
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
DeepOmicsAE: Rappresentazione dei moduli di segnalazione nella malattia di Alzheimer con analisi di deep learning di proteomica, metabolomica e dati clinici
In questo articolo
Riepilogo
DeepOmicsAE è un flusso di lavoro incentrato sull'applicazione di un metodo di deep learning (ad esempio, un autoencoder) per ridurre la dimensionalità dei dati multi-omici, fornendo una base per modelli predittivi e moduli di segnalazione che rappresentano più livelli di dati omici.
Abstract
I grandi set di dati omici stanno diventando sempre più disponibili per la ricerca sulla salute umana. Questo documento presenta DeepOmicsAE, un flusso di lavoro ottimizzato per l'analisi di set di dati multi-omici, tra cui proteomica, metabolomica e dati clinici. Questo flusso di lavoro utilizza un tipo di rete neurale chiamata autoencoder, per estrarre un insieme conciso di funzionalità dai dati di input multi-omici ad alta dimensionalità. Inoltre, il flusso di lavoro fornisce un metodo per ottimizzare i parametri chiave necessari per implementare l'autoencoder. Per mostrare questo flusso di lavoro, i dati clinici sono stati analizzati da una coorte di 142 individui sani o con diagnosi di Alzheimer, insieme al proteoma e al metaboloma dei loro campioni cerebrali post-mortem. Le caratteristiche estratte dallo strato latente dell'autoencoder conservano le informazioni biologiche che separano i pazienti sani da quelli malati. Inoltre, le singole caratteristiche estratte rappresentano moduli di segnalazione molecolare distinti, ognuno dei quali interagisce in modo univoco con le caratteristiche cliniche degli individui, fornendo un mezzo per integrare la proteomica, la metabolomica e i dati clinici.
Introduzione
Una percentuale sempre più ampia della popolazione sta invecchiando e si prevede che l'onere delle malattie legate all'età, come la neurodegenerazione, aumenterà notevolmente nei prossimi decenni1. Il morbo di Alzheimer è il tipo più comune di malattia neurodegenerativa2. I progressi nella ricerca di un trattamento sono stati lenti data la nostra scarsa comprensione dei meccanismi molecolari fondamentali che guidano l'insorgenza e il progresso della malattia. La maggior parte delle informazioni sulla malattia di Alzheimer viene ottenuta post-mortem dall'esame del tessuto cerebrale, il che ha reso difficile distinguere le cause e leconseguenze. Il Religious Orders Study/Memory and Aging Project (ROSMAP) è uno sforzo ambizioso per ottenere una comprensione più ampia della neurodegenerazione, che coinvolge lo studio di migliaia di individui che si sono impegnati a sottoporsi a esami medici e psicologici ogni anno e a contribuire con i loro cervelli alla ricerca dopo laloro morte. Lo studio si concentra sulla transizione dal normale funzionamento del cervello al morbo di Alzheimer2. Nell'ambito del progetto, i campioni cerebrali post-mortem sono stati analizzati con una pletora di approcci omici, tra cui genomica, epigenomica, trascrittomica, proteomica5 e metabolomica.
Le tecnologie omiche che offrono letture funzionali degli stati cellulari (i.e., proteomica e metabolomica)6,7 sono fondamentali per interpretare la malattia 8,9,10,11,12, a causa della relazione diretta tra l'abbondanza di proteine e metaboliti e le attività cellulari. Le proteine sono i principali esecutori dei processi cellulari, mentre i metaboliti sono i substrati e i prodotti per le reazioni biochimiche. L'analisi dei dati multi-omici offre la possibilità di comprendere le complesse relazioni tra i dati di proteomica e metabolomica invece di apprezzarli isolatamente. La multi-omica è una disciplina che studia più strati di dati biologici ad alta dimensionalità, inclusi i dati molecolari (sequenza e mutazioni del genoma, trascrittoma, proteoma, metaboloma), i dati di imaging clinico e le caratteristiche cliniche. In particolare, l'analisi dei dati multi-omici mira a integrare tali strati di dati biologici, comprendere la loro regolazione reciproca e le dinamiche di interazione e fornire una comprensione olistica dell'insorgenza e della progressione della malattia. Tuttavia, i metodi per integrare i dati multi-omici rimangono nelle prime fasi di sviluppo13.
Gli autoencoder, un tipo di rete neurale non supervisionata14, sono un potente strumento per l'integrazione dei dati multi-omici. A differenza delle reti neurali supervisionate, gli autoencoder non mappano i campioni a valori target specifici (come sano o malato), né vengono utilizzati per prevedere i risultati. Una delle loro applicazioni principali risiede nella riduzione della dimensionalità. Tuttavia, gli autoencoder offrono diversi vantaggi rispetto ai metodi di riduzione della dimensionalità più semplici, come l'analisi delle componenti principali (PCA), l'inclusione stocastica dei vicini t-distribuiti (tSNE) o l'approssimazione e la proiezione uniforme della varietà (UMAP). A differenza della PCA, gli autoencoder possono acquisire relazioni non lineari all'interno dei dati. A differenza di tSNE e UMAP, sono in grado di rilevare relazioni gerarchiche e multimodali all'interno dei dati poiché si basano su più livelli di unità computazionali, ciascuno contenente funzioni di attivazione non lineari. Pertanto, rappresentano modelli interessanti per catturare la complessità dei dati multi-omici. Infine, mentre l'applicazione principale di PCA, tSNE e UMAP è quella di raggruppare i dati, gli autoencoder comprimono i dati di input in funzionalità estratte che sono adatte per le attività predittive a valle15,16.
In breve, le reti neurali comprendono diversi livelli, ognuno contenente più unità computazionali o "neuroni". Il primo e l'ultimo livello sono indicati rispettivamente come livelli di input e di output. Gli autoencoder sono reti neurali con una struttura a clessidra, costituita da uno strato di input, seguito da uno a tre strati nascosti e da un piccolo strato "latente" che contiene tipicamente da due a sei neuroni. La prima metà di questa struttura è nota come encoder ed è combinata con un decoder che rispecchia l'encoder. Il decodificatore termina con un livello di output contenente lo stesso numero di neuroni del livello di input. Gli autoencoder prendono l'input attraverso il collo di bottiglia e lo ricostruiscono nel livello di output, con l'obiettivo di generare un output che rispecchi il più fedelmente possibile le informazioni originali. Ciò si ottiene riducendo matematicamente al minimo un parametro chiamato "perdita di ricostruzione". L'input consiste in una serie di caratteristiche, che nell'applicazione qui illustrata saranno l'abbondanza di proteine e metaboliti e le caratteristiche cliniche (ad esempio, sesso, istruzione ed età alla morte). Il livello latente contiene una rappresentazione compressa e ricca di informazioni dell'input, che può essere utilizzata per applicazioni successive come i modelli predittivi17,18.
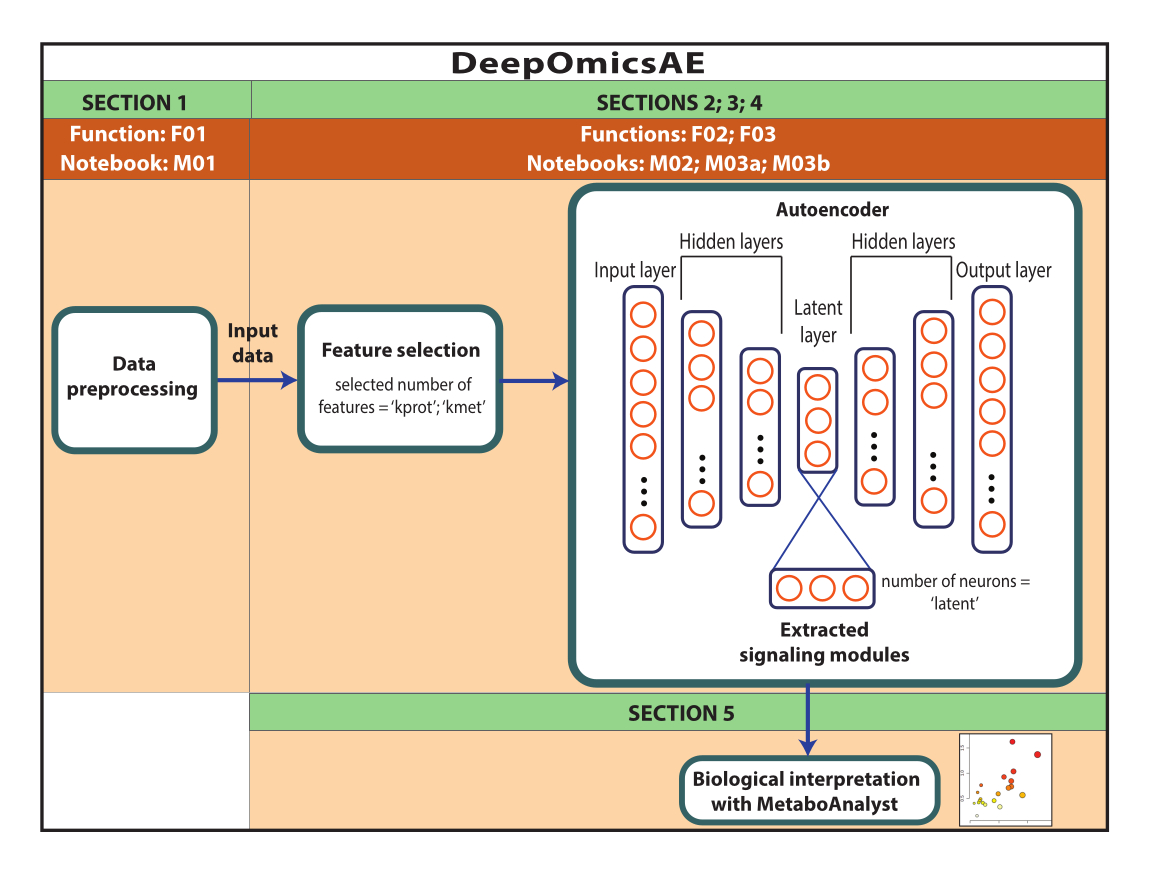
Questo protocollo presenta un flusso di lavoro, DeepOmicsAE, che prevede: 1) la pre-elaborazione di dati proteomici, metabolomici e clinici (ad esempio, normalizzazione, ridimensionamento, rimozione dei valori anomali) per ottenere dati con una scala coerente per l'analisi dell'apprendimento automatico; 2) selezionare le caratteristiche di ingresso dell'autoencoder appropriate, poiché il sovraccarico di funzionalità può oscurare i modelli di malattia rilevanti; 3) l'ottimizzazione e l'addestramento dell'autoencoder, compresa la determinazione del numero ottimale di proteine e metaboliti da selezionare, e di neuroni per lo strato latente; 4) estrazione di elementi dallo strato latente; e 5) utilizzare le caratteristiche estratte per l'interpretazione biologica identificando i moduli di segnalazione molecolare e la loro relazione con le caratteristiche cliniche.
Questo protocollo mira ad essere semplice e applicabile da biologi con limitata esperienza computazionale che hanno una conoscenza di base della programmazione con Python. Il protocollo si concentra sull'analisi dei dati multi-omici, tra cui proteomica, metabolomica e caratteristiche cliniche, ma il suo utilizzo può essere esteso ad altri tipi di dati di espressione molecolare, inclusa la trascrittomica. Un'importante nuova applicazione introdotta da questo protocollo è la mappatura dei punteggi di importanza delle caratteristiche originali sui singoli neuroni nello strato latente. Di conseguenza, ogni neurone nello strato latente rappresenta un modulo di segnalazione, che descrive in dettaglio le interazioni tra specifiche alterazioni molecolari e le caratteristiche cliniche dei pazienti. L'interpretazione biologica dei moduli di segnalazione molecolare è ottenuta utilizzando MetaboAnalyst, uno strumento disponibile al pubblico che integra dati genetici/proteici e metaboliti per derivare vie metaboliche e di segnalazione cellularearricchite 17.
Protocollo
NOTA: i dati utilizzati in questo caso sono dati ROSMAP scaricati dal portale AD Knowledge. Il consenso informato non è necessario per scaricare e riutilizzare i dati. Il protocollo qui presentato utilizza il deep learning per analizzare i dati multi-omici e identificare i moduli di segnalazione che distinguono specifici pazienti o gruppi di campioni in base, ad esempio, alla loro diagnosi. Il protocollo fornisce anche un piccolo set di funzionalità estratte che riepilogano i dati originali su larga scala e possono essere utilizzate per ulteriori analisi, ad esempio per l'addestramento di un modello predittivo utilizzando algoritmi di apprendimento automatico (Figura 1). Fare riferimento al file supplementare 1 e alla tabella dei materiali per informazioni sull'accesso al codice e sulla configurazione dell'ambiente computazionale prima di eseguire il protocollo. I metodi devono essere eseguiti seguendo l'ordine specificato di seguito.

Figura 1: Schema del flusso di lavoro di DeepOmicsAE. Rappresentazione schematica del flusso di lavoro per l'analisi dei dati multi-omici utilizzando il flusso di lavoro. Nella rappresentazione dell'autoencoder, i rettangoli rappresentano i livelli della rete neurale e i cerchi rappresentano i neuroni all'interno dei livelli. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
1. Pre-elaborazione dei dati
NOTA: l'obiettivo di questa sezione è la pre-elaborazione dei dati, inclusa la gestione dei dati mancanti; normalizzazione e ridimensionamento dell'espressione proteomica, metabolomica e dei dati clinici; e rimuovendo i valori anomali. Il protocollo è progettato per un set di dati che include dati di proteomica espressi come log2 (rapporto); dati metabolomici espressi come fold change; e caratteristiche cliniche, comprese le caratteristiche continue e categoriche. I pazienti o i campioni devono essere raggruppati in base alla diagnosi o ad altri parametri simili. I campioni o i pazienti devono trovarsi tra le righe e le caratteristiche nelle colonne.
- Per avviare una nuova istanza di Jupyter Notebook nel browser, aprire una nuova finestra del terminale, digitare quanto segue e premere Invio.
Notebook Jupyter - Nella home page di Jupyter del browser fare clic sul notebook M01 - Expression Data Pre-processing.ipynb per aprirlo in una nuova scheda (File supplementare 2, passaggio 1.1).
- Nella seconda cella del notebook digitare il nome del file del set di dati al posto di your_dataset_name.csv.
- Nell'ultima cella del notebook digitare il nome desiderato del file di dati di output al posto di M01_output_data.csv.
- Nella quinta cella del notebook, specificare la posizione delle colonne per ogni tipo di dati come segue: dati di proteomica (cols_prot), dati metabolomici (cols_met), dati clinici continui (ad esempio, età) (cols_clin_con), dati clinici binari (ad esempio, sesso) (cols_clin_bin). Immettere l'indice della prima colonna per ogni tipo di dati al posto di col_start e l'indice dell'ultima colonna al posto di col_end; Ad esempio: cols_prot = slice(0, 8817). Assicurarsi che i valori specificati negli oggetti slice corrispondano agli indici della prima e dell'ultima colonna corrispondenti a ciascun tipo di dati. Utilizzare il comando nella quarta cella dello stesso notebook (df.iloc[:, :]) per determinare la posizione iniziale e finale per ogni tipo di dati (File supplementare 2, passaggio 1.2).
- Seleziona cella | Eseguire tutto dalla barra dei menu in Jupyter per creare il file di dati di output nella cartella specificata (File supplementare 2, passaggio 1.3).
NOTA: Questi dati verranno utilizzati come input per i protocolli descritti nelle sezioni 2, 3 o 4.
2. Ottimizzazione personalizzata del flusso di lavoro (opzionale)
NOTA: la sezione 2 è facoltativa perché richiede un uso intensivo del computer. Gli utenti devono passare direttamente alla sezione 4 se decidono di non eseguire la sezione 2. Questo protocollo guiderà l'utente attraverso l'ottimizzazione del flusso di lavoro in modo automatizzato. In particolare, il metodo identifica i parametri che offrono le migliori prestazioni dell'autoencoder in termini di generazione di feature estratte che separano bene i gruppi di campioni. I parametri ottimizzati generati come output includono il numero di feature da utilizzare per la selezione delle feature (k_prot e k_met) e il numero di neuroni nel livello latente dell'autoencoder (latente). Questi parametri possono quindi essere utilizzati nel protocollo descritto nella sezione 3 per generare il modello.
- Nella home page di Jupyter del browser, fare clic sul notebook M02 - DeepOmicsAE model optimization.ipynb per aprirlo in una nuova scheda (File supplementare 2, passaggio 2.1).
- Nella seconda cella del blocco appunti digitare il nome del file di input al posto di M01_output_data.csv. L'input per questa funzione sono i dati di output della sezione 1.
- Nella quinta cella del notebook specificare la posizione delle colonne per ogni tipo di dati come segue: dati di proteomica (cols_X_prot), dati di metabolomica (cols_X_met), dati clinici (cols_clin; include tutti i dati clinici), tutti i dati di espressione molecolare, inclusi i dati di proteomica e metabolomica (cols_X_expr). Immettere l'indice della prima colonna per ogni tipo di dati al posto di col_start e l'indice dell'ultima colonna al posto di col_end; Ad esempio, cols_prot = slice(0, 8817). Assicurarsi che i valori specificati negli oggetti slice corrispondano all'indice della prima e dell'ultima colonna corrispondente a ciascun tipo di dati e utilizzare i comandi nella terza e nella quarta cella del notebook per esplorare i dati e determinare le posizioni iniziale e finale per ogni tipo di dati. Specificare il nome della colonna contenente la variabile di destinazione al posto di y_column_name come y_label (File supplementare 2, passaggio 2.2).
NOTA: i valori degli indici specificati in cols_X_prot, cols_X_met, cols_clin e cols_X_expr saranno diversi da quelli utilizzati nella sezione 1 a causa della rimodellazione del frame di dati che si verifica durante la pre-elaborazione dei dati. - Nella sesta cella del notebook specificare il numero di cicli di ottimizzazione da eseguire assegnando un valore a n_comb. I tempi di lavorazione sono di circa 4-5 min per 10 giri; 20 min per 50 round e 40 min per 100 round (File supplementare 2, passaggio 2.3).
- Seleziona cella | Esegui tutto dalla barra dei menu in Jupyter.
NOTA: le variabili di output kprot, kmet e latent verranno archiviate e sarà possibile accedervi dagli altri notebook, che verranno utilizzati per continuare il flusso di lavoro analitico. Il grafico AE_optimization_plot.pdf verrà generato e salvato nella cartella locale (Figura 2).
3. Implementazione del flusso di lavoro con parametri ottimizzati personalizzati
NOTA: Eseguire questo protocollo solo dopo l'ottimizzazione del metodo (sezione 2). Se gli utenti scelgono di non eseguire l'ottimizzazione del metodo, passare direttamente alla sezione 4. Questo protocollo guiderà l'utente attraverso la generazione di un modello utilizzando i parametri ottimizzati per l'utente derivati dalla sezione 2. L'autoencoder 1) genererà una serie di caratteristiche estratte che ricapitolano i dati originali e 2) identificherà le caratteristiche importanti che guidano ciascun neurone nello strato latente, rappresentando efficacemente moduli di segnalazione unici. I moduli di segnalazione saranno interpretati utilizzando il protocollo fornito nella sezione 5.
- Nella home page di Jupyter del browser, fare clic sul notebook M03a - Implementazione di DeepOmicsAE con parametri ottimizzati per l'utente.ipynb per aprirlo in una nuova scheda (File supplementare 2, passaggio 3.1).
- Nella seconda cella del blocco appunti digitare il nome del file di input al posto di M01_output_data.csv. L'input per questa funzione sono i dati di output della sezione 1.
- Nella quinta cella del notebook specificare la posizione delle colonne per ogni tipo di dati come indicato di seguito: dati di proteomica (cols_prot), dati metabolomici (cols_met), dati clinici (cols_clin; include tutti i dati clinici). Immettere l'indice della prima colonna per ogni tipo di dati al posto di col_start e l'indice dell'ultima colonna al posto di col_end; Ad esempio: cols_prot = slice(0, 8817). Assicurarsi che i valori specificati negli oggetti slice corrispondano agli indici della prima e dell'ultima colonna corrispondenti a ciascun tipo di dati e utilizzare i comandi nella terza e nella quarta cella del notebook per esplorare i dati e determinare le posizioni iniziale e finale per ogni tipo di dati. Specificare il nome della colonna contenente la variabile di destinazione (ad esempio, 0 o 1, corrispondente a sano o malato) al posto di y_column_name come y_label.
NOTA: il valore degli indici specificati in cols_X_prot, cols_X_met, cols_clin e cols_X_expr sarà diverso da quelli utilizzati nella sezione 1 a causa della ridefinizione del frame di dati che si verifica durante la pre-elaborazione dei dati. - Seleziona cella | Eseguire tutto dalla barra dei menu in Jupyter per generare e salvare i grafici PCA_initial_data.pdf, PCA_extracted_features.pdf e distribution_important_feature_scores.pdf nella cartella locale (Figura 3 e Figura supplementare S1). Inoltre, gli elenchi delle caratteristiche importanti per ogni modulo di segnalazione identificato verranno memorizzati in file di testo nella cartella locale, denominata module_n.txt, dove n sarà sostituito dal numero del modulo.
4. Implementazione del flusso di lavoro con parametri preimpostati
- Fare riferimento alla sezione 3 per istruzioni dettagliate su come eseguire questo metodo (File supplementare 2, passaggio 4.1). L'unica differenza tra questi due protocolli è che i parametri kprot, kmet e latente (nella settima cella del notebook) sono derivati matematicamente in base ai risultati dell'ottimizzazione eseguita come mostrato nella Figura 2.
NOTA: se la sezione 4 fornisce una separazione insufficiente dei gruppi di campioni, indicando prestazioni del modello non ottimali, si consiglia di eseguire l'ottimizzazione del modello (sezione 2) utilizzando almeno 15 iterazioni e, se possibile, fino a 50.
5. Interpretazione biologica con MetaboAnalyst
- Aprire il browser e passare al collegamento sottostante per accedere alla funzionalità Joint Pathway Analysis sul sito Web di MetaboAnalyst : https://www.metaboanalyst.ca/MetaboAnalyst/upload/JointUploadView.xhtml.
- Accedere alla cartella in cui sono stati salvati i file di output del Metodo 3 o del Metodo 4 e aprire i file di testo module_n.txt per ogni modulo di segnalazione n generato dal Metodo 3 o dal Metodo 4.
- Individua le proteine nei file di testo e copiale.
- Incollare l'elenco delle proteine nella finestra Geni/proteine con modifiche di piega opzionali nella pagina web di MetaboAnalyst.
- Ripeti il passaggio precedente per i metaboliti e incollali nella finestra Elenco composti con modifiche di piega opzionali nella stessa pagina web.
- Selezionare l'organismo e il tipo di ID appropriati, quindi fare clic su Invia nella parte inferiore della pagina (File supplementare 2, passaggio 5.1).
NOTA: Assicurarsi che gli identificatori siano riconosciuti da MetaboAnalyst. Gli identificatori riconosciuti includono l'ID Entrez, i simboli genici ufficiali e l'ID Uniprot per le proteine; nome del composto, ID HMDB e ID KEGG per i metaboliti. Se gli identificatori sono diversi da questi tipi, è necessaria una conversione appropriata prima dell'analisi. - Nella pagina seguente, controllare il mapping ID prima di fare clic su Procedi per verificare che gli identificatori vengano riconosciuti.
- Nella pagina Impostazione parametri , selezionare Vie metaboliche (integrate) o Tutte le vie (integrate) per visualizzare rispettivamente il contributo dell'input alle sole vie metaboliche o a tutte le vie di segnalazione (File supplementare 2, Passaggio 5.2). Nel pannello di selezione dell'algoritmo , scegliere Analisi di arricchimento: Test ipergeometrico, Misura topologia: Centralità del grado e Metodo di integrazione: Combina valori p (a livello di percorso). Fare clic su Invia in fondo alla pagina.
- L'ultima pagina è la visualizzazione dei risultati, che presenta i risultati dell'analisi di arricchimento. I percorsi arricchiti vengono tracciati in base al loro impatto e significato e l'elenco dei percorsi viene fornito anche in formato tabellare.
Risultati
Per mostrare il protocollo, abbiamo analizzato un set di dati che comprendeva il proteoma, il metaboloma e le informazioni cliniche derivate dai cervelli post-mortem di 142 individui sani o con diagnosi di Alzheimer.
Dopo aver eseguito la sezione 1 del protocollo per pre-elaborare i dati, il set di dati includeva 6.497 proteine, 443 metaboliti e tre caratteristiche cliniche (sesso, età alla morte e istruzione). La caratteristica target è la diagnosi di consenso clinico dello stato co...
Discussione
La struttura del set di dati è fondamentale per il successo del protocollo e deve essere attentamente controllata. I dati devono essere formattati come indicato nella sezione 1 del protocollo. Anche la corretta assegnazione delle posizioni delle colonne è fondamentale per il successo del metodo. I dati di proteomica e metabolomica vengono pre-elaborati in modo diverso e la selezione delle caratteristiche viene condotta separatamente a causa della diversa natura dei dati. Pertanto, è fondamentale assegnare correttament...
Divulgazioni
L'autore dichiara di non avere conflitti di interesse.
Riconoscimenti
Questo lavoro è stato sostenuto dalla sovvenzione NIH CA201402 e dal Cornell Center for Vertebrate Genomics (CVG) Distinguished Scholar Award. I risultati qui pubblicati si basano, in tutto o in parte, sui dati ottenuti dall'AD Knowledge Portal (https://adknowledgeportal.org). I dati dello studio sono stati forniti attraverso l'Accelerating Medicine Partnership for AD (U01AG046161 e U01AG061357) sulla base di campioni forniti dal Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. La raccolta dei dati è stata supportata attraverso il finanziamento delle sovvenzioni NIA P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, il Dipartimento di sanità pubblica dell'Illinois e l'Istituto di ricerca sulla genomica traslazionale. Il set di dati metabolomici è stato generato a Metabolon e pre-elaborato dall'ADMC.
Materiali
| Name | Company | Catalog Number | Comments |
| Computer | Apple | Mac Studio | Apple M1 Ultra with 20-core CPU, 48-core GPU, 32-core Neural Engine; 64 GB unified memory |
| Conda v23.3.1 | Anaconda, Inc. | N/A | package management system and environment manager |
| conda environment DeepOmicsAE | N/A | DeepOmicsAE_env.yml | contains packages necessary to run the worflow |
| github repository DeepOmicsAE | Microsoft | https://github.com/elepan84/DeepOmicsAE/ | provides scripts, Jupyter notebooks, and the conda environment file |
| Jupyter notebook v6.5.4 | Project Jupyter | N/A | a platform for interactive data science and scientific computing |
| DT01-metabolomics data | N/A | ROSMAP_Metabolon_HD4_Brain 514_assay_data.csv | This data was used to generate the Results reported in the article. Specifically, DT01-DT04 were merged by matching them based on the individualID. The column final consensus diagnosis (cogdx) was filtered to keep only patients classified as healthy or AD. Climnical features were filtered to keep the following: age at death, sex and education. Finally, age reported as 90+ was set to 91, then the age column was transformed to float64. The data is available at https://adknowledgeportal.synapse.org |
| DT02-TMT proteomics data | N/A | C2.median_polish_corrected_log2 (abundanceRatioCenteredOn MedianOfBatchMediansPer Protein)-8817x400.csv | |
| DT03-clinical data | N/A | ROSMAP_clinical.csv | |
| DT04-biospecimen metadata | N/A | ROSMAP_biospecimen_metadata .csv | |
| Python 3.11.3 | Python Software Foundation | N/A | programming language |
Riferimenti
- Hou, Y., et al. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 15 (10), 565-581 (2019).
- Scheltens, P., et al. Alzheimer’s disease. The Lancet. 397 (10284), 1577-1590 (2021).
- Breijyeh, Z., Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules. 25 (24), 5789 (2020).
- Bennett, D. A., et al. Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s Disease. 64 (s1), S161-S189 (2018).
- Higginbotham, L., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Science Advances. 6 (43), eaaz9360 (2020).
- Aebersold, R., et al. How many human proteoforms are there. Nature Chemical Biology. 14 (3), 206-214 (2018).
- Nusinow, D. P., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 180 (2), 387-402.e16 (2020).
- Johnson, E. C. B., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine. 26 (5), 769-780 (2020).
- Geyer, P. E., et al. Plasma proteome profiling to assess human health and disease. Cell Systems. 2 (3), 185-195 (2016).
- Akbani, R., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nature Communications. 5, 3887 (2014).
- Panizza, E., et al. Proteomic analysis reveals microvesicles containing NAMPT as mediators of radioresistance in glioma. Life Science Alliance. 6 (6), e202201680 (2023).
- Li, Z., Vacanti, N. M. A tale of three proteomes: visualizing protein and transcript abundance relationships in the Breast Cancer Proteome Portal. Journal of Proteome Research. 22 (8), 2727-2733 (2023).
- Subramanian, I., Verma, S., Kumar, S., Jere, A., Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinformatics and Biology Insights. 14, 1177932219899051 (2020).
- Wang, Y., Yao, H., Zhao, S. Auto-encoder based dimensionality reduction. Neurocomputing. 184, 232-242 (2016).
- Mulla, F. R., Gupta, A. K. A review paper on dimensionality reduction techniques. Journal of Pharmaceutical Negative Results. 13, 1263-1272 (2022).
- Shrestha, A., Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access. 7, 53040-53065 (2019).
- Pang, Z., et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Research. 49 (W1), W388-W396 (2021).
- Hinton, G. E., Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science. 313 (5786), 504-507 (2006).
- Altmann, A., Toloşi, L., Sander, O., Lengauer, T. Permutation importance: a corrected feature importance measure. Bioinformatics. 26 (10), 1340-1347 (2010).
- Lundberg, S. M., Allen, P. G., Lee, S. -. I. A unified approach to interpreting model predictions. , (2017).
- Wang, Q., et al. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Communications. 4 (1), (2021).
- Beebe-Wang, N., et al. Unified AI framework to uncover deep interrelationships between gene expression and Alzheimer’s disease neuropathologies. Nature Communications. 12 (1), 5369 (2021).
- Camandola, S., Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. The EMBO Journal. 36 (11), 1474-1492 (2017).
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 350 (6265), 1208-1213 (2015).
- Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F., Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nature Reviews Drug Discovery. 18 (5), 379-401 (2019).
- Wang, R., Reddy, P. H. Role of glutamate and NMDA receptors in Alzheimer’s disease. Journal of Alzheimer’s Disease. 57 (4), 1041-1048 (2017).
- Skaper, S. D., Facci, L., Zusso, M., Giusti, P. Synaptic plasticity, dementia and Alzheimer disease. CNS & Neurological Disorders - Drug Targets. 16 (3), 220-233 (2017).
- Reisberg, B., et al. Memantine in moderate-to-severe Alzheimer’s disease. New England Journal of Medicine. 348 (14), 1333-1341 (2003).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati