
Method Article
Quantification of Insoluble Protein Aggregation in Caenorhabditis elegans during Aging with a Novel Data-Independent Acquisition Workflow
In This Article
Summary
This novel workflow efficiently extracts and isolates SDS-insoluble proteins (insolublome) from Caenorhabditis elegans with minimal starting material for quantitative differential proteomic analysis. The protocol uses a comprehensive data-independent acquisition mass spectrometry analysis to quantify the insolublome and bioinformatic analysis to gain biological insights into aging mechanisms and pathologies.
Abstract
We and others have shown that the aging process results in a proteome-wide accumulation of insoluble proteins. Knocking down genes encoding the insoluble proteins over 40% of the time results in an extension of the lifespan in C. elegans, suggesting that many of these proteins are key determinants of the aging process. Isolation and quantitative identification of these insoluble proteins are crucial to understand key biological processes that occur during aging. Here, we present a modified and improved protocol that details how to extract and isolate the SDS-insoluble proteins (insolublome) from C. elegans more efficiently to streamline mass spectrometric workflows via a novel label-free quantitative proteomics analysis. This improved protocol utilizes a highly efficient sonicator for worm lysis that greatly increases efficiency for protein extraction and allows us to use significantly less starting material (approximately 3,000 worms) than in previous protocols (typically using at least 40,000 worms). Subsequent quantitative proteomic analysis of the insolublome was performed using data-dependent acquisition (DDA) for protein discovery and identification and data-independent acquisition (DIA) for comprehensive and more accurate protein quantification. Bioinformatic analysis of quantified proteins provides potential candidates that can be easily followed up with other molecular methods in C. elegans. With this workflow, we routinely identify more than 1000 proteins and quantify more than 500 proteins. This new protocol enables efficient compound screening with C. elegans. Here, we validated and applied this improved protocol to wild-type C. elegans N2-Bristol strain and confirmed that aged day-10 N2 worms showed greater accumulation of the insolublome than day-2 young worms.
Introduction
Protein homeostasis progressively declines with aging and results in increased protein aggregation1,2,3. Protein aggregation is associated with several neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis4. Aging is considered a principal risk factor for the onset of neurodegenerative disorders associated with protein aggregation. Proteins that are prone to form insoluble aggregates are often linked with cellular toxicity and tissue dysfunction, which might further accelerate aggregation of other proteins5,6,7. Alternatively, insoluble protein aggregates may activate cellular defense mechanisms to remove the toxic oligomeric forms of the protein from the system. Knocking down selected genes encoding insoluble proteins modulates the lifespan of Caenorhabditis elegans (C. elegans) in the context of both age-related disease and normal aging5,8,9. Thus, studying the cellular and molecular mechanisms of protein aggregation is crucial to understanding aging and ultimately may lead to approaches for treating neurodegenerative diseases.
The nematode C. elegans has become one of the most extensively used model organism to study protein aggregation in aging and age-related diseases due to its unique characteristics, such as a relative short lifespan (around 2 weeks), ease of cultivation and genetic manipulation.
The ability to extract and characterize insoluble proteins has played a critical role in determining the age-related changes associated with protein aggregation in C. elegans models. To investigate the contribution of protein aggregation to normal aging processes, we5 and others2 previously extracted and proteolytically digested the insolublome of young versus aged C. elegans, chemically labeled using iTRAQ reagents (‘isobaric tagging for relative and absolute quantification’), and then quantified using MS-based methods. Using an isobaric labeling method and 120 mg of wet worms (about 40,000 worms), we were able to gain significant protein insolublome depth and coverage5. Quantitative analysis demonstrated that 203 out of 1200 proteins identified were significantly enriched in the insolublome of aged C. elegans compared to similar insolublome fractions from young worms5. Independently, David et al. also utilized an iTRAQ LC-MS/MS workflow to examine alterations in protein aggregates with normal aging2. Starting with about 300 mg of worms, they identified ~1000 insoluble proteins using two biological replicates and determined that ~700 out of about 1000 proteins accumulated by 1.5-fold or more with age compared to young worms2. Overall, these independent results indicate that widespread protein insolubility and aggregation are an inherent part of normal aging and may affect both lifespan and the incidence of neurodegenerative disease2,5.
Studying the insolublome had allowed us to determine how environmental influences can accelerate or decelerate the aging process. Klang et al. established label-free proteomic workflows in C. elegans to investigate the role of metallostasis in longevity10. In this study, at least 40,000 worms were used to extract the insolublome10. Data showed that iron, copper, calcium and manganese levels increase with aging and that feeding worms a diet with elevated iron significantly accelerated the age-related accumulation of insoluble proteins10. Using the same workflow to examine the effects of vitamin D on the insolublome of C. elegans, 38 proteins were quantified in young worms (Day 2) and 721 proteins in aged worms (Day 8). Vitamin D feeding significantly reduced the insolublome of aged worms from 721 to 371 proteins11. Further investigation revealed that feeding vitamin D suppressed protein insolubility with age, promoted protein homeostasis, and extended the lifespan in C.elegans N2 wild type worms11. Thus, studying the insolublome can help identify novel modulators of aging and age-related diseases.
While studying the insolublome has been invaluable in progressing the understanding of the aging process, it has been hindered by the requirement for collecting large amounts of starting sample material. Groh et al. recently introduced a label-free proteomic quantification workflow to study inherent protein aggregation changes in C. elegans with aging; however, it required large amounts of starting material (350 mg of ground worms)12. In the present report, we established an improved new extraction and isolation protocol (Figure 1). Use of the highly efficient sonicator during worm lysis significantly improved the extraction efficiency and subsequently reduced the amount of starting material needed, from 40,000 to 3,000 worms. Combining this novel insolublome isolation protocol with a label-free data-independent acquisition (DIA) mass spectrometric workflow significantly improved protein depth and coverage. The protocol presented here is cost-effective and easily modified to allow the performance of insolublome analyses in other model systems.
Protocol
NOTE: For a better understanding of the experimental procedure, see Figure 1 for a schematic of the workflow.
1. Mass culture of synchronized aging C. elegans
- Plate preparation
- Pour 30 mL of NGM agar with or without 50 mM fluorodeoxyuridine (FUdR) into each 90 mm plate and let the plates dry for 2 days in culture hoods.
- On the day before seeding the NGM agar plate, inoculate a 50 µL aliquot of frozen glycerol stock of the OP50 bacterial strain into 1 L of pre-autoclaved LB medium in a 2 L flask.
- Grow the bacteria at 37 °C in a shaking incubator at 250 rpm for 16 h.
- After 16 h, centrifuge the bacterial cultures at a speed of 5,000 x g for 10 min.
- Discard the supernatant and re-suspend the bacterial pellet completely in 40 mL of autoclaved S-basal solution (containing 50 mM K3PO4, 100 mM NaCl in water) to generate the 25x concentrated OP50 E. coli culture.
- Dispense 2 mL of the 25x concentrated bacterial culture onto each NGM agar plate and spread evenly so that the bacterial culture covers the entire agar surface.
- Remove the lids from the plates in a bacterial culture hood for 2–3 h until the plates are properly dried.
NOTE: Make sure to evenly distribute the bacteria by swirling the plates every 15 min. This is important as bacteria tend to concentrate on one side of the plate resulting in regions with low bacterial density. - After the plates are dried, close the lids and leave the plates at room temperature for 48 h to ensure complete drying of the OP50 E. coli food source.
NOTE: Plates can be prepared 2 weeks in advance and stored at 4 °C before use.
- Maintenance and preparation of synchronized cohorts of C. elegans
- To obtain the required input of protein lysate (1.0–1.5 mg) for isolation of the SDS-insoluble fraction, use approximately 3,000 synchronized worms per sample.
- Transfer ~100 N2 wild-type gravid adult worms to a NGM 25x OP50 E. coli-seeded plate and maintain the worms at 20 °C for ~72 h until the plate is full with gravid adult worms.
- Pipette ~10 mL of S-basal solution onto each plate and collect all the worms into a 15 mL tube.
NOTE: It is important to use low-retention tips or Pasteur pipettes during this and all future steps involving worm collection to prevent any loss of worms due to attachment to tip surfaces. - Spin the sample at 520 x g for 30 s. Remove the supernatant after spinning. To eliminate any OP50 E. coli, add 10 mL of S-basal solution to the pellet and spin again followed by removal of the supernatant. Repeat this step.
- Add 10 mL of sodium hypochlorite bleaching solution (containing 0.5 M KOH and 0.48% sodium hypochlorite in water) to the worm pellet in a 15 mL conical tube and vortex vigorously for 2 min at room temperature.
- Centrifuge at 520 x g for 30 s at room temperature to spin down any incompletely dissolved worm bodies and eggs. Then remove the supernatant.
- Repeat steps 1.2.5 and 1.2.6 to ensure complete removal of all worm bodies and release of eggs (view tube under microscope to assure).
- Wash the egg pellet at least 4 times with 10 mL of S-basal solution to ensure complete removal of bleach solution.
NOTE: Complete removal of bleach is important as remaining bleach may hinder the hatching of eggs. - Leave eggs resuspended in 5 mL of S-basal media in a 15 mL tube and place in rotor at 20 °C for 24 h to allow eggs to hatch.
- After 24 h, take 10 µL of S-basal larvae solution to count the number of worms in it. Use at least three separate aliquots of 10 µL of S-basal larvae solution for replication of the worm count. Calculate the average of the three larval concentrations. Based on the average concentration, dilute the worm solution to obtain ~3,000 larvae per 200 µL of solution.
NOTE: Make sure to shake the tube gently by inverting as the larvae tend to settle if left undisturbed and that can affect the larva count. - Mix well and add 200 µL of S-basal larvae solution onto each of the 90-mm NGM bacteria-seeded plates.
NOTE: Add drops of S-basal larvae solution throughout the plate for even distribution of larvae to avoid bacterial depletion from certain spots. - Allow the S-basal solution to dry and then place plates inverted in a 20 °C incubator for 48 h.
- After 48 h, collect the worms from the plate in S-basal solution and transfer them to a fresh NGM-seeded plate containing 50 mM FUDR and incubate plates at 20 °C.
NOTE: It is important to transfer worms early in the L4 stage to ensure FUDR efficiency. - Collect day-2 young adult samples after 48 h by adding 10 mL of S-basal solution onto the plate and transferring the solution into a 15 mL tube. Allow worms to settle by gravity and remove the S-basal solution after it becomes clear.
- Wash the worm pellet 2-3x in 5 mL of S-basal solution to remove any attached bacteria. Remove as much S-basal solution as possible and freeze the worm pellet (in 15 mL tubes) in a dry ice/ethanol bath. Frozen samples are stored at -80 °C.
- Transfer sample worms to a fresh NGM OP50 E. coli plate containing FUDR every alternate day until day 10. Collect and freeze worms as described in steps 1.2.14 and 1.2.15.
NOTE: Start with a higher number of worms for day-10 samples to account for any loss in worm number due to transfer and burrowing.
2. Extraction of SDS-insoluble fraction from worms
- Thaw the frozen worm pellet on ice in the presence of 900 µL of ice-cold worm lysis buffer cocktail containing 20 mM Tris base, pH 7.4, 100 mM NaCl, 1 mM MgCl2 and EDTA-free protease inhibitor. Briefly vortex samples to ensure complete resuspension of worm pellet in the lysis buffer.
- Place the 15 mL tubes in the sonication bath and set for 10 sonication cycles (30 s on and 30 s off for each cycle) at high intensity. Repeat for up to five cycles (make sure worms are fully lysed).
- Spin the lysate at 3,000 x g for 4 min in the cold room. After centrifugation, discard the pellet containing any worm debris and carefully transfer the supernatant to fresh pre-cooled 1.5 mL centrifuge tubes on ice.
- Quantify the protein concentration using the BCA assay. Transfer lysate aliquots, typically containing about 1 mg of protein, into fresh pre-cooled 1.5 mL centrifuge tubes.
- Centrifuge the protein lysate for 15 min at 20,000 x g in the cold room.
- Without disturbing the pellet, transfer the supernatant into a fresh 1.5 mL centrifuge tube and save it as the aqueous-soluble protein fraction.
- Wash the pellet from the previous step in 500 µL of worm lysis buffer containing 1% SDS at room temperature and centrifuge at 20,000 x g for 15 min at room temperature. Remove the supernatant and save as the SDS-soluble fraction. Repeat this washing step two times to remove any SDS-soluble fraction. The remaining pellet after the third round of washing is defined as the 1% SDS-insoluble protein fraction.
- Resuspend the SDS-insoluble protein pellet in 60 µL of 70% formic acid and vortex vigorously to dissolve the proteins. Repeat vigorous vortexing as many times as necessary to dissolve the pellet.
- Sonicate the pellet for 30 min in an ultrasonicator water bath at room temperature.
NOTE: The SDS- insoluble pellet is typically hard to dissolve in the buffer. However, in the end the entire pellet should be dissolved in this step. - Dry the samples in a vacuum concentrator for 1 h to completely remove the formic acid solution.
- Add 40 µL of 1x LDS sample gel buffer to the dried pellet and heat the sample to 95 °C for 10 min. Briefly vortex and spin down the samples. Load 13 µL onto a 4-12% NUPAGE Bis-Tris gel and run the gel. Stain the gel with a fluorescent protein stain for imaging. Save the rest of the sample for mass spectrometry (MS) analysis.
- Load the remaining sample into a 4-12% Bis-Tris gel and run for about 20 min for MS in-gel digestion.
3. In-gel digestion with trypsin protease to isolate proteins for MS analysis
- Prepare the following solutions (fresh): 25 mM NH4HCO3 (pH 7–8), 25 mM NH4HCO3 in 50% acetonitrile (ACN, pH 7–8), and 50% ACN in 5% formic acid.
- Dice each gel slice into small pieces (typically <1 mm2) and place these into 0.65 mL siliconized tubes.
- Add about 100 µL (or enough to cover) of a 25 mM NH4HCO3/50% ACN solution and vortex at room temp for 10 min. Extract the supernatant and transfer to a separate tube (to be discarded). Repeat this step two more times.
- Dry the gel pieces completely in a vacuum concentrator (~20 min).
- Prepare fresh solutions and add ~100 µL (or enough to cover) of 10 mM DTT in 25 mM NH4HCO3 to the dried gel pieces (use freshly prepared NH4HCO3). Vortex and briefly spin. Allow the reaction to proceed at 56 °C for 1 h and with shaking at 1400 rpm on a mixer.
- Remove the supernatant and add 100 µL of 55 mM iodoacetamide (IAA) in 25 mM NH4HCO3 to the gel pieces. Vortex and briefly spin. Allow reaction to proceed in the dark for 45 min at RT.
- Remove the supernatant and discard. Wash the gel pieces by adding ~100 µL of 25mM NH4HCO3 and vortexing for 10 min. Briefly spin and remove the supernatant, discarding the latter.
- Dehydrate the gel pieces by adding ~100 µL (or enough to cover) of 25 mM NH4HCO3 in 50% ACN to the gel pieces and vortex for 10 min. Then briefly spin to remove and discard the supernatant. Repeat this step twice or even a third time if the gel pieces are not quite dry as indicated by smaller size of the gel pieces and cloudy white color.
- Dry the gel pieces completely in a vacuum concentrator (~20 min).
- Initially add 15 µL of trypsin solution (250 ng trypsin) to each sample, and then add enough 25 mM NH4HCO3 to cover the gel pieces (~100 µL). Vortex for 10 min. Then briefly spin and incubate at 4 °C in the cold room for 30 min without mixing.
- Add 25 mM NH4HCO3 as needed to completely cover the gel pieces. Spin and incubate at 37 °C overnight for 16–20 h at 1400 rpm on a mixer.
- The next day, briefly vortex and spin the digestion. Add about 100 µL of HPLC grade water, spin, parafilm the tube and sonicate continuously for 10 min. Spin briefly after sonication.
- Transfer the digestion solution, representing the aqueous extraction, into a clean 0.65 mL siliconized tube.
- Add ~100 µL of 50% ACN/5% formic acid to the gel pieces (enough to cover them), vortex 10 min at room temperature, spin briefly and collect the solution, and repeat once. Pool all solutions containing the extracted peptides from this step and the previous step 3.13 together into a single tube.
- Vortex the extracted digestions. Dry the peptides completely in a vacuum concentrator (~2 h).
- Add 30 µL of 0.2% formic acid to resuspend the peptides on a mixer in the cold room for 10 min.
- Spin the samples at 1850 x g at room temperature for 5 min. Aspirate the peptide solution and move into a new clean 0.65 mL siliconized tube. Then desalt the peptides solution using C18 desalting tips (see below).
4. Desalting digested peptides with C18 desalting tip
- Set the pipette to 10 µL and attach the C18 desalting tip. Wet the desalting tip by pipetting up 10 µL of 100% ACN and then discarding it. Repeat this step 2x.
- Wash the C18 desalting tip by pipetting up 10 µL of 50% ACN, 49.8% water, and 0.2% FA and then discarding it. Repeat this step 2x.
- Equilibrate the C18 desalting tip by pipetting up 10 µL of 0.2% FA in water and then discarding it. Repeat this step 2x.
- Set the pipet to 10 µL and load the peptides from the solution to the resin by pipetting the digested peptides through the resin up and down 15x.
NOTE: The repeated action will ensure binding of all peptides to the resin of the desalting tips. - Desalt the digested peptides bound to the resin by pipetting up 10 µL of 0.2% FA in water and then discarding it. Repeat this step 4x to complete the desalting step.
- In a new tube, elute the peptides with 10 µL of 50% ACN, 49.8% water, and 0.2% FA by pipetting up and down 10x. Repeat this step once to elute the protein digest for a second time in the same tube.
- Dry the desalted peptides completely in a vacuum concentrator (~20 min).
- Resuspend peptides in 15 µL of 0.2% FA + 1 µL iRT (indexed Retention Time) peptides. Voxtex for 10 min, centrifuge at 12,000 x g for 2 min, and then transfer to an autosampler vial for MS analysis (see below).
5. Mass spectrometry analysis of digested peptides using DDA and DIA
NOTE: Samples can be analyzed using either DDA or DIA LC-MS/MS methods. In this study, the samples were analyzed using a nano-LC 2D HPLC system coupled to a high-resolution mass spectrometer.
- Use a HPLC system combined with a chip-based HPLC system directly connected to a quadrupole time-of-flight mass spectrometer (other LC-MS configurations and systems can also be used).
- Analyze samples using reverse-phase HPLC-ESI-MS/MS.
- Build the chromatography protocol to load peptides sample onto a C18 pre-column chip. Wash and desalt the loaded peptides with loading solvent (0.1% formic acid) for 10 min at a flow rate of 2 µL/min.
- Transfer the peptides to an analytical column C18 chip and elute at a flow rate of 300 nL/min with a 3 hour gradient using mobile phase A (2% acetonitrile, 0.1% formic acid) and B (98% acetonitrile, 0.1% formic acid). The first elution step consists of a linear gradient from 5% B to 35% B over a period of 80 min.
- Ramp the mobile phase B to 80% over 5 min, and then maintain the mobile phase B at 80% for 8 min before changing into 5% B to re-equilibrate the column for 25 min.
- Set up a MS instrument method for DDA and define the instrument parameters as following.
- Use the following parameters for Experiment 1:MS1 precursor ion scan from m/z 400–1500 (accumulation time of 250 ms); set the intensity threshold to trigger MS/MS scans for ions of charge states 2–5 to 200 counts; set the dynamic exclusion of precursor ions to 60 s).
- Use the following parameters for Experiment 2: MS/MS product ion scans with a MS2 scan range from m/z 100–1500 (accumulation time of 100 ms per each of the 30 product ion scans per cycle); set the collision energy spread to CES=5, and checkmark ‘high sensitivity product ion scan mode’.
NOTE: The DDA acquisition method is used to build spectral libraries as described in step 6.3 (see below). Here, it will acquire MS/MS spectra for the 30 most abundant precursor ions after each MS1 scan per cycle; the total cycle time is about 3.3 s.
- Obtain DIA. Set up a MS instrument method for DIA and define the instrumental parameters as follows.
- Use the following parameters for Experiment 1: MS1 precursor ion scan from m/z 400-1250 (accumulation time of 250 ms).
- Use the following parameters for Experiment 2: MS/MS product ion scans for 64 variable SWATH segments with a MS2 scan range from m/z 100-1500 (accumulation time of 45 msec per each of the 64 product ion scans per cycle); set the collision energy spread to CES=10, checkmark ‘high sensitivity product ion scan mode’.
NOTE: Use the 64-variable window DIA/SWATH acquisition method as described by Schilling et al.13 to perform label-free quantification with a total cycle time of about 3.2 s. For DDA acquisition, instead of using the Q1 quadrupole to transmit precursor ion of a narrow mass range to the collision cell, a range of variable window width (for example m/z 5–90) is used to incrementally step over the full m/z range (m/z 400–1250) with 64 SWATH segments, 45 ms accumulation time for each segment, resulting in a cycle time of 3.2 sec, which includes one MS1 scan with 250 ms accumulation time. The variable window width is adjusted according to the complexity of the typical MS1 ion current observed within a certain m/z range using a SCIEX ‘variable window calculator’ algorithm13 (narrower windows are applied to ‘busy’ m/z ranges, wider windows are applied to m/z ranges with few eluting precursor ions). On other MS platforms, alternative DIA window selection strategies can also be implemented.
6. Data analysis
NOTE: Certain data analysis settings should be tailored to specific experimental conditions. For example, the protein database (fasta file) selected will depend on the species that the sample was prepared from (in this protocol we used C. elegans).
- Use a MS database search engine to analyze DDA acquisitions and identify proteins. Generate a darabase search engine method as follows:
- For Sample Description Parameters, select Identification; under Sample Type, select Iodoacetic Acid; under Cysteine Alkylation, select Trypsin; under Digestion (assuming C-terminal cleavage at lysine and arginine), select the name of the mass spectrometer under Instrument; and select Caenorhabditis elegans under Species.
- For Specific Processing parameters, check Biological modifications; under ID Focus, select SwissProt; under Database, check Thorough ID; under Search Effort, select 0.05 (10%) under Detected Protein Threshold; and check Run False Discovery Rate Analysis under Results Quality. Save the search engine method and submit the MS raw files to the database search engine for processing, using the generated method.
NOTE: In an iterative process, all MS and MS/MS scans were automatically recalibrated by the search engine based on initial annotations and results.
- Click on Export Peptide Summary upon completion of the search and filter all peptide identification results by the confidence threshold of 99 in Excel (false discovery rate (FDR) = 1%).
- Build MS/MS spectral libraries for further processing of the DIA raw data file, and for further relative data quantification.
- Open the DIA Quantitative Analysis Software. Select Library tab and then (at the bottom of the page) click Generate Spectral Library from “Database Search Engine” and open a “Database Search Engine” FDR report (the *FDR.xlsx spreadsheet file) that was automatically generated as part of the DDA raw data file database search process.
- Click next, select the Library Settings Schema and click Next. Select Uniprot_Caenorhabditis elegans _proteome as the database then click Finish, the spectral library will be generated.
- Use the DIA Quantitative Analysis Software to analyze DIA acquisitions for comprehensive relative protein quantification. DIA data analysis pipeline is described as follows.
- To analyze and quantify peptides, open the DIA Quantitative Analysis Software to set up an analysis schema by using the template schema. The template schema is available in the software by clicking Settings, then DIA Analysis, and finally BGS Factory Settings.
- Set up the parameters in the template schema as the following:
1) under identification select PTM localization (probability cutoff = 0.75).
2) under quantification, select Major Group Quantity as Sum peptide quantity, set up Major Group N as Max 7 and Min 1, select Minor Group Quantity as Sum precursor quantity, and set up Minor Group N as Max 10 and Min 1, and selected Data Filtering as Qvalue sparse and do not select Cross Run Normalization.
- Perform Relative Quantification Analysis.
- Select the Pipeline tab, click Set up a DIA Analysis from File, open the MS DIA raw files of interest (for example: sample and control), click Assign Spectral Library, select the library that was generated in 6.3, click Load, and lastly click Next.
- Select the analysis schema set up in 6.5.1 and click Next. Select the appropriate database fasta file (in this protocol: Uniprot_Caenorhabditis elegans_proteome) and click Next. Define the condition setup, and assign the different conditions according to the samples and click Next.
- Review the analysis overview (summary of the experiment set-up) and browse to the Output Directory and click Finish. Finally click Run Pipeline to perform the label-free quantitative analysis.
NOTE: Statistical modules in the ‘DIA Quantitative Analysis Software’ automatically perform FDR analysis, generate heat maps and volcano plots comparing the different conditions, generate lists of identified and quantified peptides and proteins and provide Q-values along with relative fold-changes comparing different conditions. Here sample conditions set-up included Day 10 aged worms with two biological replicates and Day 2 young worms with two biological replicates as control.
Results
Traditional worm lysis methods have various disadvantages. For example, probe-based sonication and bead-beater methods produce excessive heat by allowing contact of the metal tip or beads directly with the samples, resulting in variable protein recoveries and protein denaturization. Liquid nitrogen grinding followed by sonication in lysis buffer, can be time-consuming and requires a large number of worms. Due to the limitations of traditional worm lysis methods, previous MS workflows, such as the labeling methods iTRAQ or label-free methods that have been historically used in the C. elegans model system to gain quantitative information about the insolublome, require large input of starting material (at least 40,000 worms). Laborious worm culture work is required to obtain these numbers of worms. Moreover, the labelling methods require expensive isobaric chemical labels. Label-free quantification methods are cost-effective and have easier and more straightforward sample preparation and labeling methods, but require significantly large numbers of worms to achieve sufficient MS analysis coverage.
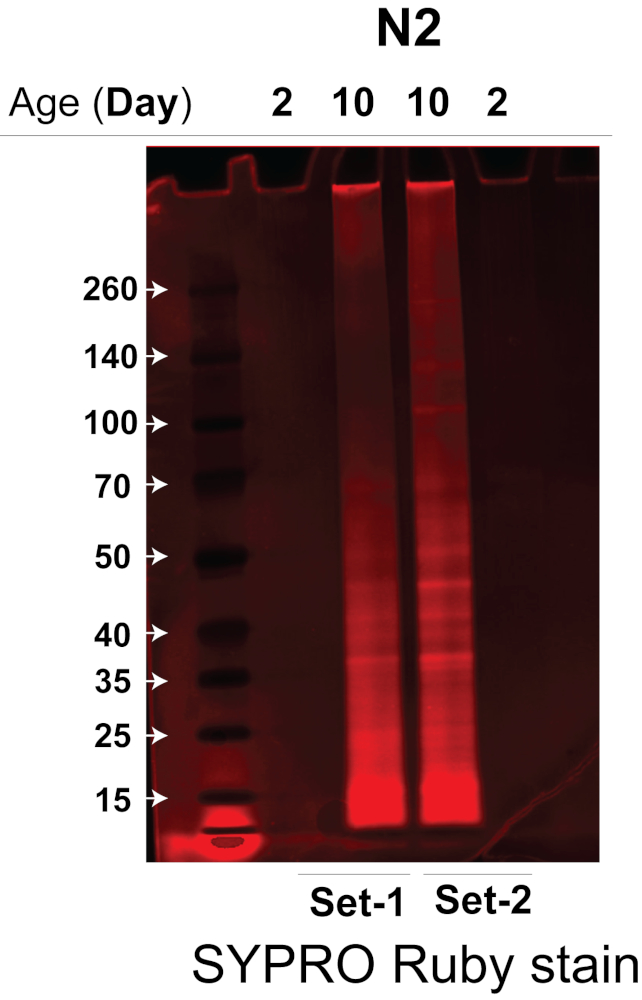
The sonicator that we used greatly increases the efficiency and reproducibility of worm lysis by lysing multiple worm samples simultaneously in a temperature-controlled water bath sonicator without cross-contamination14, thus significantly reducing the amount of starting worm material required. Combining the highly efficient sonication method and the quantitative DIA label-free MS approach, we were able to robustly quantify the insolublome of aged and young worms using ~3,000 worms. Here we tested and validated the efficiency of the protocol and compared the insolublome of aged and young worms from a wild-type worm strain, N2-Bristol C. elegans. We applied this protocol to extract and isolate the insolublome from ~3,000 aged and young N2 C. elegans (two biological replicates for each condition), followed by MS analysis with a quadrupole time-of-flight mass spectrometer or other MS systems using a combination of data-dependent acquisition (DDA) and data-independent acquisitions (DIA/SWATH) for protein identification and quantification. The insoluble proteins were first analyzed on a Bis-Tris 4-12% gradient gel to determine the amount of protein in each insolublome sample. As demonstrated in Figure 2, the insolublome sample from N2 aged worms (lanes 2 and 3, biological replicate experiments) has significantly more protein than samples from the N2 young worms (lanes 1 and 4, biological replicate experiments).
After in-gel digestion, the protein profiles of the insolublome were analyzed by HPLC-MS. Using this workflow, we can generally identify 1000–1500 proteins and quantify 500–1,000 proteins from the SDS-insoluble fraction with high reproducibility (unpublished data). Here we were able to quantify 989 proteins from the insolublome of N2-Bristol C. elegans by analyzing the DIA data and removing redundancy: 768 proteins were significantly enriched and 27 proteins were significantly decreased in the insolublome of aged N2 worm (Day 10) compared with young (day 2) using a fold-change of at least 1.5 and a Q value of less than 0.01 (Figure 3A). As seen on the histogram plot (Figure 3B), the fold-change of significantly altered proteins shows a normal distribution. Aged worms were demonstrated to be significantly enriched for the insolublome: The largest change observed showed the relative protein abundance in the insolublome to be 592 times higher in the old versus young worms; and for 32 proteins the relative protein abundance in the insolublome was >250 times higher in the old versus young worms indicating dramatic insolublome changes with age.
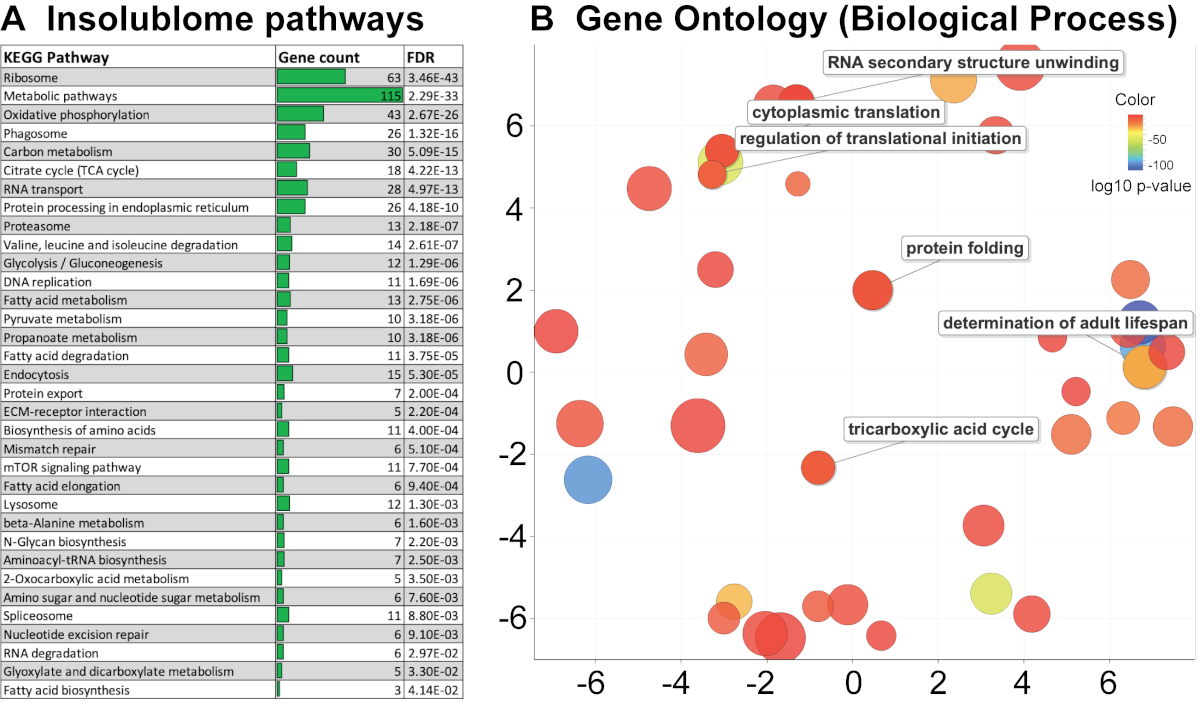
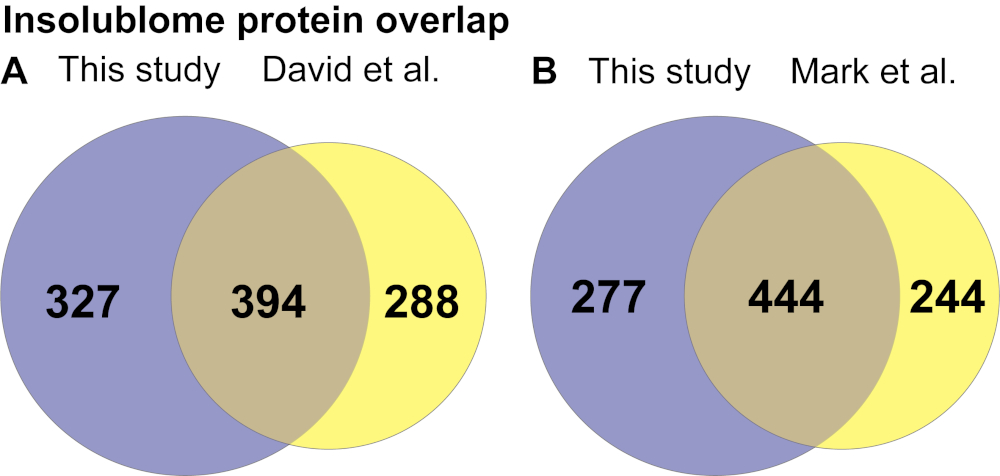
After extracting the list of insoluble proteins that are significantly increased in the aged worms and identified by the wormbase (WS271), KEGG pathway and Gene Ontology (GO) analysis were performed to determine the pathways that are enriched in the aged insolublome to gain biological insights into how these relate to aging. The KEGG pathway analysis of proteins identified in this study shows enrichment of several pathways involving ribosomes, mitochondria, proteasome and spliceosome (Figure 4A). The gene ontology analysis shows that the insolublome from aged worms comprises many proteins in particular categories including mitochondrial, developmental, determinants of adult lifespan, and ribosomal proteins (Figure 4B and Supplementary Table 1A). We then compared the list of proteins identified in this study with previously published work from David et al.2 and Mark et al.11 as demonstrated in Venn diagrams (Figure 5A,5B). The comparison showed significant overlap of identified proteins 394/721 and 444/721 with David et al (Figure 5A) and Mark et al. (Figure 5B) study, respectively. The biological pathways revealed by the KEGG analysis of insolublome from this study have also been identified in the past thus validating our methodology (Supplementary Table 1B). Identification of these pathways and proteins suggests that they may serve as candidates for further biological investigation in regards to their function in the context of aging.
In summary, the use of the efficient sonication method enables the lysis of multiple worm samples at the same time in an environment with well-controlled temperatures and reduced cross-contamination to achieve high protein coverage with significantly less starting worm material. Combining the efficient sonication method with a DIA label-free protein quantification workflow has provided reliable and reproducible results for the quantification of insoluble worm proteins.

Figure 1. Experimental workflow of the protocol. C. elegans were cultured and collected on different days. After worm lysis with a sonicator, the 1% SDS-insoluble protein fraction (insolublome) was extracted and isolated from the lysate. The insolublome was then digested via in-gel trypsin digestion and quantified via DIA mass spectrometry, followed by bioinformatic analysis. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. SDS-PAGE gel of the insolublome isolated young versus old worms of the N2-Bristol strain. The insolublomes of young versus old worms of the N2-Bristol strain were analyzed by SDS-PAGE to determine the amount of protein present. The SDS-PAGE gel was stained with a fluorescent protein stain to visualize protein bands. Lanes 1 and 4: Insolublome from two biological replicate experiments of young N2 worms (Day 2). Lanes 2 and 3: Insolublome from two biological replicate experiments of aged N2 worms (Day 10). Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Protein candidates identified as showing significant alteration in the aged versus young insolublome and their fold-change distributions. (A) Volcano plot for quantification of the insolublome of aged versus young N2-Bristol worms. Candidates with an absolute fold change >=1.5 and Q value <0.01 are shown as red dots. (B) Histogram plot for fold-change distribution of significantly enriched SDS-insoluble proteins in aged versus young worm samples. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. KEGG pathway and Gene Ontology (GO) analysis. (A) KEGG pathway analysis of the day-10 insolublome arranged according to p-value with highly significant pathway shown at the top. (B) Gene Ontology analysis shows that insolublome of aged worms is enriched for many proteins in particular categories including mitochondrial, developmental, determinants of adult lifespan, and ribosomal proteins. The scatterplot view visualizes the GO terms in a “semantic space” where the more similar terms are positioned closer together. The color of the bubble reflects the p-value obtained in the STRING analysis, while its size reflects the generality of the GO term in the UniProt-GOA database. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Insolublome protein overlap identified in day-10 insolublome comparing this study with (A) David et al.2 and (B) Mark et al.11 studies. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1 (related to Figure 4 and Figure 5). (A) Gene ontology (biological process) analyzed with STRING database. (B) Detailed list of proteins and KEGG pathways identified in this study with color codes depicting their overlap with the published work. Please click here to download this file.
Discussion
In this protocol, we report an improved sample preparation method for the extraction of insoluble proteins from C. elegans. By replacing traditional worm lysis (e.g., probe sonication or bead beater techniques) with the efficient sonicator, we increased the yield of the insoluble protein extraction and reduced the number of worms needed for label-free MS analysis from 40,000 worms to 3,000 worms. A database search engine was used for protein identification from DDA data and the C. elegans spectral library was built using the ‘DIA Quantitative Analysis Software’ and the corresponding DDA database search results (importing FDR file reports generated by the database search engine). Relative quantification of aged and young C. elegans insolublome data was carried out using the ‘DIA Quantitative Analysis Software’ to process the novel DIA dataset and the generated spectral library.
Several steps are critical in the protocol. The short lifespan of C. elegans makes it an ideal system to study aging, compared to other eukaryotes such as mammalian cells, but it is crucial to isolate a homogenous population of worms when studying aging-related phenomenon. FUDR was used in this protocol to obtain synchronized aging worms. It is important to transfer worms early in their L4 stage onto the NGM-seeded plate containing FUDR to ensure its effectiveness. During worm lysis using the efficient sonicator, water bath temperature must be set at 4 °C and sonication at 30 s ON and 30 s OFF to prevent overheating of the samples. After the first round of sonication for 10 cycles (10 min), it is important to check under the microscope to assure that all worms were efficiently lysed. If not, more cycles of sonication are needed. During the process of in-gel digestion, each gel slice must be diced into pieces of the proper size (<1 mm2)—if too small, they may be lost in the sample preparation process, and if too large, the digestion may be inadequate.
The need for much less starting material significantly reduces the laborious work associated with worm culture to obtain samples for insolubolome analyses. However, extraction and isolation of the 1% SDS-insoluble protein fraction involves multiple washing steps and careful sample handling is required to avoid sample loss and to ensure reproducible results. The amount of material generated for MS analysis is sufficient for ~3 injections for subsequent DDA and DIA analysis but not to save for future experiments. Furthermore, despite its potentially confounding effects15, we used the lowest possible concentration of FUdR to sterilize worms during the aging process. Future studies may circumvent the use of FUdR by using sterile mutants or by manually transferring and collecting worms.
Use of the highly efficient sonicator for worm lysis allows efficient extraction of the insolublome allowing good protein coverage and cost-effective label-free DIA MS analysis to quantify the insolublome using a greatly reduced number of worms. It significantly reduces the workload allowing for the screening for more conditions per experiment. In addition, the label-free MS DIA workflow is cost-effective and provides protein depth and coverage at comparable levels to labeling methods including iTRAQ, TMT or SILAC. The C. elegans model is a fast screening system for aging research. The workflow can be easily modified and applied to study aging and age-related disease research in this and other organisms. For example, in ongoing studies we are applying this workflow to investigate protein profiles of insolublome and proteostasis in various Alzheimer’s disease (AD) C. elegans models including Abeta, tau, and dual Abeta/tau worms with or without different drug interventions for future high-throughput drug screening.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by a NIH shared instrumentation grant for a TripleTOF system (1S10 OD016281, Buck Institute), NIH grant, RF1 AG057358 (GJL, JKA) and NIH grant U01AG045844 (GJL). XX is supported by a T32 postdoctoral fellowship (NIH grant 5T32AG000266, PI: Judith Campisi and Lisa Ellerby). MC is supported by a postdoctoral fellowship from the Larry L. Hillblom Foundation.
Materials
| Name | Company | Catalog Number | Comments |
| Strains used | |||
| Esherichia coli OP50 | Caenorhabditis Genetics Center (CGC) | ||
| N2 (Bristol) | Caenorhabditis Genetics Center (CGC) | ||
| Buffer/Solution | |||
| NGM (Nematode Growth Media) | Recipe: 3 g/L NaCl, 23 g/L agar; 2.5 g/L peptone; 1 mM CaCl2, 5 mg/L cholesterol, 1 mM MgSO4, 25 mM KH2PO4 | ||
| S-basal solution | Recipe: 5.85 g/L NaCl, 1g/L K2HPO4, 6 g/L KH2PO4, H2O to 1 L | ||
| Sodium hypochlorite bleach solution | Recipe: Mix 0.5 mL 5 N NaOH with 1 ml Sodium hypochlorite (5%) and make volume to 5 mL with H20. | ||
| Material/ Equipment | |||
| Agar | Difco Granulated Agar, BD Biosciences | 90000-782 | |
| Bioruptor Plus sonication device | Diagenode, USA | B01020001 | |
| Cholesterol | Sigma | c8503 | |
| 2'-deoxy-5-fluorouridine | VWR | TCD2235 | |
| Glycerol | Millipore Sigma | 356350-1000ML | |
| LB broth, Miller | Millipore Sigma | 60801-450 | |
| Sodium dodecyl sulfate (SDS? | Sigma | L4509-250G | |
| Sodium chloride | Sigma | 59888 | |
| M880 Ultrasonic bath, 117 V, holds 5.5 gallons | VWR, USA | 89375-458 | |
| Magnesium sulphate | Sigma | M506 | |
| Magnesium chloride | Sigma | 208337 | |
| NGM agar plate | VWR Disposable Petri Dishes | 25384-342 | |
| NuPAGE LDS Sample Buffer (4X) | Thermo Fisher Scientific | NP0007 | |
| NuPAGE protein gels, 4-12% | Invitrogen | NP 0335BOX | |
| Protease inhibiotr cocktail (PIC) | Roche | 11836170001 | |
| Pierce BCA Assay | Thermo Fisher Scientific | 23225 | |
| Sodium hypochlorite 5% | VWR | JT9416-1 | |
| SYPRO Ruby Protein Gel Stain | Thermo Fisher Scientific | S12000 | |
| MS Section | |||
| Acetonitrile, Burdick and Jackson LC-MS | Honeywell International Inc., Charlotte, NC, USA | 36XL66 | |
| Agilent Zorbax 300Extend C18 column | Agilent Technologies Inc., Santa Clara, CA, USA | 770995-902 | |
| Ammonium bicarbonate | Sigma Aldrich, St. Louis, MO, USA | 9830 (1 kg) | |
| Dithiothreitol (DTT) | Sigma Aldrich, St. Louis, MO, USA | D9779-5G | |
| Eppendorf Thermomixer Compact | Eppendorf AG, Hamburg, Germany | T1317-1EA | |
| Formic acid | Sigma Aldrich, St. Louis, MO, USA | F0507-500ML | |
| Indexed retention time (iRT) normalization peptide standard | Biognosys AG, Schlieren, Zurich, Switzerland | Ki-3002-2 | |
| Iodoacetamide (IAA) | Sigma Aldrich, St. Louis, MO, USA | I1149-25G | |
| Methanol, HPLC Grade | Honeywell International Inc., Charlotte, NC, USA | 34885 | |
| Nano cHiPLC Trap ChromXP C18-CL, 200 um x 6 mm, 3 um, 120A. (pre-column chip) (200 um x 6 mm ChromXP C18-CL chip, 3 um, 300 A) | Sciex LLC, Framingham, MA, USA | 804-00006 | |
| Nano cHiPLC ChromXP 75 um by 15cm, C18-CL, 3 um, 120 A (analytical column chip) | Sciex LLC, Framingham, MA, USA | 804-00001 | |
| Orthoganol quadrupole time-of-flight (QqTOF) TripleTOP 6600 mass spectrometer | Sciex LLC, Framingham, MA, USA | Per quote | |
| ProteinPilot 5.0 | Sciex LLC, Framingham, MA, USA | software download Sciex | |
| Savant SPD131DDA Speedvac Concentrator | Thermo Fisher Scientific, Waltham, MA, USA | SPD131DDA-115 | |
| Sequencing-grade lyophilized trypsin | Life Technologies | 23225 | |
| Spectronaut | Biognosys AG, Schlieren, Zurich, Switzerland | Sw-3001 | |
| SWATH 2.0 plugin into PeakView 2.2 | Sciex LLC, Framingham, MA, USA | software download Sciex | |
| Ultra Plus nano-LC 2D HPLC system | Sciex LLC, Eksigent Division, Framingham, MA, USA | Model # 845 | |
| Water, Burdick and Jackson LC-MS | Honeywell International Inc., Charlotte, NC, USA | 600-30-76 | |
| Waters 1525 binary HPLC pump system | Waters Corp., Milford, MA, USA | WAT022939 | |
| Waters 2487 Dual Wavelength UV detector | Waters Corp., Milford, MA, USA | WAT081110 | |
| Waters 717plus Autosampler | Waters Corp., Milford, MA, USA | WAT022939 | |
| Waters Fraction Collector III | Waters Corp., Milford, MA, USA | 186001878 |
References
- Walther, D. M., et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell. , 919-932 (2015).
- David, D. C., et al. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biology. 8 (8), 47-48 (2010).
- Labbadia, J., Morimoto, R. I. The Biology of Proteostasis in Aging and Disease. Annual Review of Biochemistry. (1), 435-464 (2013).
- Ross, C. A., Poirier, M. A. Protein aggregation and neurodegenerative disease. Nature Medicine. , 10-17 (2004).
- Reis-Rodrigues, P., et al. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell. 11 (1), 120-127 (2012).
- Ayyadevara, S., et al. Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging. 8 (12), 3486-3497 (2016).
- Huang, C., et al. Intrinsically aggregation-prone proteins form amyloid-like aggregates and contribute to tissue aging in Caenorhabditis elegans. eLife. 8, (2019).
- Morimoto, R. I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes and Development. 22 (11), 1427-1438 (2008).
- Morimoto, R. I., Cuervo, A. M. Protein homeostasis and aging: Taking care of proteins from the cradle to the grave. Journals of Gerontology - Series A Biological Sciences and Medical Sciences. 64 (2), 167-170 (2009).
- Klang, I. M., et al. Iron promotes protein insolubility and aging in C. elegans. Aging. 6 (11), 975-991 (2014).
- Mark, K. A., et al. Vitamin D Promotes Protein Homeostasis and Longevity via the Stress Response Pathway Genes skn-1, ire-1, and xbp-1. Cell Reports. 17 (5), 1227-1237 (2016).
- Groh, N., et al. Methods to study changes in inherent protein aggregation with age in caenorhabditis elegans. Journal of Visualized Experiments. (129), 1-12 (2017).
- Schilling, B., Gibson, B. W., Hunter, C. L. Generation of High-Quality SWATH((R)) Acquisition Data for Label-free Quantitative Proteomics Studies Using TripleTOF((R)) Mass Spectrometers. Proteomics: Methods and Protocols, Methods in Molecular Biology. 1550, 223-233 (2017).
- Walther, D. M., et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 161 (4), 919-932 (2015).
- Angeli, S., et al. A DNA synthesis inhibitor is protective against proteotoxic stressors via modulation of fertility pathways in Caenorhabditis elegans. Aging. 5 (10), 759-769 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved