
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Морфологический и композиционный анализ внеклеточных ловушек нейтрофилов, индуцированных микробными и химическими раздражителями
В этой статье
Резюме
Здесь представлен протокол индукции и анализа внеклеточных ловушек нейтрофилов in vitro (NETs). Количественная оценка ДНК, кателицидина (LL37) и активности ферментов дала данные, которые показывают изменчивость состава и морфологии NETs, индуцированных микробными и химическими стимулами в аналогичных контролируемых условиях.
Аннотация
Нейтрофилы функционируют как первая линия клеточной защиты во врожденном иммунном ответе, используя различные механизмы, такие как формирование внеклеточных ловушек нейтрофилов (NETs). В этом исследовании анализируются морфологические и композиционные изменения в НЭТ, индуцированные микробными и химическими стимулами, с использованием стандартизированных методологий in vitro для индукции и характеристики NET с клетками человека. Описанные здесь процедуры позволяют проанализировать морфологию NET (литическую или нелитическую) и состав (ДНК-белковые структуры и ферментативную активность), а также влияние растворимых факторов или клеточного контакта на такие характеристики. Кроме того, методы, описанные здесь, могут быть модифицированы для оценки влияния экзогенных растворимых факторов или клеточного контакта на композицию NET.
Применяемые методы включают очистку полиморфноядерных клеток из периферической крови человека с использованием градиента двойной плотности (1,079-1,098 г / мл), гарантируя оптимальную чистоту и жизнеспособность (≥ 95%), что продемонстрировано окрашиванием Райта, исключением трипан-синего и проточной цитометрией, включая анализ FSC против SSC и окрашивание 7AAD. Образование NET индуцируется микробными (Pseudomonas aeruginosa, Staphylococcus aureus и Candida albicans) и химическими (форбол миристата ацетат, HOCl) стимулами, а NETs характеризуются окрашиванием ДНК-DAPI, иммуноокрашиванием антимикробного пептида кателицидина (LL37) и количественной оценкой ферментативной активности (нейтрофильная эластаза, катепсин G и миелопероксидаза). Изображения получены с помощью флуоресцентной микроскопии и проанализированы с помощью ImageJ.
Введение
Нейтрофилы являются наиболее распространенными лейкоцитами в кровотоке, играя существенную роль во время клиренса патогенных агентов несколькими механизмами, включая высвобождение крупных структур хроматина, состоящих из ДНК и нескольких ядерных, цитоплазматических и зернистых антибактериальных белков 1,2. Прямой предшественник, описывающий эту антимикробную роль нейтрофилов, был сделан Takei et al.3 в 1996 году. Эти авторы сообщили о новой форме смерти, отличной от апоптоза и некроптоза у нейтрофилов, показали морфологические изменения, демонстрирующие ядерный разрыв с последующим выплескиванием из нуклеоплазмы в цитоплазму и увеличением проницаемости мембраны от 3 ч инкубации с форбол миристатом ацетатом (PMA)2,3. Однако только в 2004 году был использован термин «внеклеточные ловушки нейтрофилов (NETs)»4.
Образование NET наблюдалось в различных условиях, таких как бактериальные, грибковые5, вирусные6 и паразитарные инфекции, для нейтрализации, уничтожения и предотвращения распространения микробов7. Другие исследования показывают, что он также может возникать в непатогенных условиях стерильными стимулами, такими как цитокины, мононатриевая мочевая кислота или кристаллы холестерина, аутоантитела, иммунные комплексы и активированные тромбоциты7. Липополисахарид (ЛПС), интерлейкин-8 (IL-8) и ПМА были одними из первых стимулов in vitro, описанных как индукторы NET, а участие IN VIVO NET в патогенных процессах было продемонстрировано в двух моделях острого воспаления: экспериментальной дизентерии и спонтанного аппендицита человека4. ДНК является важным компонентом NET. Его соответствующая структура и состав необходимы для связывания и уничтожения микроорганизмов путем доставки высокой локальной концентрации антимикробных молекул к пойманным микробам, о чем свидетельствует кратковременная обработка дезоксирибонуклеазой (ДНКаза), которая разрушает НЭТ и их микробицидные свойства4. Помимо ДНК, NETs содержат присоединенные белки, такие как гистоны, нейтрофильная эластаза (NE), катепсин G (CG), протеиназа 3, лактоферрин, желатиназа, миелопероксидаза (MPO) и антимикробные пептиды (AMPs), такие как катионный провоспалительный пептидный кателицидин LL-37 среди других 8,9. Такие агрегаты могут образовывать более крупные нити диаметром до 50 нм. Эти факторы могут нарушать микробные факторы вирулентности или целостность клеточной мембраны патогена; кроме того, AMP могут стабилизировать ПОЛУЧЕННУЮ ИЗ NET ДНК против деградации бактериальными нуклеазами10.
Конкретные механизмы, регулирующие образование NET, до сих пор полностью не выяснены. Наиболее характеризуемым путем, ведущим к высвобождению NET, является передача сигналов ERK, которая приводит к активации NADPH-оксидазы и производству активных форм кислорода (АФК), а также к увеличению внутриклеточного кальция, который вызывает активацию пути MPO. Это, в свою очередь, превращает перекись водорода в хлорноватистую кислоту, активируя NE путем окисления11,12. NE отвечает за деградацию актиновых нитей цитоскелета для блокирования фагоцитоза и перемещения их в ядро для обработки протеолитическим расщеплением и дезаминированием PAD4, которые управляют десенсибилизацией хроматиновых волокон, которые связываются с гранулами и цитоплазматическими белками, а затем высвобождаются внеклеточнымобразом 7. Эти протеазы включают те, которые высвобождаются из азуросомного комплекса гранул азурофилов и других протеаз, таких как катепсин G13.
В зависимости от морфологических изменений нейтрофилов, НЭТ классифицируются на два типа: суицидальное или литическое образование NET, приводящее к гибели клеток4, и жизненно важное или нелитическое ОБРАЗОВАНИЕ NET, продуцируемое жизнеспособными клетками, опосредованными везикулярным высвобождением ядерной или митохондриальной ДНК, с остатком ануклеированного цитопласта с фагоцитарной способностью14,15. Как правило, НЭТ, состоящие из митохондриальной ДНК, имеют удлиненную морфологию волокна14, в то время как те, которые структурированы из ядерной ДНК, имеют облачный вид3. Однако неизвестно, как нейтрофил выбирает свое ДНК-происхождение. В отличие от предыдущих исследований, которые описывали канонические пути НЭТ как требующие нескольких часов, жизненно важный путь быстро активируется всего за 5-60 минут15.
Несмотря на эти достижения, состав NET варьируется в зависимости от стимула; например, различные мукоидные и немукоидные штаммы P. aeruginosa индуцируют образование NETs, содержащих 33 общих белка и до 50 переменных белков7. Таким образом, необходимо гомогенизировать методики, позволяющие формировать объективные выводы в исследовательских группах. В данной работе описан протокол с различными методами, позволяющими сравнивать и оценивать состав, структуру и морфологию НЭТ, индуцированных различными микроорганизмами: золотистым стафилококком (грамположительная бактерия), Pseudomonas aeruginosa (грамотрицательная бактерия) и Candida albicans (гриб), а также химическими стимулами (PMA, HOCl) в нейтрофилах человека от здоровых людей. Репрезентативные результаты демонстрируют гетерогенность НЭТ в зависимости от их индуцирующего стимула в сопоставимых условиях in vitro, характеризующихся окрашиванием ДНК-DAPI, иммуноокрашиванием для LL37 и количественной оценкой ферментативной активности (NE, CG и MPO).
протокол
Образцы крови были получены в качестве донорства от клинически здоровых участников после информированного согласия. Все эксперименты проводились с разрешения Комитета по этике исследований человека факультета биохимических наук Автономного университета «Бенито Хуарес» в Оахаке.
ПРИМЕЧАНИЕ: Критериями включения в исследование были нечеткий пол и возраст, а также клинически здоровые в соответствии с ответами участников на вопросник до взятия образца крови. Был проведен гематологический анализ для определения количества клеток и исключения инфекций или анемии, а также тест на С-реактивный белок, чтобы исключить воспаление у донора.
1. Забор периферической крови и получение пакета эритроцитов и лейкоцитов
- Собрать 10 мл периферической крови путем венипункции в пробирках с 1,8 мг/мл K2· ЭДТА в качестве антикоагулянта (см. Таблицу материалов) от клинически здоровых лиц после получения информированного согласия. Затем выполните стандартную биометрию крови и тест на С-реактивный белок, чтобы исключить инфекцию или воспаление, обеспечивая качество образца.
- Центрифугируют образец периферической крови при 82 х г в течение 15 мин для удаления обогащенной тромбоцитами плазмы с последующим вторым центрифугированием при 630 х г в течение 5 мин. Выбросьте оставшуюся плазму для получения эритроцита и лейкоцитарного пакета.
- Разбавьте его в соотношении 1:1 (v/v) с 1x фосфатно-буферным физиологическим раствором Dulbecco (DPBS).
2. Полиморфноядерная очистка нейтрофилов (ПМН) с использованием градиента двойной плотности
ПРИМЕЧАНИЕ: Выполняют очистку нейтрофилами сразу после сбора крови, потому что они имеют ограниченное время жизни in vitro около 8 ч.
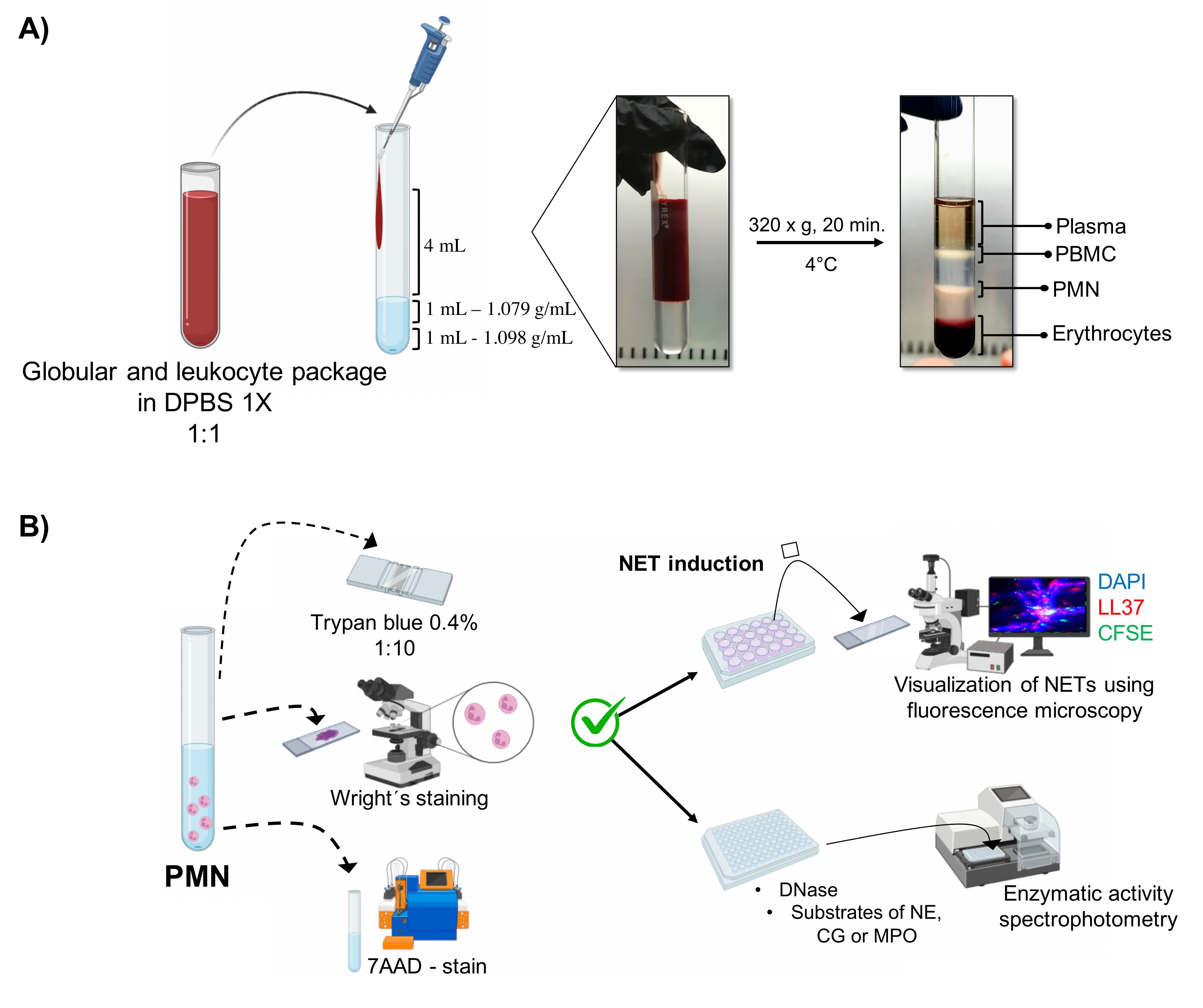
- Поместите в стерильную стеклянную трубку объемом 10 мл (см. Таблицу материалов) в следующем порядке: 1 мл раствора плотности 1,098 г/мл, 1 мл раствора плотности 1,079 г/мл (см. Таблицу материалов), а затем 4 мл разбавленного пакета эритроцитов и лейкоцитов. Перелить по стенам, не нарушая поверхностного натяжения между слоями, чтобы предотвратить их перемешивание.
- Центрифуга при 320 х g в течение 20 мин при 4 °C, избегая ускорения/замедления, чтобы высокие силы центрифуги не нарушали градиент.
- Аспирировать фазу, соответствующую гранулоцитам (фиг.1А) путем пипетирования, и перенести в другую стерильную стеклянную трубку объемом 10 мл. Промывайте 4 мл 1x DPBS при 300 x g в течение 10 мин при 4 °C.
- Выбросьте супернатант и обработайте клетки осмотическим шоком, чтобы удалить оставшиеся эритроциты. Добавить 4 мл 0,2% физиологического раствора в течение 2 мин при 4 °C и центрифугу при 300 х г в течение 10 мин при 4 °C. Выбросьте супернатант. Затем добавляют 4 мл изотонического раствора (0,65% физиологического раствора) в течение 5 мин при 4 °C для восстановления целостности мембраны и центрифугу при 300 х г в течение 10 мин при 4 °C.
ПРИМЕЧАНИЕ: 0,2% физиологический раствор представляет собой гипотоническую среду с более низкой концентрацией растворенного вещества по сравнению с концентрацией внутриклеточной среды эритроцитов. Контакт с гипотоникмедией позволяет воде диффундировать в эритроциты, что приводит к их отеку и гемолизу. Это удаление эритроцита из супернатанта было подтверждено микроскопическим наблюдением. - Удалите супернатант. Повторно суспендируют клетки в 4 мл 1x DPBS для удаления клеточного мусора, а затем центрифугу при 300 х г в течение 10 мин при 4 °C. Наконец, повторно суспендируйте клеточную гранулу в 2 мл холодного буфера сбалансированного солевого раствора Хэнка (HBSS).
3. Морфология и жизнеспособность нейтрофилов (Рисунок 1В)
- Тест на исключение синего цвета Трипана
- Развести 5 мкл клеточной суспензии в 20 мкл 0,4% трипан синего (соотношение 1:5). Подсчитайте клетки в камере Нойбауэра и определите жизнеспособность клеток с помощью теста на исключение. Рассмотрим клетки, которые поддерживают целостность своей мембраны без проникновения красителя, как жизнеспособные.
- Установите 5 мкл клеточной суспензии на затвор; сухость и окрашивание пятном Райта в течение 15 с. Немедленно зафиксируйте образец с фосфатным буфером pH 6,4 в течение 30 с. Промыть достаточным количеством дистиллированной воды и понаблюдать за морфологией под оптическим микроскопом (100x).
- 7AAD-окрашивание и анализ проточной цитометрии
- Добавьте 1 х 105 клеток в проточные трубки цитометрии и окрашивайте 1 мкл 7AAD в 100 мкл буфера FACS (1x DPBS, 0,1% азида натрия и 10% аутологичной дезкомплементированной плазмы) в течение 15 мин при 4 °C в темноте.
- Промывайте 500 мкл буфера FACS при 300 х г в течение 10 мин. Зафиксируйте клетки 500 мкл 2% параформальдегида и храните при 4 °C до их анализа в проточном цитометре.
- Для контроля мертвой клетки зафиксируйте 1 x 105 клеток с 200 мкл 4% параформальдегида в течение 30 мин и промывайте 500 мкл 1x PBS при 300 x g в течение 10 мин при 4 °C. Снимите супернатант и выбросьте. Затем добавляют 200 мкл 0,1% Тритона Х-100 в течение 1 ч при 4 °С. Промывайте 500 мкл 1x PBS и окрашивайте 7AAD, как на шаге 3.2.1.
- Используя проточный цитометр (см. Таблицу материалов), выполните анализ FSC против SSC для анализа чистоты клеток и окрашивания SSC против 7AAD для анализа жизнеспособности клеток. Считывание 3 x 104 событий в 100 мкл объема поглощения при среднем потоке (1000 ячеек/с) в полиморфноядерных установках (FSC, 400-490 и SSC, 300-320).
- Проанализируйте полученные данные в программном обеспечении проточного цитометра (см. Таблицу материалов) и определите процент чистоты и положительных клеток для 7AAD в полиморфноядерной популяции, представленный с помощью точечных графиков и гистограмм.
4. CfSE окрашивание микроорганизмов
- Добавьте 1 х 108 бактерий или 1 х 106 грибковых псевдогифа в 1,5 мл микротрубок и окрашивание 200 мкл 5 мкМ карбоксифлуоресцеина сукцинимидилового эфира (CFSE), растворенного в 1x PBS. Перемешать в течение нескольких секунд и инкубировать при 37 °C в течение 10 мин в темноте.
- Остановить реакцию, добавив 500 мкл дезкомплементированной плазмы и центрифугу при 620 х г в течение 10 мин для псевдогифай или при 1,800 х г в течение 10 мин для бактерий.
- Выбросьте супернатанты и промыть гранулы 1 мл 1x PBS центрифугированием, как на этапе 4.2. Наконец, повторно суспендируют микроорганизмы в 250 мкл 1x PBS.
- Приготовьте 50 мкл аликвот в микротрубках по 1,5 мл с 2 х 107 бактериями (MOI: 100) или 2 х 105 псевдогифай (MOI: 1) для индукции NET.
5. Индукция NET
- Поместите стерильные стеклянные крышки размером 10 мм х 10 мм в 24-луночную пластину и накройте 10 мкл 0,001% поли-L-лизина в течение 1 ч при комнатной температуре. Дважды промыть 100 мкл 1x PBS, высушить на воздухе и облучить ультрафиолетовым светом в течение 15 мин.
- Замените раствор HBSS суспензии нейтрофилов на этапе 2.5 средой RPMI 1640, дополненной 10% аутологичной плазмой. К 24-луночной пластине (стадия 5.1) добавляют 350 мкл этой клеточной суспензии, для конечной концентрации 2 х 105 нейтрофилов/лунка.
- Дайте ячейкам прилипнуть к дну лунок путем инкубации в течение 20 мин при 37 °C с 5% CO2.
- Добавьте стимулы для индуцирования образования NET в 50 мкл: микробные стимулы-грамположительная бактерия S. aureus (ATCC 25923), грамотрицательная бактерия P. aeruginosa (ATCC 10145) при MOI 100 и псевдогифы C. albicans (ATCC 10231) при MOI:1; биохимические раздражители - PMA (200 нМ) и HOCl (4,5 мМ) и контроль с отсутствующим стимулом (50 мкл HBSS).
- Получите конечный объем 400 мкл на скважину. Смешайте на пластинчатом шейкере при 140 об/мин в течение 30 с и инкубируйте в течение 4 ч при 37 °C и 5% CO2.
6. Визуализация НЭТ методом флуоресцентной микроскопии
- Иммуноразрашивание ДНК и LL37
- После индукции NET удалите супернатанты из скважин путем тщательного пипетирования и зафиксируйте ячейки 300 мкл 4% параформальдегида в течение 30 мин.
- Промыть клетки 200 мкл 1x PBS без центрифугирования и добавить 200 мкл блокирующего буфера (10% декомплементированной плазмы в 1x PBS) в течение 30 мин.
- Для окрашивания LL-37 пермеабилизируйте клетки 200 мкл 0,2% Triton X-100 в 1x PBS в течение 10 мин, чтобы позволить антителу проникнуть в клетки. Тщательно вымойте 2x с 1x PBS, чтобы удалить излишки моющего средства.
- Установите крышки на стеклянные слайды (четыре крышки на каждом слайде). ДНК окрашивает клетки 2 мкл DAPI (см. Таблицу материалов), запечатывает покровы и хранит при -20 °C до их анализа с помощью конфокальной флуоресцентной микроскопии.
- Получение и анализ флуоресцентных изображений
- Возьмите изображения NET для количественной оценки их компонентов и используйте соответствующие фильтры в конфокальном флуоресцентном микроскопе (см. Таблицу материалов) для получения изображений с помощью программного обеспечения компьютера.
ПРИМЕЧАНИЕ: Учтите, что ДНК окрашена DAPI (синий цвет), показывая возбуждение при 360 нм и излучение при 460 нм. Микроорганизмы окрашивают CFSE (зеленый цвет), который имеет возбуждение 492 нм и излучение 521 нм. Пептид LL37 помечен антителом против LL37 Alexa Fluor 594 (красный цвет), которое имеет возбуждение 594 нм и излучение 614 нм. - Откалибруйте микроскоп. Поместите слайд и фокусировку с помощью дифференциального интерференционного контраста (DIC) при включенном нормальном свете. Выберите Live , чтобы проецировать изображение на монитор.
- Выключите свет и выберите соответствующий фторхромный канал. Например, выберите фильтр 365 нм/синий для DAPI, 43 HE DsRed для Alexa 594 или 38 HE GFP для CFSE.
- Настройте настройки с помощью контрольного антитела изотипа для LL37 и незапятнанных клеток для DAPI и CFSE. Установите одинаковое время экспозиции, напряжение, контрастность и настройки объектива, чтобы захватывать все изображения в одинаковых условиях.
ПРИМЕЧАНИЕ: В этом исследовании время экспозиции, напряжение и контрастность были установлены на уровне 1,0 мс, 4,0 В и 0,0 соответственно с целью 40x. Эти значения могут быть скорректированы для облегчения наилучшего захвата изображения для образцов. - Выберите «Привязать », чтобы записать изображение. Сохраните пять изображений (четыре крайности и центр) на скважину, а также колокализацию (слияние) ДНК/LL37/CFSE.
- Определите три класса пикселей в качестве фона с независимыми изображениями каждого цвета и проанализируйте значение среднего серого сигнала для каждой области с помощью программного обеспечения Image J.
- Возьмите изображения NET для количественной оценки их компонентов и используйте соответствующие фильтры в конфокальном флуоресцентном микроскопе (см. Таблицу материалов) для получения изображений с помощью программного обеспечения компьютера.
7. Количественная оценка ферментативной активности
- В 96-луночную пластину добавляют 90 мкл клеточной суспензии в HBSS, содержащей 1 х 105 нейтрофилов для индукции NET, и инкубируют в течение 20 мин при 37 °C и 5% CO2.
- Немедленно добавляют 10 мкл соответствующих стимулов (концентрация, как на стадии 5.4) и инкубируют в течение 4 ч при 37 °C с 5% CO2.
- Выбросьте супернатанты и промыть клетки 100 мкл HBSS. Обрабатывать 1 ЕД/мл ДНКазы в течение 10 мин при 37 °C, чтобы способствовать высвобождению ДНК-белковых структур, и центрифугу при 1 800 х г в течение 10 мин.
- Восстановите супернатанты и оцените активность фермента в супернатанте с помощью колориметрических реакций, как описано ранее White et al.17.
- Определить максимальную активность ферментов NE, CG и MPO в нейтрофилах в тех же экспериментальных условиях без добавления каких-либо стимулов для индукции NET. Затем заморозьте образец клетки при -70 °C и разморозьте при 37 °C на водяной бане, создавая температурный шок в пользу высвобождения внутриклеточных белков путем лизиса клеток. Центрифуга при 1 800 х г в течение 10 мин и восстановление супернатантов.
- Добавьте 50 мкл супернатанта к каждой скважине в 96-луночных пластинах, а затем добавьте 50 мкл каждого субстрата, как указано на этапе 7.7.
- Добавьте 0,5 М N-метоксисукцинил-Ала-Ала-Про-Валь--нитроанилина в качестве субстрата для NE и 1 мМ N-сукцинил-Ала-Ала-Про-Фе--нитроанилида для CG. Инкубировать в течение 3 ч при комнатной температуре. Для МПО добавляют 1,6 мМ 3,3', 5,5'-тетраметилбензидина (ТМБ) и инкубируют в течение 30 мин при комнатной температуре.
- После инкубации добавляют 50 мкл стоп-раствора (0,5 МH2SO4) для МПО и измеряют поглощение при 405 нм для NE и CG и 450 нм для МПО, используя спектрофотометр.
- Сравните полученные значения с соответствующими калибровочными кривыми и покажите результаты каждого состояния относительно максимальной активности фермента (100%).
8. Статистический анализ
- Анализируйте данные измерений в трех экземплярах для каждого независимого эксперимента (n = 10) и выполняйте ANOVA для статистического анализа, сравнивая группы с уровнем достоверности 95%.

Рисунок 1: Очистка ПМН и протокол индукции NET. (А) Плазму удаляли из периферической крови для получения пакета эритроцитов и лейкоцитов и разбавляли 1:1 (v/v) 1x DPBS. Затем 4 мл разбавления добавляли вдоль стенки к трубке градиента двойной плотности и центрифугировали при 320 х г в течение 20 мин при 4 °С, получая разделение различных слоев клеток и восстанавливая тот, который соответствует ПМН. (B) Очищенные клетки были подсчитаны, и их морфология была проанализирована окрашиванием Райта. Жизнеспособность определяли путем исключения трипанового синего цвета и окрашивания 7AAD с использованием проточной цитометрии. После того, как оптимальная чистота и жизнеспособность нейтрофилов были проверены, образование NET индуцировалось добавлением микробов (S. aureus, P. aeruginosa и C. albicans) или химических веществ (PMA, HOCl) в 24-луночные пластины для анализа с помощью флуоресцентной микроскопии с DAPI-ДНК, анти-LL37 Alexa Fluor 594 и окрашивания микроорганизмами CFSE. Для количественной оценки ферментов НЭТ индуцировали в 96-луночных пластинах в течение 3 ч и обрабатывали ДНКазой с последующим добавлением субстратов для каждого фермента: NE, CG и MPO; изменения цвета были количественно определены с помощью спектрофотометрии. DPBS = фосфатно-буферный физиологический раствор Дульбекко; PBMC = мононуклеарные клетки периферической крови; PMN = полиморфноядерные нейтрофилы; NE = нейтрофильная эластаза; CG = Катепсин G; МПО = миелопероксидаза; PMA = формола миристата ацетат; HOCl = Хлорноватисто-кислота. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Результаты
Чистота и жизнеспособность нейтрофилов
Динамические клеточные фазы визуализируются в трубке от очистки градиента двойной плотности. Внутри этих слоев слой, соответствующий гранулоцитам, находится выше слоя плотности 1,079 г/мл, что отличается от фаз мононуклеоцитов перифери...
Обсуждение
Высокочистая популяция жизнеспособных нейтрофилов должна быть получена, чтобы индуцировать высвобождение НЭТ, поскольку эти клетки имеют ограниченное время жизни ex vivo в среднем 8 ч, период, в течение которого должны быть выполнены все эксперименты. С этой целью идеальной методоло...
Раскрытие информации
Авторы заявляют, что у них нет конфликта интересов.
Благодарности
Эта работа была поддержана грантом фундаментальной науки (#285480) от CONACyT и Департаментом клинических иммунологических исследований факультета биохимических наук Автономного университета «Бенито Хуарес» де Оахака. A.A.A, S.A.S.L и W.J.R.R. имеют докторские стипендии номеров CONACyT #799779, #660793 и #827788 соответственно.
Материалы
| Name | Company | Catalog Number | Comments |
| 24 Well plate for cell culture | Corning | 3526 | |
| 7-aminoactinomycin D (7-AAD) | BD Pharmingen | 51-668981E | |
| 96 Well plate for cell culture | Costar | 3596 | Flat bottom |
| Agitator | CRM Globe | CRM-OS1 | |
| Antibody LL37 | Santa Cruz Biotechnology | sc-166770 | |
| Blood collection tubes | BD VACUTAINER | 368171 | K2 EDTA 7.2 mg |
| Carboxyfluorescein succinimidyl ester (CFSE) | Sigma-Aldrich | 21878 | |
| Centrifuge | Hettich | 1406-01 | |
| Coverslip | Madesa | M03-CUB-22X22 | 22 mm x 22 mm |
| Dulbecco´s phosphate-buffered saline (DPBS) | Caisson | 1201022 | |
| Falcon tubes 50 mL | CORNING | 430829 | |
| Flow Cytometry Tubes | Miltenyi Biotec | 5 mL - Without caps | |
| FlowJo Software | BD Biosciences | Analyze flow cytometry data | |
| Fluorescence microscope | DM 2000 | LEICA | |
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | |
| Incubator | NUAIRE | UN-4750 | |
| MACSQuant Analyzer | Miltenyi Biotec | Flow cytometer | |
| Microplate reader photometer | Clarkson Laboratory - CL | ||
| Microtubes 1.5 mL | Zhejiang Runlab Tech | 35200N | wire snap |
| Minitab Software | Minitab | Statistical analysis | |
| Needles | BD VACUTAINER | 301746 | Diameter 1.34 mm |
| Optical microscope | VELAB | VE-B50 | |
| Percoll | GE Healthcare | 17-0891-01 | Solution for density gradient |
| Phosphate Buffered Saline (10x) | Caisson | PBL07-500ML | |
| Pyrex culture tubes | CORNING | CLS982025 | N°9820 |
| RPMI 1640 1x | Corning | 10-104-CV | contains Glutagro |
| Slides | Madesa | PDI257550 | 22 mm x 75 mm |
| Trypan Blue solution 0.4% | SIGMA | T8154-100ML |
Ссылки
- De Buhr, N., Maren, K. B. How neutrophil extracellular traps become visible. Journal of Immunology Research. 2016, 4604713 (2016).
- Karlsson, A., Nixon, J. B., McPhail, L. C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. Journal of Leukocyte Biology. 67 (3), 396-404 (2000).
- Takei, H., Araki, A., Watanabe, H., Ichinose, A., Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. Journal of Leukocyte Biology. 59 (2), 229-240 (1996).
- Brinkmann, V., et al. Neutrophil extracellular traps kill bacteria. Science. 303 (5663), 1532-1535 (2004).
- Kenny, E. F., et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 6, 24437 (2017).
- Schultz, B. M., Acevedo, O. A., Kalergis, A. M., Bueno, S. M. Role of extracellular trap release during bacterial and viral infection. Frontiers in Microbiology. 13, 798853 (2022).
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology. 18 (2), 134-147 (2018).
- Delgado-Rizo, V., et al. Neutrophil extracellular traps and its implications in inflammation: An overview. Frontiers in Immunology. 8, 81 (2017).
- Petretto, A., et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 14 (7), 0218946 (2019).
- Neumann, A., et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. Journal of Innate Immunity. 6 (6), 860-868 (2014).
- Hakkim, A., et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nature Chemical Biology. 7 (2), 75-77 (2011).
- Sabbatini, M., Magnelli, V., Renò, F. NETosis in wound healing: When enough Is enough. Cells. 10 (3), 494 (2021).
- Metzler, K. D., Goosmann, C., Lubojemska, A., Zychlinsky, A., Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 8 (3), 883-896 (2014).
- Clark, S. R., et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Medicine. 13 (4), 463-469 (2007).
- Pilsczek, F. H., et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. Journal of Immunology. 185 (12), 7413-7425 (2010).
- Sosa, S. A., et al. Structural differences of neutrophil extracellular traps induced by biochemical and microbiologic stimuli under healthy and autoimmune milieus. Immunologic Research. 69 (3), 264-274 (2021).
- White, P. C., et al. Characterization, quantification, and visualization of neutrophil extracellular traps. Methods in Molecular Biology. 1537, 481-497 (2017).
- Boeltz, S., et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death and Differentiation. 26 (3), 395-408 (2019).
- Yousefi, S., Mihalache, C., Kozlowski, E., Schmid, I., Simon, H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 16 (11), 1438-1444 (2009).
- Brinkmann, V., Zychlinsky, A. Neutrophil extracellular traps: is immunity the second function of chromatin. The Journal of Cell Biology. 198 (5), 773-783 (2012).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены