
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
微生物・化学刺激による好中球細胞外トラップの形態・組成解析
要約
ここに提示されるのは、 in vitro 好中球細胞外トラップ(NET)の誘導と分析のためのプロトコルです。DNA、カテリシジン(LL37)、および酵素活性の定量により、同様の制御条件下で微生物および化学刺激によって誘導されるNETの組成および形態の変動性を示すデータが得られました。
要約
好中球は、好中球細胞外トラップ(NET)の形成など、さまざまなメカニズムを採用することにより、自然免疫応答における細胞防御の第一線として機能します。この研究では、ヒト細胞でのNET誘導と特性評価のための標準化された in vitro 方法論を使用して、微生物および化学的刺激によって誘発されるNETの形態学的および組成的変化を分析します。ここで説明する手順により、NET形態(溶解性または非溶解性)および組成(DNA-タンパク質構造および酵素活性)、およびそのような特性に対する可溶性因子または細胞接触の影響の分析が可能になります。さらに、ここで説明する手法を変更して、NET組成に対する外因性可溶性因子または細胞接触の影響を評価することができます。
適用される技術には、二重密度勾配(1.079-1.098 g / mL)を使用したヒト末梢血からの多形核細胞の精製が含まれ、ライト染色、トリパンブルー排除、およびフローサイトメトリー(FSC対SSC分析および7AAD染色を含む)によって実証されるように、最適な純度と生存率(≥95%)を保証します。NET形成は微生物(緑膿菌、黄色ブドウ球菌、カン ジダ・アルビカンス)および化学的刺激(ホルボールミリスチン酸酢酸塩、HOCl)刺激によって誘導され、NETはDNA-DAPI染色、抗菌ペプチドカテリシジン(LL37)の免疫染色、および酵素活性の定量(好中球エラスターゼ、カテプシンG、およびミエロペルオキシダーゼ)によって特徴付けられます。画像は蛍光顕微鏡で取得され、ImageJで分析されます。
概要
好中球は血流中に最も豊富な白血球であり、DNAといくつかの核、細胞質、および粒状の抗菌タンパク質で構成される大きなクロマチン構造の放出を含む、いくつかのメカニズムによる病原体の除去中に重要な役割を果たします1,2。好中球のこの抗菌的役割を記述する直接の前例は、1996年に武井らによってなされた3。これらの著者らは、好中球のアポトーシスおよびネクロトーシスとは異なる新しい形態の死を報告し、核破裂を示す形態学的変化を示し、その後、核質から細胞質にこぼれ、ホルボールミリスチン酸酢酸塩(PMA)との3時間のインキュベーションから膜透過性の増加を示しました2,3。しかし、「好中球細胞外トラップ(NET)」という用語が使用されたのは2004年のことでした4。
NET形成は、細菌、真菌5、ウイルス6、寄生虫感染などのさまざまな条件で観察されており、微生物の拡散を中和、死滅、および防止します7。他の研究は、サイトカイン、尿酸一ナトリウムまたはコレステロール結晶、自己抗体、免疫複合体、および活性化血小板などの滅菌刺激によって非病原性状態でも起こり得ることを示している7。リポ多糖(LPS)、インターロイキン-8(IL-8)、およびPMAは、NET誘導物質として記載された最初のin vitro刺激の1つであり、病原性プロセスへのin vivoNETの関与は、急性炎症の2つのモデル、実験的赤痢と自然発生性ヒト虫垂炎で実証されました4。DNAは不可欠なNETコンポーネントです。NETとその殺菌特性を分解する短時間のデオキシリボヌクレアーゼ(DNase)処理によって実証されるように、その適切な構造と組成は、捕獲された微生物に向かって高濃度の抗菌分子を送達することにより、微生物の隔離と死滅に必要です4。DNAに加えて、NETは、ヒストン、好中球エラスターゼ(NE)、カテプシンG(CG)、プロテイナーゼ3、ラクトフェリン、ゼラチナーゼ、ミエロペルオキシダーゼ(MPO)、およびカチオン性炎症誘発ペプチドカテリシジンLL-37などの抗菌ペプチド(AMP)などの付着タンパク質を含む8,9。そのような凝集体は、最大50nmの直径を有するより大きな糸を形成し得る。これらの要因は、微生物の病原性因子または病原体細胞膜の完全性を破壊する可能性があります。さらに、AMPは、細菌ヌクレアーゼによる分解に対してNET由来のDNAを安定化させることができる10。
NET形成を制御する具体的なメカニズムはまだ完全には解明されていません。NET放出につながる最も特徴のある経路は、NADPHオキシダーゼの活性化と活性酸素種(ROS)産生につながるERKシグナル伝達、およびMPO経路の活性化を引き起こす細胞内カルシウムの増加によるものです。これにより、過酸化水素が次亜塩素酸に変換され、酸化によってNEが活性化されます11,12。NEは、細胞骨格のアクチンフィラメントを分解して食作用をブロックし、それらを核に移動して、顆粒および細胞質タンパク質と会合し、細胞外に放出されるクロマチン線維の脱感作を促進するPAD4によるタンパク質分解切断および脱アミノ化による処理を行います7。これらのプロテアーゼとしては、アズロフィル顆粒のアズロソーム複合体やカテプシンG13などの他のプロテアーゼから放出されるものが挙げられる。
好中球の形態学的変化に応じて、NETは2つのタイプに分類されます:細胞死につながる自殺または溶解性のNET形成4、および貪食能力を備えた核細胞またはミトコンドリアDNAの小胞放出によって媒介される生存細胞によって生成されるバイタルまたは非溶解性のNET形成14,15。一般に、ミトコンドリアDNAからなるNETは細長い繊維14形態を示し、一方核DNAから構成されたものは雲のような外観3を有する。しかしながら、好中球がそのDNA起源をどのように選択するかは知られていない。NETの標準的な経路が数時間かかると説明していた以前の研究とは対照的に、バイタル経路はわずか5〜60分で急速に活性化されます15。
これらの進歩にもかかわらず、NET組成は刺激によって異なります。例えば、緑膿菌の異なるムコイド株および非ムコイド株は、33個の共通タンパク質および最大50個の可変タンパク質を含むNETの形成を誘導する7。したがって、研究グループで客観的な結論を生み出すことを可能にする技術を均質化する必要があります。本稿では、黄色ブドウ球菌(グラム陽性菌)、緑膿菌(グラム陰性菌)、カンジダ・アルビカンス(真菌)、および健常人のヒト好中球の化学刺激(PMA、HOCl)など、さまざまな微生物で誘導されたNETの組成、構造、形態を比較および評価できるさまざまな手法によるプロトコルについて説明します。代表的な結果は、DNA-DAPI染色、LL37の免疫染色、および酵素活性(NE、CG、およびMPO)の定量を特徴とする、同等のin vitro条件下での誘導刺激に依存するNETの不均一性を示しています。
プロトコル
血液サンプルは、インフォームドコンセントの後、臨床的に健康な参加者からの寄付として取得されました。すべての実験は、オアハカのベニート・フアレス大学生化学科学部の人間研究倫理委員会の許可を得て実施されました。
注:研究の選択基準は、性別と年齢が不明瞭であり、血液サンプルを採取する前の質問票への参加者の回答によると臨床的に健康でした。血液学的分析を実施して細胞数を決定し、感染症または貧血を除外し、ドナーの炎症を除外するためのC反応性タンパク質検査を実施しました。
1.末梢血採取と赤血球および白血球パッケージの入手
- 1.8 mg/mLのK2·インフォームドコンシデントコンセントを得た後の臨床的に健康な個人からの抗凝固剤としてのEDTA( 材料の表を参照)。次に、標準的な血液バイオメトリーとC反応性タンパク質検査を実行して、感染または炎症を除外し、サンプルの品質を確保します。
- 末梢血サンプルを82 x gで15分間遠心分離して多血小板血漿を除去し、続いて630 x gで5分間2回目の遠心分離を行います。残りの血漿を廃棄し、赤血球および白血球パッケージを得る。
- 1xダルベッコのリン酸緩衝生理食塩水(DPBS)で1:1の比率(v / v)で希釈します。
2. 二重密度勾配を用いた多形核好中球(PMN)精製
注:好中球はin vitro での寿命が約8時間に制限されているため、採血後すぐに好中球の精製を実行してください。
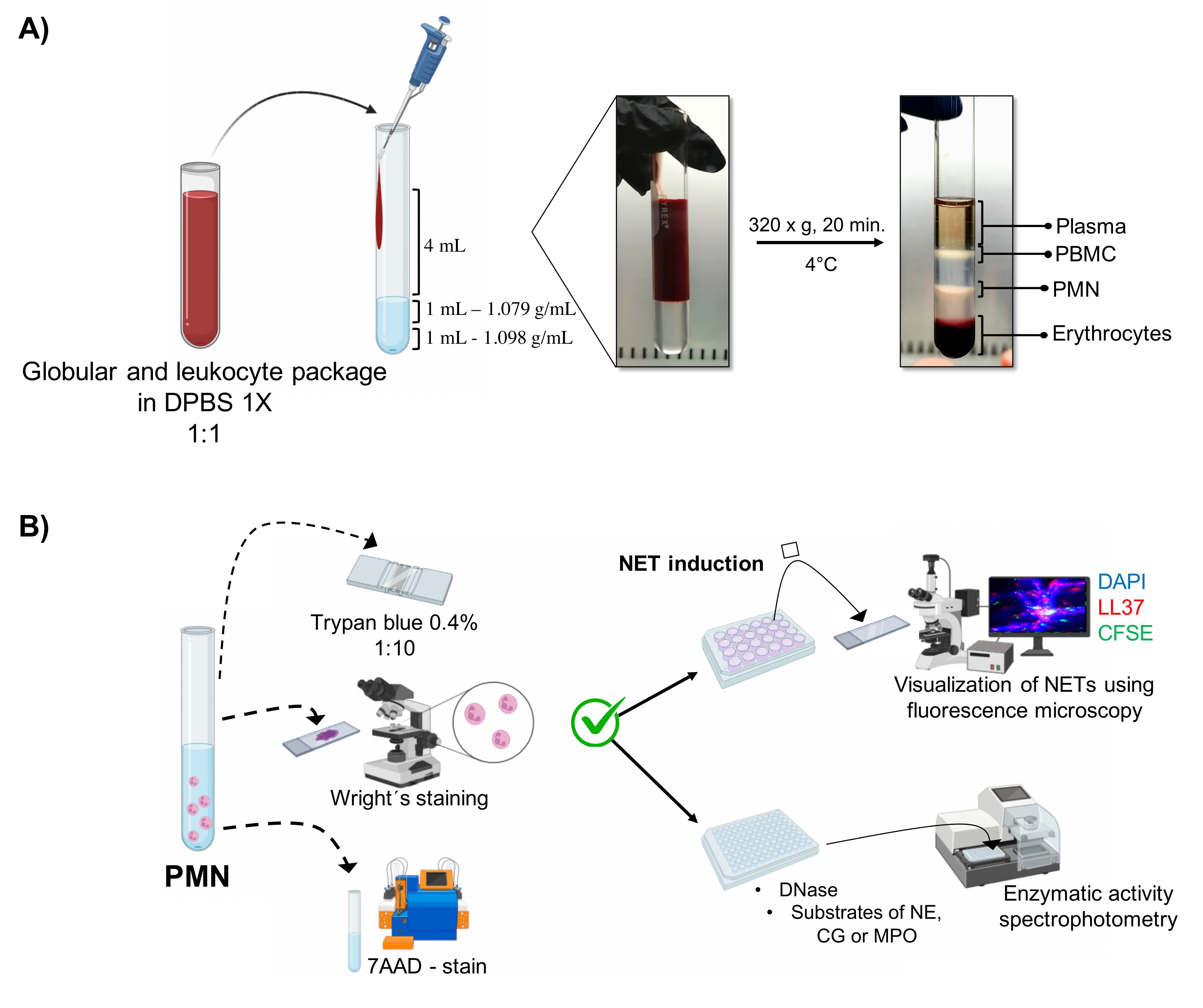
- 滅菌済みの10 mLガラスチューブ( 材料表を参照)に、1 mLの密度1.098 g / mL溶液、1 mLの1.079 g / mL密度溶液( 材料表を参照)、次に4 mLの希釈赤血球および白血球パッケージを順番に入れます。層間の表面張力を壊さずに壁に注ぎ、層が混ざらないようにします。
- 320 x g で4°Cで20分間遠心分離し、遠心分離機の高力が勾配を乱さないように加速/減速を回避します。
- ピペッティングにより顆粒球(図1A)に対応する相を吸引し、別の滅菌10mLガラス管に移す。4 mLの1x DPBSで300 x g で4°Cで10分間洗浄します。
- 上清を捨て、浸透圧ショックで細胞を処理して残りの赤血球を除去します。4 mLの0.2%生理食塩水を加えて4°Cで2分間、300 x g で4°Cで10分間遠心分離します。 上澄み液を捨てる。次に、4 mLの等張溶液(0.65%生理食塩水)を加えて4°Cで5分間添加して膜の完全性を回復し、300 x g で4°Cで10分間遠心分離します。
注:0.2%生理食塩水は低張培地であり、赤血球細胞内培地に比べて溶質濃度が低くなっています。低張性媒体との接触により、水が赤血球に拡散し、それらの腫脹および溶血を引き起こす。この上清からの赤血球の除去は顕微鏡観察により確認した。 - 上清を取り除きます。細胞を4 mLの1x DPBSに再懸濁して細胞の破片を除去した後、300 x g で4°Cで10分間遠心分離します。 最後に、細胞ペレットを2 mLの冷たいハンク平衡塩溶液(HBSS)バッファーに再懸濁します。
3.好中球の形態と生存率(図1B)
- トリパンブルー排除試験
- 5 μLの細胞懸濁液を20 μLの0.4%トリパンブルー(1:5の比率)で希釈します。ノイバウアーチャンバー内の細胞をカウントし、排除試験を使用して細胞生存率を決定します。色素を透過することなく膜の完全性を維持する細胞を生存可能と考えてください。
- 5 μLの細胞懸濁液をスライドにマウントします。ライトの染みで15秒間乾かして染みます。直ちにサンプルをリン酸緩衝液pH 6.4で30秒間固定します。十分な蒸留水で洗浄し、光学顕微鏡(100倍)で形態を観察します。
- 7AAD染色およびフローサイトメトリー解析
- 1 x 105 細胞をフローサイトメトリーチューブに加え、100 μLのFACSバッファー(1x DPBS、0.1%アジ化ナトリウム、および10%自己脱補血漿)中の1 μLの7AADで、4°C暗所で15分間染色します。
- 500 μLのFACSバッファーで300 x g で10分間洗浄します。細胞を500 μLの2%パラホルムアルデヒドで固定し、フローサイトメーターで分析するまで4°Cで保存します。
- 死細胞コントロールの場合は、1 x 105 細胞を200 μLの4%パラホルムアルデヒドで30分間固定し、500 μLの1x PBSで300 x g で4°Cで10分間洗浄します。 上清を引き抜いて捨てます。次に、200 μLの0.1%トリトンX-100を4°Cで1時間加えます。 500 μLの1x PBSで洗浄し、ステップ3.2.1と同様に7AADで染色します。
- フローサイトメーター( 材料表を参照)を使用して、FSC対SSC分析を実行して細胞純度を分析し、SSC対7AAD染色を実行して細胞生存率を分析します。多形核設定(FSC、400-490およびSSC、300-320)で、培地フロー(1,000細胞/秒)での取り込み量100 μLの3 x 104 イベントを読み取ります。
- キャプチャしたデータをフローサイトメーターソフトウェア( 材料表を参照)で分析し、ドットプロットとヒストグラムで提示された多形核集団における7AADの純度と陽性細胞の割合を決定します。
4. 微生物のCFSE染色
- 1.5 mLマイクロチューブに1 x 108 細菌または1 x 106 真菌偽菌糸を加え、1x PBSに溶解した200 μLの5 μMカルボキシフルオレセインスクシンイミジルエステル(CFSE)で染色します。数秒間混合し、暗所で37°Cで10分間インキュベートします。
- 脱補血漿を500 μL加えて反応を停止し、偽菌糸の場合は620 x gで10分間、細菌の場合は1,800 x gで10分間遠心分離します。
- 上清を廃棄し、ステップ4.2と同様に遠心分離しながら1 mLの1x PBSでペレットを洗浄します。最後に、微生物を250 μLの1x PBSに再懸濁します。
- NET誘導のために、2 x 107 細菌(MOI:100)または2 x 10 5偽菌糸(MOI:1)を含む1.5 mLのマイクロチューブに50 μLアリコートを調製します。
5. ネット誘導
- 10 mm x 10 mmの滅菌ガラスカバーガラスカバーガラスを24ウェルプレートに入れ、10 μLの0.001%ポリL-リジンで室温で1時間覆います。100 μLの1x PBSで2回洗浄し、風乾し、UV光を15分間照射します。
- ステップ2.5の好中球懸濁液のHBSS溶液を、10%自己血漿を添加したRPMI 1640培地と交換します。24ウェルプレート(ステップ5.1)に、この細胞懸濁液を350 μL添加し、最終濃度2 x 105 好中球/ウェルにします。
- 5%CO2を用いて37°Cで20分間インキュベートすることにより、細胞をウェルの底に接着させます。
- 微生物刺激グラム陽性菌黄色ブドウ球菌(ATCC 25923)、グラム陰性菌緑膿菌(ATCC 10145)をMOI 100に、偽菌糸を偽菌糸(ATCC 10231)をMOI:1で、50μLでNET形成を誘導する刺激を加える:1; 生化学的刺激-PMA(200 nM)およびHOCl(4.5 mM)、および刺激がない場合のコントロール(50 μLのHBSS)。
- ウェルあたり400 μLの最終容量を取得します。プレートシェーカーで140rpmで30秒間混合し、37°Cおよび5%CO2で4時間インキュベートします。
6. 蛍光顕微鏡によるNETの可視化
- DNAおよびLL37免疫染色
- NET誘導後、慎重にピペッティングしてウェルから上清を除去し、細胞を300 μLの4%パラホルムアルデヒドで30分間固定します。
- 遠心分離せずに200 μLの1x PBSで細胞を洗浄し、200 μLのブロッキングバッファー(1x PBS中の10%脱補血漿)を30分間加えます。
- LL-37染色の場合、1x PBS中の0.2%Triton X-100を200 μLで10分間透過処理し、抗体が細胞に入るようにします。1x PBSで2回注意深く洗浄し、余分な洗剤を取り除きます。
- カバーガラスをスライドガラスに取り付けます(各スライドに4つのカバーガラス)。DNAを2 μLのDAPI( 材料表を参照)で細胞を染色し、カバーガラスを密封し、共焦点蛍光顕微鏡による分析まで-20°Cで保存します。
- 蛍光画像の取得と解析
- NET画像を撮影してその成分を定量化し、共焦点蛍光顕微鏡( 材料表を参照)の対応するフィルターを使用して、コンピューターのソフトウェアで画像を取得します。
注:DNAがDAPI(青色)で染色され、360 nmで励起、460 nmで発光を示していると考えてください。微生物は、492nmの励起および521nmの発光を有するCFSE(緑色)で染色される。LL37ペプチドは、594 nmの励起および614 nmの発光を有する抗LL37 Alexa Fluor 594抗体(赤色)で標識されています。 - 顕微鏡を校正します。スライドを配置し、通常のライトを点灯した状態で微分干渉コントラスト(DIC)を使用して焦点を合わせます。 ライブ を選択して、画像をモニターに投影します。
- ライトを消し、蛍光色素に対応するチャンネルを選択します。たとえば、DAPIの場合は365 nm/blue、Alexa 594の場合は43 HE DsRed、CFSEの場合は38 HE GFPを選択します。
- LL37の場合はアイソタイプコントロール抗体、DAPIおよびCFSEの場合は非染色細胞を使用して設定を調整します。同じ露出時間、電圧、コントラスト、レンズ設定を設定して、同じ条件ですべての画像をキャプチャします。
注:この研究では、露光時間、電圧、コントラストをそれぞれ1.0ミリ秒、4.0V、0.0に設定し、対物レンズを40倍にしました。これらの値は、サンプルに最適な画像キャプチャを容易にするために調整できます。 - [ スナップ ] を選択して画像をキャプチャします。ウェルごとに5つの画像(4つの極値と中央)と、DNA/LL37/CFSEの共局在(マージ)を保存します。
- 各色の独立した画像を使用して、3つのクラスのピクセルを背景として定義し、Image Jソフトウェアを使用して領域ごとの平均グレー信号値を分析します。
- NET画像を撮影してその成分を定量化し、共焦点蛍光顕微鏡( 材料表を参照)の対応するフィルターを使用して、コンピューターのソフトウェアで画像を取得します。
7. 酵素活性の定量化
- 96ウェルプレートに、NET誘導用の1 x 10 5好中球を含むHBSS中の細胞懸濁液90 μLを加え、37°Cおよび5 %CO2で20分間インキュベートします。
- 直ちに、対応する刺激を10 μL(ステップ5.4と同様の濃度)を加え、5%CO2と共に37°Cで4時間インキュベートします。
- 上清を捨て、100 μLのHBSSで細胞を洗浄します。1 U/mLのDNaseで37°Cで10分間処理してDNAタンパク質構造の遊離を促進し、1,800 x g で10分間遠心分離します。
- 上清を回収し、Whiteらによって前述されたように比色反応を用いて上清中の酵素活性を評価する17。
- NET誘導のための刺激を加えることなく、同じ実験条件下で好中球中のNE、CG、およびMPOの最大酵素活性を決定します。次いで、細胞サンプルを-70°Cで凍結し、水浴中で37°Cで解凍し、細胞溶解による細胞内タンパク質の放出を促進する温度ショックを発生させる。1,800 x g で10分間遠心分離し、上清を回収します。
- 96ウェルプレートの各ウェルに50 μLの上清を加え、次にステップ7.7に示すように各基質50 μLを加えます。
- NEの基質として0.5 MのN-メトキシスクシニル-アラ-アラ-プロ-バル-p-ニトロアニリンを、CGの基質として1 mMのN-スクシニル-アラ-プロ-Phe-p-ニトロアニリドを追加します。室温で3時間インキュベートします。MPOの場合は、1.6 mMの3,3',5,5'-テトラメチルベンジジン(TMB)を加え、室温で30分間インキュベートします。
- インキュベーション後、MPO用のストップ溶液(0.5 M H2SO4)を50 μL加え、分光光度計を使用して、NEおよびCGの場合は405 nm、MPOの場合は450 nmの吸光度を測定します。
- 得られた値を対応する検量線と比較し、最大酵素活性(100%)に対する各条件の結果を示す。
8.統計分析
- 独立した実験ごとに測定データを3連で分析し(n = 10)、95%の信頼水準を持つグループを比較することにより、統計分析のANOVAを実行します。

図1:PMN精製とNET誘導プロトコル 。 (A)血漿を末梢血から除去して赤血球と白血球のパッケージを取得し、1xDPBSで1:1(v / v)に希釈しました。次に、4 mLの希釈液を壁に沿って二重密度勾配チューブに加え、320 x g で4°Cで20分間遠心分離し、異なる細胞層の分離を求め、PMNに対応するものを回収しました。(B)精製した細胞を計数し、その形態をライト染色により解析した。生存率は、フローサイトメトリーを用いたトリパンブルー排除および7AAD染色によって決定した。最適な好中球の純度と生存率が検証されたら、DAPI-DNA、抗LL37 Alexa Fluor 594、および微生物-CFSE染色による蛍光顕微鏡による分析のために、微生物(黄色ブドウ球、緑膿菌、およびC.アルビカンス)または化学物質(PMA、HOCl)を24ウェルプレートに添加することによりNET形成を誘導しました。酵素定量のために、NETを96ウェルプレートで3時間誘導し、DNaseで処理した後、各酵素の基質(NE、CG、およびMPO)を添加しました。色の変化は分光光度法によって定量化した。DPBS = ダルベッコのリン酸緩衝生理食塩水;PBMC =末梢血単核細胞;PMN =多形核好中球;NE =好中球エラスターゼ;CG = カテプシンG;MPO = ミエロペルオキシダーゼ;PMA = ホルボールミリスチン酸アセテート;HOCl = 次亜塩素酸。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
結果
好中球の純度と生存率
動的な細胞相は、倍密度勾配精製からチューブ内で視覚化されます。これらの層の中で、顆粒球に対応する層は1.079 g/mL密度層の上にあり、末梢血単核球(PBMC)および赤血球の相とは区別されます(図1A)。精製された細胞の形態は、成熟好中球に対応する糸状のフィラメントで接続されたセグメント化された核を持つ細胞を観察するこ?...
ディスカッション
NETの放出を誘導するには、これらの細胞の 生体外 寿命が平均8時間に制限されているため、生存可能な好中球の高純度集団を取得する必要があり、その期間内にすべての実験を実行する必要があります。この目的のために、理想的な方法論は、Ficoll-Histopaque勾配またはデキストラン沈降技術とは対照的に、外因性刺激に対してより応答性の高い非活性化細胞を単離することによって精?...
開示事項
著者は、利益相反がないことを宣言します。
謝辞
この研究は、CONACyTからの基礎科学助成金(#285480)と、オアハカ自治大学生化学学部の臨床免疫学研究部門によってサポートされました。A.A.A、S.A.S.L、W.J.R.R.は、それぞれCONACyT番号#799779、#660793、#827788の博士号を取得しています。
資料
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
参考文献
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved