiore del coprislip e assicurati che il collegamento con lo stub sia soddisfacente. Evitare qualsiasi agglomerato di vernice.
NOTA: La striscia di vernice argentata deve essere sottile per evitare qualsiasi contatto tra il campione e la lente dell'obiettivo del SEM quando si lavora ad alta risoluzione (cioè a bassa distanza di lavoro). - Attendere che il solvente evapori completamente.
NOTA: La durata di questo passaggio può variare a seconda della quantità e dello spessore del deposito di vernice argentata. Questo passaggio può essere abbreviato utilizzando una campana di vetro o il rivestimento e un aspirapolvere primario per 10-30 minuti. - Utilizzare un apparecchio dotato di una testa di sputtering al plasma-magnetron e di uno stadio planetario rotante e seguire i protocolli standard forniti dal produttore. Qui, l'apparecchio è stato evacuato a 2,5 x 10-5 mbar, spurgato 1x con argon di alta qualità e quindi regolato a 8,0 x 10-3 mbar.
- Eseguire il pre-sputtering (120 mA per 60 s) per rimuovere lo strato di ossido sulla superficie. Quindi, depositare 1,5 nm di tungsteno (90 mA, distanza di lavoro = 50 mm) con l'aiuto di un monitor dello spessore del film.
NOTA: Il rivestimento deve essere perfettamente pulito per garantire la riproducibilità dell'evaporazione del film. Il rivestimento deve essere fermato una volta raggiunto lo spessore target. Lo spessore finale del film viene calcolato e corretto successivamente. Le pellicole di circa 1,5 nm hanno, in media, una post-correzione di 0,7 nm utilizzando il nostro apparato. Poiché tali valori di correzione sono vicini al target, viene eseguita una serie di rivestimenti per valutare e quindi sottrarre questa correzione dal valore target. - Conservare i campioni sotto vuoto per proteggerli dall'aria ambiente fino a tutte le analisi SEM.
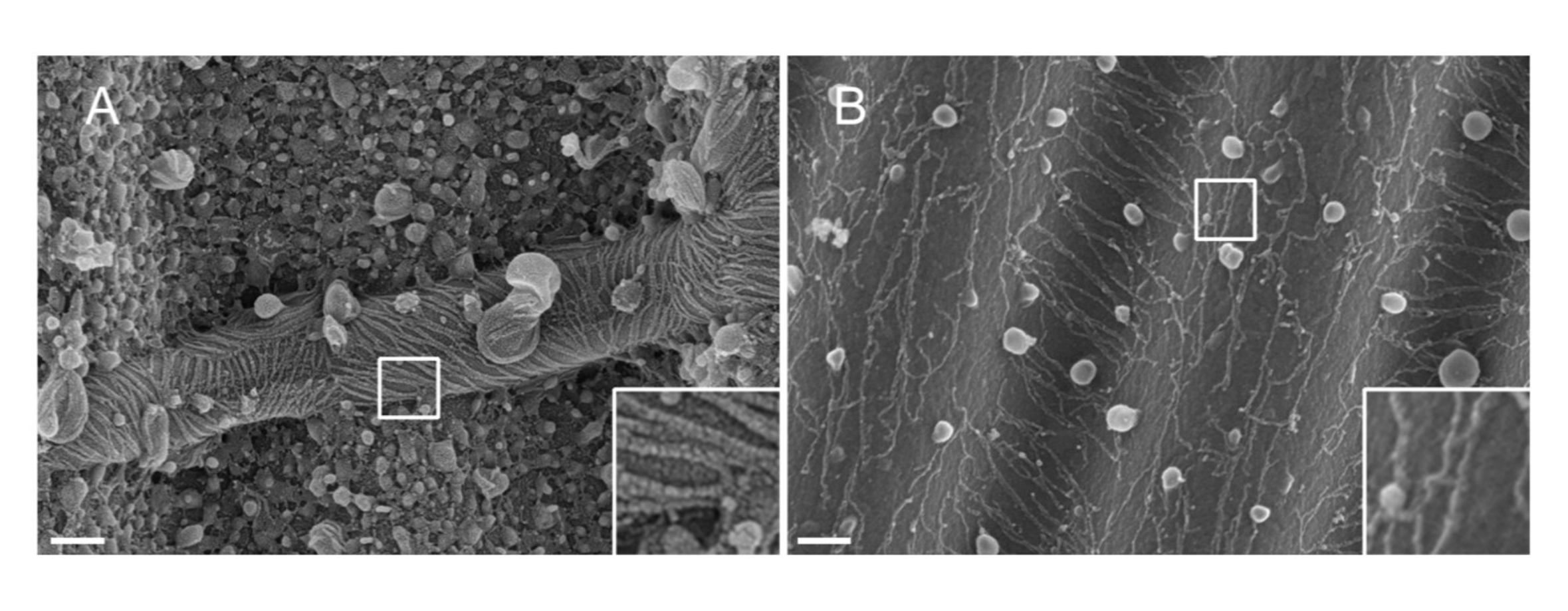
NOTA: La natura del metallo utilizzato per lo sputtering è importante. Il Pt, nonostante sia un materiale comune, si traduce in un rivestimento di scarsa qualità con uno spessore del film Pt adattato ai filamenti di settina (1,5 nm). Ad alta risoluzione, un film Pt da 1,5 nm manca di coesione; le dimensioni dei cluster di Pt e dei filamenti di settina diventano quindi simili, portando a interpretazioni errate durante il processo di segmentazione del filamento33 (vedi figura 2A, riquadro). Il tungsteno è una buona alternativa al Pt perché mostra una granulometria più piccola, appena visibile a SEM ad alta risoluzione (vedi Figura 2B, riquadro). Tuttavia, il tungsteno puro è facilmente ossidabile, portando a forti artefatti dell'effetto di carica durante l'osservazione SEM se la procedura descritta nel passaggio 3.3.4. non è strettamente seguito.
- Allegare la copertina agli stub che verranno utilizzati per future osservazioni SEM. Utilizzare la vernice argentata a causa della sua migliore conduttività elettrica, rispetto ai dischi di carbonio. Aggiungi una striscia di vernice argentata sulla faccia superiore del coprislip e assicurati che il collegamento con lo stub sia soddisfacente. Evitare qualsiasi agglomerato di vernice.
- Preparazione di reagenti e soluzioni madre
- Acquisire le immagini utilizzando un microscopio SESEM (Field-Emission SESEM).
NOTA: le tecnologie SEM sono state recentemente aggiornate per migliorare la risoluzione e le tecnologie dipendono dal produttore (ad esempio, ottica elettronica, decelerazione del fascio, lente magnetica). Questo guadagno di risoluzione (accessibile con diversi produttori), specialmente a bassa tensione di accelerazione (vicino al nanometro a 1 kV), è necessario per risolvere strutture nanometriche simili alle reti di settini.- Ottenere immagini ad alta risoluzione attraverso il rilevamento di elettroni secondari primari (SE1) con il rilevatore "in-lens" utilizzando le seguenti impostazioni.
- Impostare la tensione di accelerazione su 3 kV. Fissare la corrente del fascio con l'apertura di 20 μm (per le colonne Zeiss Gemini I, o equivalente a 34 pA) o con l'apertura di 15 μm (per le colonne Zeiss Gemini I, o equivalente a 18,5 pA), se è necessaria la soppressione degli effetti di carica.
- Per le osservazioni, utilizzare risoluzioni che vanno da 21,25 nm / pixel a 1,224 nm / pixel e per l'analisi dei dati, utilizzare una risoluzione di ~ 5,58 nm / pixel (ingrandimento 20.000x secondo il riferimento Polaroid 545).
- Impostare la distanza di lavoro tra 1 mm e 2 mm per l'osservazione ad alta risoluzione e circa 3 mm se è richiesta una maggiore profondità di campo. Regolare continuamente la velocità di scansione e l'integrazione della linea per garantire un rapporto segnale-rumore costante con un tempo di acquisizione di circa 30-45 s per immagine.

Figura 2: Effetto del materiale depositato sui filamenti di settina su modelli PDMS ondulati. SEM di filamenti di settina rivestiti da sputtering con (A) 1,5 nm di platino, che mostrano il modello "terreno arido fessurato" tipico di una mancanza di coesione tra i gruppi di nuclei di platino, o (B) 1,5 nm di tungsteno coperto da uno strato liscio e coesivo. Barra di scala = 200 nm. Le caselle quadrate bianche rappresentano le viste ingrandite in basso a destra. I globuli sferici sono piccole vescicole lipidiche che interagiscono con le settine. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Risultati
Deformazioni dei GUV
Le tipiche immagini di fluorescenza confocale dei GUV rimodellati dopo essere stati incubati con settine sono visualizzate nella Figura 3, in condizioni in cui le settine polimerizzano. I GUV nudi (Figura 3A) erano perfettamente sferici. Dopo l'incubazione con più di 50 filamenti di settina di lievito in erba nM, le vescicole apparivano deformate. Fino a una concentrazione di ottameri settini di lievito in erba 100 nM, l...
Discussione
Come detto sopra, è stata utilizzata una miscela lipidica che migliora l'incorporazione di PI(4,5)P2 all'interno del doppio strato lipidico e quindi facilita le interazioni settina-membrana. In effetti, abbiamo dimostrato altrove25 che le settine di lievito in erba interagiscono con le vescicole in modo PI(4,5)P2-specifico. Questa composizione lipidica è stata regolata empiricamente dallo screening di composizioni multiple ed è ora ampiamente utilizzata dagli autori. I lip...
Divulgazioni
Gli autori non hanno conflitti di interesse.
Riconoscimenti
Ringraziamo Patricia Bassereau e Daniel Lévy per i loro utili consigli e discussioni. Questo lavoro ha beneficiato del sostegno dell'ANR (Agence Nationale de la Recherche) per il finanziamento del progetto "SEPTIME", ANR-13-JSV8-0002-01, ANR SEPTIMORF ANR-17-CE13-0014, e del progetto "SEPTSCORT", ANR-20-CE11-0014-01. B. Chauvin è finanziato dall'Ecole Doctorale "ED564: Physique en Ile de France" e dalla Fondation pour lea Recherche Médicale. K. Nakazawa è stato sostenuto dalla Sorbonne Université (AAP Emergence). G.H. Koenderink è stato sostenuto dalla Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO/OCW) attraverso il "BaSyC-Building a Synthetic Cell". Sovvenzione per gravitazione (024.003.019). Si ringraziano Labex Cell(n)Scale (ANR-11-LABX0038) e Paris Sciences et Lettres (ANR-10-IDEX-0001-02). Ringraziamo il Cell and Tissue Imaging (PICT-IBiSA), Institut Curie, membro dell'Infrastruttura Nazionale di Ricerca France-BioImaging (ANR10-INBS-04).
Materiali
| Name | Company | Catalog Number | Comments |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | 850725 | |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine | Avanti Polar Lipids | 840035 | |
| Bath sonicator | Elma | Elmasonic S10H | |
| Bodipy-TR-Ceramide | invitrogen, Thermo Fischer scientific | 11504726 | |
| Chemicals: NaCl, Tris-HCl, sucrose, KCl, MgCl2, B-casein, chloroform, sodium cacodylate, tannic acid, ethanol | Sigma Aldrich | ||
| Confocal microscope | nikon | spinning disk or confocal | |
| Critical point dryer | Leica microsystems | CPD300 | |
| Deionized water generator | MilliQ | F1CA38083B | MilliQ integral 3 |
| Egg L-α-phosphatidylcholine | Avanti Polar Lipids | 840051 | |
| Field Emission Gun SEM (FESEM) | Carl Zeiss | Gemini SEM500 | |
| Glutaraldehyde 25 %, aqueous solution | Thermo Fischer scientific | 50-262-19 | |
| High vacuum grease, Dow corning | VWR | ||
| IMOD software | https://bio3d.colorado.edu/imod/ | software suite for tilted series image alignment and 3D reconstruction | |
| Lacey Formvar/carbon electron microscopy grids | Eloise | 01883-F | |
| Lipids | Avanti Polar Lipids | ||
| L-α-phosphatidylinositol-4,5-bisphosphate | Avanti Polar Lipids | 840046 | |
| Metal evaporator | Leica microsystems | EM ACE600 | |
| NOA (Norland Optical Adhesives), NOA 71 and NOA 81 | Norland Products | NOA71, NOA81 | |
| Osmium tetraoxyde 4% | delta microscopies | 19170 | |
| Osmometer | Löser | 15 M | |
| Plasma cleaner | Alcatel | pascal 2005 SD | |
| Plasma generator | Electron Microscopy Science | ||
| Plunge freezing equipment | leica microsystems | EMGP | |
| Transmission electron microscope | Thermofischer | Tecnai G2 200 kV, LaB6 | |
| Uranyl acetate | Electron Microscopy Science | 22451 | this product is not available for purchase any longer |
| Wax plates, Vitrex | VWR |
Riferimenti
- Finger, F. P. Reining in cytokinesis with a septin corral. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 27 (1), 5-8 (2005).
- Barral, Y., Kinoshita, M. Structural insights shed light onto septin assemblies and function. Current Opinion in Cell Biology. 20 (1), 12-18 (2008).
- Hu, Q., et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329 (5990), 436-439 (2010).
- Lin, Y. -. H., Kuo, Y. -. C., Chiang, H. -. S., Kuo, P. -. L. The role of the septin family in spermiogenesis. Spermatogenesis. 1 (4), 298-302 (2011).
- Addi, C., Bai, J., Echard, A. Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Current Opinion in Cell Biology. 50, 27-34 (2018).
- Spiliotis, E. T., Kesisova, I. A. Spatial regulation of microtubule-dependent transport by septin GTPases. Trends in Cell Biology. 31 (12), 979-993 (2021).
- Spiliotis, E. T., Nakos, K. Cellular functions of actin- and microtubule-associated septins. Current Biology: CB. 31 (10), 651-666 (2021).
- Salameh, J., Cantaloube, I., Benoit, B., Poüs, C., Baillet, A. Cdc42 and its BORG2 and BORG3 effectors control the subcellular localization of septins between actin stress fibers and microtubules. Current Biology: CB. 31 (18), 4088-4103 (2021).
- Ewers, H., Tada, T., Petersen, J. D., Racz, B., Sheng, M., Choquet, D. A septin-dependent diffusion barrier at dendritic spine necks. PloS One. 9 (12), 113916 (2014).
- Myles, D. G., Primakoff, P., Koppel, D. E. A localized surface protein of guinea pig sperm exhibits free diffusion in its domain. The Journal of Cell Biology. 98 (5), 1905-1909 (1984).
- Luedeke, C., Frei, S. B., Sbalzarini, I., Schwarz, H., Spang, A., Barral, Y. Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. The Journal of Cell Biology. 169 (6), 897-908 (2005).
- Gilden, J. K., Peck, S., Chen, Y. -. C. M., Krummel, M. F. The septin cytoskeleton facilitates membrane retraction during motility and blebbing. The Journal of Cell Biology. 196 (1), 103-114 (2012).
- Dolat, L., Hu, Q., Spiliotis, E. T. Septin functions in organ system physiology and pathology. Biological Chemistry. 395 (2), 123-141 (2014).
- Angelis, D., Spiliotis, E. T. Septin mutations in human cancers. Frontiers in Cell and Developmental Biology. 4, 122 (2016).
- Takehashi, M., et al. Septin 3 gene polymorphism in Alzheimer's disease. Gene Expression. 11 (5-6), 263-270 (2004).
- Shuman, B., Momany, M. Septins from protists to people. Frontiers in Cell and Developmental Biology. 9, 824850 (2022).
- Bertin, A., et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105 (24), 8274-8279 (2008).
- Iv, F., et al. Insights into animal septins using recombinant human septin octamers with distinct SEPT9 isoforms. Journal of cell science. 134 (15), (2021).
- Beber, A., et al. Membrane reshaping by micrometric curvature sensitive septin filaments. Nature communications. 10 (1), 420 (2019).
- Bridges, A. A., Jentzsch, M. S., Oakes, P. W., Occhipinti, P., Gladfelter, A. S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. The Journal of Cell Biology. 213 (1), 23-32 (2016).
- Patzig, J., et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 5, 17119 (2016).
- Szuba, A., et al. Membrane binding controls ordered self-assembly of animal septins. eLife. 10, 63349 (2021).
- Tanaka-Takiguchi, Y., Kinoshita, M., Takiguchi, K. Septin-mediated uniform bracing of phospholipid membranes. Current Biology: CB. 19 (2), 140-145 (2009).
- Bertin, A., et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of Molecular Biology. 404 (4), 711-731 (2010).
- Beber, A., et al. Septin-based readout of PI(4,5)P2 incorporation into membranes of giant unilamellar vesicles. Cytoskeleton. 76 (4,5), 92-103 (2019).
- Mastronarde, D. N., Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology. 197 (2), 102-113 (2017).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Nania, M., Foglia, F., Matar, O. K., Cabral, J. T. Sub-100 nm wrinkling of polydimethylsiloxane by double frontal oxidation. Nanoscale. 9 (5), 2030-2037 (2017).
- Nania, M., Matar, O. K., Cabral, J. T. Frontal vitrification of PDMS using air plasma and consequences for surface wrinkling. Soft Matter. 11 (15), 3067-3075 (2015).
- Svitkina, T. M., Borisy, G. G. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods in Enzymology. 298, 570-592 (1998).
- Franck, A., et al. Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Molecular Biology of the Cell. 30 (5), 579-590 (2019).
- Elkhatib, N., et al. Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science. 356 (6343), (2017).
- Stokroos, I., Kalicharan, D., Van Der Want, J. J., Jongebloed, W. L. A comparative study of thin coatings of Au/Pd, Pt and Cr produced by magnetron sputtering for FE-SEM. Journal of Microscopy. 189, 79-89 (1998).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati