
Method Article
드문 이벤트 감지 오류 수정 DNA와 RNA 시퀀싱을 사용 하 여
요약
차세대 시퀀싱 (NGS) 플랫폼 (~0.5–2.0%)의 높은 오류 속도 의해 제한 되는 genomic 특성 분석을 위한 강력한 도구입니다. NGS 오류율을 제거 하 고 변형 대립 유전자 분수 0.0001 희귀에서 변이 감지 수 있는 오류 수정 시퀀싱의 우리의 방법을 설명 합니다.
초록
기존의 차세대 시퀀싱 기술 (NGS) 10 년 이상에 대 한 거 대 한 게놈 특성에 대 한 수 있다. 특히, NGS 악성에 클론 돌연변이의 스펙트럼을 분석 하 사용 되었습니다. 비록 전통적인 Sanger 방법, 식별 하는 ~0.5–2.0 %의 그것의 높은 에러율으로 인해 드문 클론와 subclonal 돌연변이와 NGS 투쟁 보다 훨씬 더 효율적. 따라서, 표준 NGS는 돌연변이 대 한 검색의 제한이 > 0.02 변형 대립 유전자 분수 (VAF). 이 환자에서 희소 한 돌연변이 대 한 임상 중요성 동안 알려진된 질병 불분명, 남아 없이 백혈병 치료 하는 환자 크게 향상 결과 잔여 질병 때 < 0.0001 cytometry에 의해. NGS의이 artefactual 배경, 완화 하기 위해 수많은 방법이 개발 되었습니다. 여기 오류 수정 DNA와 RNA 시퀀싱 (ECS), 개별 분자 임의의 인덱스 오류 수정에 대 한 혈압의 16와 다중화 8 혈압 환자 특정 인덱스를 태그를 포함 하는 방법을 설명 합니다. 우리의 방법은 감지 하 고 변형 대립 유전자 분수 (VAFs) 두 배나 NGS의 검출 한계 보다 낮은 희귀 0.0001 VAF에 클론 변이 추적할 수 있습니다.
서문
우리가 나이, mutagens, 게놈, 그리고이에 체세포 착오의 축적에서 세포 분열 결과 중 확률 오류 노출에 기초가 악성 변화의 근본적인 병 인 신경 발달 질환, 소아 질환과 정상적인 노화1,2. 질병-운전 잠재력 체세포 돌연변이 조기 발견 및 위험 관리3,,45에 대 한 중요 한 진단 및 예 후 바이오 마커. 더 나은 physiologic clonogenesis 이해, 어떤 임상 통보 되며 결정, 정확한 정량화 및이 돌연변이의 특성화 연구 기본 중요성 이다. 차세대 시퀀싱 (NGS)은 현재 이기종 DNA 샘플; 클론 변이 공부 하는 데 사용 됩니다. 그러나, NGS 식별에서 돌연변이로 제한 됩니다 > 0.02 변형 대립 유전자 분수 (VAF)-0.5-2.0의 고유한 오류 비율 때문 시퀀싱 플랫폼6,7,8%. 그 결과, 추적 진단 및 prognostically 중요 한 체세포 변종 낮은 VAF에 얻을 수 없다 표준 NGS를 사용 하 여.
최근, 다양 한 방법은 NGS8,9,,1011의 오류율을 회피 하기 위해 개발 되었습니다. 이러한 방법을 활용 분자 태그, 시퀀싱 후 오류 수정 가능 하 게 합니다. 각 분자 또는 시퀀싱 라이브러리에서 게놈 단편으로는 임의의 독특한 분자 식별자 (우미) 그 분자에 관련 된 태그입니다. Umi는 무작위 뉴클레오티드 (8-16 N)의 문자열의 순열에 의해 구성 됩니다. 두 번째 샘플 전용 바코드 또한 동일한 NGS 시퀀싱 실행에 여러 샘플을 멀티플렉싱 있도록 워크플로에 통합 된다. PCR 증폭 분자로 태그 라이브러리에서 수행 되 고 그 후 라이브러리 시퀀싱에 대 한 전송 됩니다. 라이브러리 준비 동안 PCR 증폭 및 시퀀싱8동안 오류 게놈 단편을 무작위로 소개 것 이다는 예상 된다. 임의의 연속 오류를 제거 하려면 원시 시퀀싱 읽기 우미에 따라 그룹화 됩니다. 시퀀싱에서 아티팩트 하지 진정한 variant를 충실 하 게 증폭 하 고 같은 바다를 공유 하는 모든 읽기에 시퀀싱 것 소개의 확률적 특성으로 인해 동일한 게놈 위치에 동일한 우미와 모든 읽기에 있을 예정 이다. 아티팩트는 제거 하는 bioinformatically. 여기, 우리가 설명 하는 세 가지 방법의 오류 수정 시퀀싱 (ECS) 단일 뉴클레오티드 변종 (SNVs) 및 작은 삽입-삭제 (Indels), 식별 하기 위해 dna 그리고 RNA 유전자 발현 아래의 정량화를 촉진 하기 위해 실험실에 최적화 된 NGS 오류 임계값입니다.
첫 번째 방법은 연구자에 의해 설계 된 유전자 특정 뇌관을 사용 하 여 희귀 체세포 이벤트에 대 한 보고 하는 방법을 설명 합니다. 라이브러리 준비, 이전 연구자 관심의 파편을 대상으로 프라이 머를 설계 해야 합니다. 우리가 사용 하는 웹 애플 리 케이 션 Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). 200-250 bp의 Amplicons 연쇄 반응 (PCR)에 대 한 이상적인 Umi 통합 되었습니다 일단이 것으로, 150 bp 쌍 간 읽기와 쌍 간 읽기 중복 생성. 사용 될 최적의 뇌관 디자인 조건: 최소 뇌관 크기 = 19; 최적의 뇌관 크기 = 25; 최대 뇌관 크기 = 30; 최소 Tm = 64 ° C; 최적의 Tm = 70 ° C; 최대 Tm = 74 ° C; 최대 Tm 차이 = 5 ° C; 최소 GC 내용 = 45; 최대 GC 내용 = 80; 반환 번호 = 20; 최대 3' 끝 안정성 = 100.
방법 2에서 우리가 ECS DNA 프로토콜 클론 SNVs 및 작은 Indels 희귀 0.0001 VAF amplicons의 수백을 포함 하는 상업적으로 이용 가능한 유전자 패널을 사용 하 여 조사를 Illumina 화학 결합 하는 방법을 설명 합니다. 우리는 우리의 실험에 대 한 TruSight 골수성 시퀀싱 패널 (Illumina)를 사용 하 고 소아 골수성 질환에 대 한 관심의 추가 유전자를 포함 하는 확장 된 패널을 설계. 이러한 패널 팬 들은 그래서 우리 자신의 어댑터 전략이이 패널에 추가 했습니다 오류 정정을 촉진 것이 독특한 분자 식별자 (Umi)의 정보를 제공 하지는. ECS 작동 한다 다른 질병와 관련 된 유전자에 대 한 풍부 하 게 설계 된 다른 패널의 동등 하 게 잘. DNA 분리 및 조직 또는 관심의 샘플에서 후속 정량화, 다음 것이 좋습니다 적어도 500 재고 DNA 견본 당 ng. 우리는 정기적으로 250를 사용 하 여 단일 시퀀싱 라이브러리 확인 중복 및 VAF 계산으로 가능한 한 많은 독특한 게놈 단편으로 하류 캡처하기 위해 DNA의 ng 읽습니다. 선택적 복제 시퀀싱 라이브러리 나머지 250으로 만들어질 수 있다 DNA의 ng. 우리는 항상 표본, 당 2 개의 복제 라이브러리 만들고 참 긍정으로 둘 다 복제에 독립적으로 검색 하는 이벤트에만 생각. 우리는 또한4,13전화 이체의 정확도를 높이 genomic 위치 특정 이항 오류 모델 구현.
마지막으로,는 상용 QIAseq 대상 RNA 패널 (Qiagen)를 사용 하 여 사본 정량화에 대 한 RNA 시퀀싱에 ECS를 연결 하는 방법에 설명 합니다. 중복에는 Umi 필요한 오류 수정 키트에 통합 되었습니다 및 연구원은 제조 업체의 권장 사항에 따라 라이브러리를 만들 수 있습니다. Bioinformatically, 연구자는 ECS-DNA, 프로토콜 섹션에서 자세히 설명 될 것 이다를 위한 파이프라인을 따를 수 있습니다.
프로토콜
1. 타겟 dna 시퀀싱 오류 수정
- 관심의 게놈 조각의 PCR 증폭
- 고 충실도 DNA 중 합 효소를 사용 하 여 증폭 (자료 테이블, 항목 1) amplicons. 증폭 PCR 반응 열 cycler에 다음 조건: 30 s 98 ° c; 10의 18-40 주기 98 ° C, 30에서 s 66 ° c, s 및 30 s 72 ° c; 72 ° C에서 2 분 4 ° c.에서 개최
- 상자성 구슬 (자료 테이블, 항목 2) PCR 제품을 정화. 1: 1.8 비율로 구슬에 PCR 반응 추가 (PCR 반응 볼륨: 볼륨 구슬) 제조 업체의 프로토콜에 따라. 20 µ L ddH2오의와 함께 elute
- 계량 (재료 표, 항목 3) DNA의 최종 농도 결정 하는 DNA의 농도.
- amplicons의 크기를 확인 하려면 2 %agarose 젤 (자료 테이블, 항목 4)에 DNA의 약 수를 실행 합니다.
참고: 또는, 연구원 증폭 게놈 조각의 크기 뿐만 아니라 제품의 농도 결정 하기 위해 PCR 제품에 Bioanalyzer 분석을 수행 하기 위해 선택할 수 있습니다.
- 어댑터는 어 닐 링 시퀀싱
- (자료 테이블, 항목 5) i7 어댑터를 얻을. 그들은 후속 단계에 대 한 제공 되는 그들을 사용 합니다.
- 16N i5 어댑터 다음 올리고 시퀀스 (자료 테이블 항목 6) 상업 구매: AATGATACGGCGACCACCGAGATCTACAC(N1:25252525)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1)(N1) (N1) ACACTCTTTCCCTACACGACGCTCTTCCGATCT
참고: 16N i5 어댑터는 표준 i5 어댑터를 교체 하 고 ECS를 촉진 하기 위하여 16 임의의 뉴클레오티드의 문자열로 어댑터. - 16N i5 어댑터 작업 솔루션: 주식의 100 µ M 16N i5 어댑터 40 µ L, 10 µ L의 TE 버퍼, 그리고 10 µ L 500 µ M NaCl 솔루션의.
- 별도 PCR 웰 스 1.2.3 단계에서 준비 i5 작업 솔루션의 aliquot 7.5 µ L.
- 해당 우물에 샘플 특정 i7 어댑터의 5 µ L를 추가 합니다.
- 5 분 동안 95 ° C에 품 어 다음 모든 30 1 ° C로 냉각 열 cycler에서 4 ° c s.
- 4 ° c.에서 개최
- 끝-수리 및 라이브러리의 다-미행
참고: 어댑터 어 닐 링와 병렬로 하나 수행할 수 최종 수리 및 다 미행 단계 1.1에서 PCR amplicons. 단계 완료, 다음 최종 수리 및 PCR amplicons 다 꼬리에 단계 1.2에서 단련 된 어댑터의 결 찰이 수행 됩니다. 다음 어댑터 결 찰, ECS 도서관 건설 완료 됩니다.- 최대 1 µ g의 DNA를 시작으로 시작 (최소 ~ 200 ng)
- (자료 테이블, 항목 7) amplicons에 최종 수리 및 다 꼬리를 수행 합니다.
- 최종 준비 효소 혼합의 3.0 µ L 및 최종 수리 버퍼의 6.5 µ L를 추가 합니다.
- 65 ° C에서 30 분 동안 다음 20 ° C에서 30 분 동안 혼합 품 어와 4 ° c.에서 개최
- (자료 테이블, 항목 8) 단련된 어댑터에 결 찰을 수행 합니다.
- 2 단계, 블런트/TA 리가 Mastermix의 15 µ L 및 결 찰 증강의 1 µ L에서 단련 된 어댑터의 2.5 µ L를 추가 합니다.
- 다음 37 ° c.에 15 분 동안 20 ° C에서 15 분 동안 혼합을 품 어
- 마그네틱 구슬 (자료 테이블 항목 2) 라이브러리 정리: 구슬에 PCR 반응 수정된 1: 0.75 비율에 추가 (PCR 반응 볼륨: 마그네틱 비드 볼륨):
- 마그네틱 비드 솔루션의 62.6 µ L 단계 1.2.7에서에서 PCR 제품의 83.5 µ L로 플라스틱.
- 1.5 mL 낮은 바인딩 튜브에 혼합물을 전송.
- 적어도 10 배를 위아래로 pipetting으로 철저 하 게 혼합.
- 5 분 동안 실내 온도에 서 서 혼합물을 두십시오.
- 자석 홀더에 튜브를 놓습니다. 실내 온도에 또는 상쾌한까지 2 분 동안 품 어 분명 하다.
- 상쾌한을 제거 합니다.
- 구슬 200 µ L의 70% 에탄올으로 씻는 다.
- 30 미 제거 에탄올에 대 한 품 어.
- 한 번 에탄올 세척 단계를 반복 합니다.
- 구슬 건조
- 20 µ L ddH2오의와 함께 elute
참고:이 수정 자석 구슬 비율에 PCR 반응에서 우선적으로 200 보다 작은 DNA 조각 제거할 것 이다 혈압.
- 물방울에 의해 정량화 디지털 PCR
참고: 정확한 돌연변이 정량화 시퀀서에 로드 되는 각 라이브러리의 분자의 수의 엄격한 준수를 요구 한다. 이 위해 QX200 방울 디지털 PCR (ddPCR) 플랫폼을 사용 하 여 수행은 단위 부피 당 개별 라이브러리에 대 한 분자의 수를 측정-정량 PCR 대체 옵션입니다. DdPCR 분석에 따라 판독 라이브러리 당 µ L 당 분자의 수를 지정 합니다.- 증분 PCR 스트립-튜브에 10 배 희석 하 여 ECS 라이브러리 1:1,000을 희석.
- 1.5 mL 튜브에 ddPCR에 대 한 다음 mastermix 준비: PCR 혼합 (자료 테이블, 항목 9), P5 뇌관, P7 뇌관, 단계 1.4.1에서에서 ECS 청소 제품의 5 µ L의 0.2 µ L의 0.2 µ L의 10 µ L., 그리고 4.5 µ L ddH2o.
- 각 mastermix의 aliquot 20 µ L 샘플 잘 8의 배수는 다는 것을 확신 합니다.
- Aliquot 70 µ L 기름의 작은 물방울 생성 (자료 테이블, 항목 10) 각 유 정에. 고무 가스 켓으로 카세트 커버.
- 방울 방울 발생기 (자료 테이블, 항목 11)를 사용 하 여 확인 합니다.
- 멀티 채널 피 펫을 사용 하 여 생성 단계에서 1.4.4 PCR 플레이트 보장으로 DNA를 기울이기 방지 하려면 5 초 기간 동안 천천히 수행 되는 샘플의 pipetting 방울 로드 합니다.
- 다음과 같은 조건을 사용 하 여 열 cycler에서 40 주기 위한 작은 물방울에서 신호를 증폭: 95 ° C에서 5 분 30의 40 주기 1 분 63 ° C에서 95 ° C에서 s 4 ° C, 90 ° C에서 5 분에서 5 분 다음 4 ° c.에서 개최
- DdPCR 템플릿 드롭릿 리더 기계 (자료 테이블, 항목 11)를 준비 합니다. 절대 정량화 를 사용 하 여 매개 변수에 대 한 사양을 확인은 QX200 ddPCR에 바 그린 Supermix.
- DdPCR 분석 완료 되 면 모든 샘플에서 동일한 분열 임계값을 설정 해야 합니다.
- QX200 물방울 리더, 약 분자의 원하는 수 다음 단계를 적절 한 볼륨 수에서에서 농도 판독을 사용 하 여.
- 시퀀싱에 대 한 라이브러리의 PCR 증폭
- 1.4.9 단계에서 분자의 원하는 수에 대 한 다음 mastermix 준비: Q5 Mastermix (자료 테이블, 항목 1), P5 뇌관의 2.5 µ L의 25 µ L (10 µ M), P7 뇌관의 2.5 µ L (10 µ M), DNA, ddH2O. X 20 µ L의 X µL
- 증폭 단계 1.5.1 다음 조건을 사용 하 여 열 cycler에서에서 라이브러리: 30 s 98 ° c; 10의 20 주기 98 ° C, 30에서 s 63 ° C, 30에서 s s 72 ° c; 72 ° C에서 2 분 다음 4 ° c.에서 개최
-
마그네틱 구슬 (자료 테이블, 항목 2) 라이브러리 정리: 자석에 PCR 반응에는 수정 비즈 1: 0.75 비율 추가 (PCR 반응 볼륨: 마그네틱 비드 볼륨).
- 마그네틱 비드 솔루션의 37.5 µ L 단계 1.5.2에서에서 50 µ L PCR 제품으로 플라스틱.
- 1.5 mL 낮은 바인딩 튜브에 혼합물을 전송.
- 적어도 10 배를 위아래로 pipetting으로 철저 하 게 혼합.
- 5 분 동안 실내 온도에 서 서 혼합물을 두십시오.
- 자석 홀더에 튜브를 놓습니다. 실내 온도에 또는 상쾌한까지 2 분 동안 품 어 분명 하다.
- 상쾌한을 제거 합니다.
- 구슬 200 µ L의 70% 에탄올으로 씻는 다.
- 30 미 제거 에탄올에 대 한 품 어.
- 한 번 에탄올 세척 단계를 반복 합니다.
- 구슬 건조
- 20 µ L ddH2오의와 함께 elute
- amplicons의 크기를 확인 하는 2 %agarose 젤에서 DNA의 약 수를 실행 합니다.
- 계량 (재료 표, 항목 3) 별도 ECS 라이브러리의 농도 결정 하는 DNA의 농도.
- 아데닌 양의 라이브러리를 풀.
참고: 예를 들어 연구자 수 풀까지 400 백만 읽기 출력 시퀀싱 플랫폼을 사용 하 여 연속에 대 한 분자를 시작 하는 4 백만 아데닌 그룹4 에 8 개의 라이브러리. 보수적으로, 분자 당 오류 수정에 대 한 10 개의 원시 읽기의 평균을 사용 하는 것이 좋습니다. 이 360 백만 읽기를 걸릴 것 이다 (4 백만 분자 * 8 라이브러리 * 10 읽는 오류 수정에 대 한). 라이브러리 당 4 백만 고유 분자로, 연구원은 이론적인 의미 합의 amplicon (유전자 패널에서 4 백만/568 amplicons) 당 7042 x의 범위를 기대할 수 있습니다. - 계량 (재료 표, 항목 3) 풀링된 ECS 라이브러리의 농도 결정 하는 DNA의 농도.
- 대략 4에서 풀링된 ECS 도서관 제출 nM.
- Illumina의 시퀀싱 플랫폼 (MiSeq, HiSeq 또는 NextSeq) 다음 시퀀싱 설정을 제공: 2 x 144 한 쌍-엔드 읽습니다, 8 인덱스 1 주기와 16 주기 인덱스 2.
2. 유전자의 DNA 시퀀싱 오류 수정 패널
- 유전자 패널에서 oligos의 교 잡
참고:이 단계에서 Umi (자료 테이블, 항목 17)를 통합 하는 수정된 Illumina TruSight 또는 TruSeq 프로토콜을 사용 하 여 시퀀싱 라이브러리를 건설할 것 이다 하나.- 제조 업체의 프로토콜을 따르고 게놈 조각에 oligos 교배 DNA (또는 시작 물자의 원하는 금액)의 사용 250 ng.
- 언바운드 oligos 다음 제조 업체의 프로토콜을 제거 합니다.
- 확장-결 찰을 수행 다음 제조 업체의 프로토콜.
참고: 제조업체의 프로토콜에 대 한 수정 아래 시작합니다.
- PCR 통해 i5 및 i7 어댑터의
- 적절 한 볼륨 크기의 관으로 다음 시 약 pipetting으로 PCR mastermix 준비: Q5 Mastermix (자료 테이블, 항목 1), 6 µ L 10 µ M 16N i5 어댑터 (에 자세한 방법 1, 단계 1.2.2), i7 어댑터 (사용 다른 i7의 6 µ L의 37.5 µ L 다중화에 대 한 별도 샘플에 대 한 어댑터) 및 22 µ L 단계의 2.1.3 구슬과 확장 결 찰 솔루션의.
참고: Q5 Mastermix Illumina에서 제공 하는 중 합 효소 mastermix를 대체 합니다. Q5 중 합 효소는 높은 충실도와 적은 도입된 오류 게놈 단편을 증폭 시키고 있다. - PCR 프로그램을 실행 하는 다음 매개 변수를 사용 하 여 열 cycler에: 30 s 98 ° C에, 10의 4-6 주기 98 ° C, 30에서 s 66 ° C, 30에서 s s 72 ° c; 72 ° C, 및 4 ° c.에 대기에서 2 분
참고: 주기 수 패널 크기에 따라 달라 집니다. 우리의 경험에서 oligos의 500-600 쌍 패널 PCR의 6 주기를 요구 하는 반면 4 사이클 PCR 유전자 패널 유전자 특정 oligos의 약 1500 다른 쌍 있으면 충분 하다. -
마그네틱 구슬 (자료 테이블, 항목 2) PCR 반응 정리: 수정된 1 PCR 반응에서 PCR 반응 자석 구슬 추가: 0.75 자석 구슬 비율:
- 마그네틱 비드 솔루션의 56.25 µ L 단계 2.2.2에서에서 PCR 제품의 75 µ L로 플라스틱.
- 1.5 mL 낮은 바인딩 튜브에 혼합물을 전송.
- 적어도 10 배를 위아래로 pipetting으로 철저 하 게 혼합.
- 5 분 동안 실내 온도에 서 서 혼합물을 두십시오.
- 자석 홀더에 튜브를 놓습니다. 실내 온도에 또는 상쾌한까지 2 분 동안 품 어 분명 하다.
- 상쾌한을 제거 합니다.
- 구슬 200 µ L의 70% 에탄올으로 씻는 다.
- 30 미 제거 에탄올에 대 한 품 어.
- 한 번 에탄올 세척 단계를 반복 합니다.
- 구슬 건조
- 20 µ L ddH2오의와 함께 elute
- 적절 한 볼륨 크기의 관으로 다음 시 약 pipetting으로 PCR mastermix 준비: Q5 Mastermix (자료 테이블, 항목 1), 6 µ L 10 µ M 16N i5 어댑터 (에 자세한 방법 1, 단계 1.2.2), i7 어댑터 (사용 다른 i7의 6 µ L의 37.5 µ L 다중화에 대 한 별도 샘플에 대 한 어댑터) 및 22 µ L 단계의 2.1.3 구슬과 확장 결 찰 솔루션의.
- QX200 ddPCR 플랫폼을 사용 하 여 라이브러리를 계량.
- 1.4 방법 1의에서 단계를 따릅니다.
참고: 4 백만 분자 7,042 고유 하 게 인덱싱된 분자 (568 유전자 특정 oligos 나눈 4 백만)의 이론적 의미를 얻으려면 샘플 라이브러리4 는 대표적인 결과 (그림 2)에서 당 정상화 했다.
- 1.4 방법 1의에서 단계를 따릅니다.
- 증폭 하 고 시퀀싱에 대 한 라이브러리를 정상화.
- 다음 mastermix를 사용 하 여 최종 PCR 총 50 µ L에 대 한 분자의 원하는 수 증폭: Mastermix Q5, P5 뇌관의 2 µ L의 25 µ L (1 µ M), P7 뇌관의 2 µ L (1 µ M), 그리고 DNA 분자의 21 µ L.
- 다음 매개 변수를 사용 하 여 열 cycler에 PCR 프로그램 실행: 30 s 98 ° c; 10의 16 주기 98 ° C, 30에서 s 66 ° C, 30에서 s s 72 ° c; 72 ° C에서 2 분 다음 4 ° c.에서 개최
- 마그네틱 구슬 (자료 테이블, 항목 2)를 사용 하 여 시퀀싱 라이브러리 정리: 수정된 1 PCR 반응에서 PCR 반응 자석 구슬 추가: 0.75 자석 구슬 비율:
- 2.4.2 단계에서 50 µ L PCR 제품으로 자석 구슬 솔루션의 37.5 µ L 플라스틱.
- 1.5 mL 낮은 바인딩 튜브에 혼합물을 전송.
- 적어도 10 배를 위아래로 pipetting으로 철저 하 게 혼합.
- 5 분 동안 실내 온도에 서 서 혼합물을 두십시오.
- 자석 홀더에 튜브를 놓습니다. 실내 온도에 또는 상쾌한까지 2 분 동안 품 어 분명 하다.
- 상쾌한을 제거 합니다.
- 구슬 200 µ L의 70% 에탄올으로 씻는 다.
- 30 미 제거 에탄올에 대 한 품 어.
- 한 번 에탄올 세척 단계를 반복 합니다.
- 구슬 건조
- 20 µ L ddH2오의와 함께 elute
- 달리 eluted DNA의 약 수 (~ 3 µ L)는 amplicons의 크기를 확인 하는 2 %agarose 젤에.
- 계량 (재료 표, 항목 3) 별도 ECS 라이브러리의 농도 결정 하는 DNA의 농도.
- 아데닌 양의 라이브러리를 풀. 1.5.6 방법 1 단계를 참조 하십시오. 그리고 또한 토론에 대 한 자세한 내용은 풀링입니다.
- 대략 4에서 풀링된 ECS 도서관 제출 nM.
- Illumina의 시퀀싱 플랫폼 (MiSeq, HiSeq 또는 NextSeq) 다음 시퀀싱 설정을 제공: 2 x 144 한 쌍-엔드 읽습니다, 8 인덱스 1 주기와 16 주기 인덱스 2.
- ECS Bioinformatic 처리 및 분석
- 시퀀서에서 샘플 역다중화 읽기를 얻거나 다른 샘플 i7 어댑터 시퀀스 bioinformatically를 사용 하 여 사용자 지정 스크립트에 원시 시퀀스 읽기 디 멀티플렉싱 수행.
- 유전자 패널에서 올리고 시퀀스를 제거 하려면 각 역다중화 읽기의 처음 30 뉴클레오티드 떨어져 트림.
- 읽기 읽기 가족을 형성 하기 위하여 서로 동일한 Umi를 공유 하는 정렬.
참고: 연구원 읽기 가족 추출 MAGERI13 등 우미 인식 소프트웨어를 사용할 수 있습니다. 아니 hamming 거리 방법의 특이성을 증가이 실험에서 우미 시퀀스 내에서 허용 되었다. - 중복 및 오류 수정 사용 하 여 매개 변수를 권장 하는 다음을 수행 합니다.
- 사용 ≥5 읽기 같은 쌍 가족을 읽고. 3 읽기 쌍의 최소 것이 좋습니다.
- 같은 읽기 가족에 모든 읽기에 걸쳐 모든 포지션에서 뉴클레오티드를 비교 하 고 90% 이상의 경우 합의 뉴클레오티드를 생성 특정 염기에 대 한 읽기 사이에서 일치. 90% 계약 미만 경우 N 뉴클레오티드 위치에 대 한 호출 합니다.
- 버리고 있는 합의 읽기 > 명으로 호출 되 고 합의 세포핵의 총 수의 10%
- Bowtie2 및 BWA 등 연구원의 기본 aligner(s)를 사용 하 여 hg19 또는 hg38 인간 참조 게놈을 로컬로 모든 유지 합의 읽기를 맞춥니다.
- 프로세스 Mpileup 매개 변수를 사용 하 여 읽기 정렬-BQ0-d VAF에 적절 한 탑 쌓기 출력 되도록 범위 임계값을 제거 하는 10,000,000,000,000.
- 미만 1000 x 합의 범위와 위치 밖으로 필터링.
참고: 각 뉴클레오티드 위치에 대 한 최소 범위를 임의로 결정 하는 연구원이 좋습니다 적어도 500 x 합의 다운스트림 분석에 대 한 범위를가지고. - 다음 매개 변수 2.5.7 단계에서 보관 된 데이터에 단일 뉴클레오티드 변종 (Snp) 전화를 이항 분포를 사용 합니다. 이항 통계 genomic 위치 특정 오류 모델을 기반으로 합니다. 각 게놈 위치는 특정 위치에 대 한 모든 샘플의 오류 요금 요약 후 독립적으로 모델링 됩니다. 다음 예제에서는:
지정 된 genomic 위치, p 뉴클레오티드 프로필의 확률
∑ 변형 RF2 ∑ 총 RFs
26/255505 =
0.000101759 =
24 변종 35911 총 RFs, P중 RFs의 이항 확률 (X ≥ x) 샘플 K에
= 1-binomial(24, 35911, 0.000101759)
2.26485E =-13
참고: 쿼리 각 게놈 위치에 대 한 것 세 가지 가능한 mutational 변화 (즉,A > T, A > C, A > G), 각각의 배경 유물으로 나타낼 것 이다. 체세포 이벤트는 Bonferroni 보정 후 크게 다른 배경 유지 됩니다. 표 1에 표시 된 예제에서 수행 하는 테스트의 수는 11, 따라서는 Bonferroni 수정 p-≤0.00454545 값 (0.05/11) 중요 한 통계로 이벤트를 호출 하는 데 필요한 했다. - 체세포 이벤트는 모두 복제에; 동일한 견본에서 존재 하는 데 필요한 그렇지 않으면 가양성으로 그들을 간주 한다.

표 1: 예제 위치 특정 이항 오류 모델을 구성 하는 방법을 설명 합니다.
3. RNA의 연속 오류 수정
-
DNA 수준에서 돌연변이 대 한 평가 뿐만 아니라 RNA 수준에서 드문 또는 낮은 풍부 대 본을 검출 하기 위하여 다양 한 타겟된 RNA 시퀀싱 패널 ECS 통합. 상용 Qiagen RNA 시퀀싱 패널 ECS를 결합 하 여 우리는 몇 가지 내부 관리 유전자에 대 한 정규화를 위한 필요 없이 10 부로와 성적 증명서에 대 한 유전자 발현의 디지털 정량화 시연. Umi 오류 정정에 필요한 패널에 통합 되어 있다.
- 총 RNA 추출 (자료 테이블, 항목 20)을 수행 합니다.
- ECS RNA 라이브러리 준비 제조 업체의 프로토콜 (자료 테이블, 항목 19)에 따라 실시 합니다.
- 생물 정보학 파이프라인 단계 2.5.1–2.5.6에 따라 수행 합니다. 이전 섹션에서 설명 하는 방법 2. 2.5.6 단계, 후 진 당 정렬된 합의 읽기 수 유전자 길이 정규화에 대 한 필요 없이 유전자의 식 수준을 나타냅니다.
결과
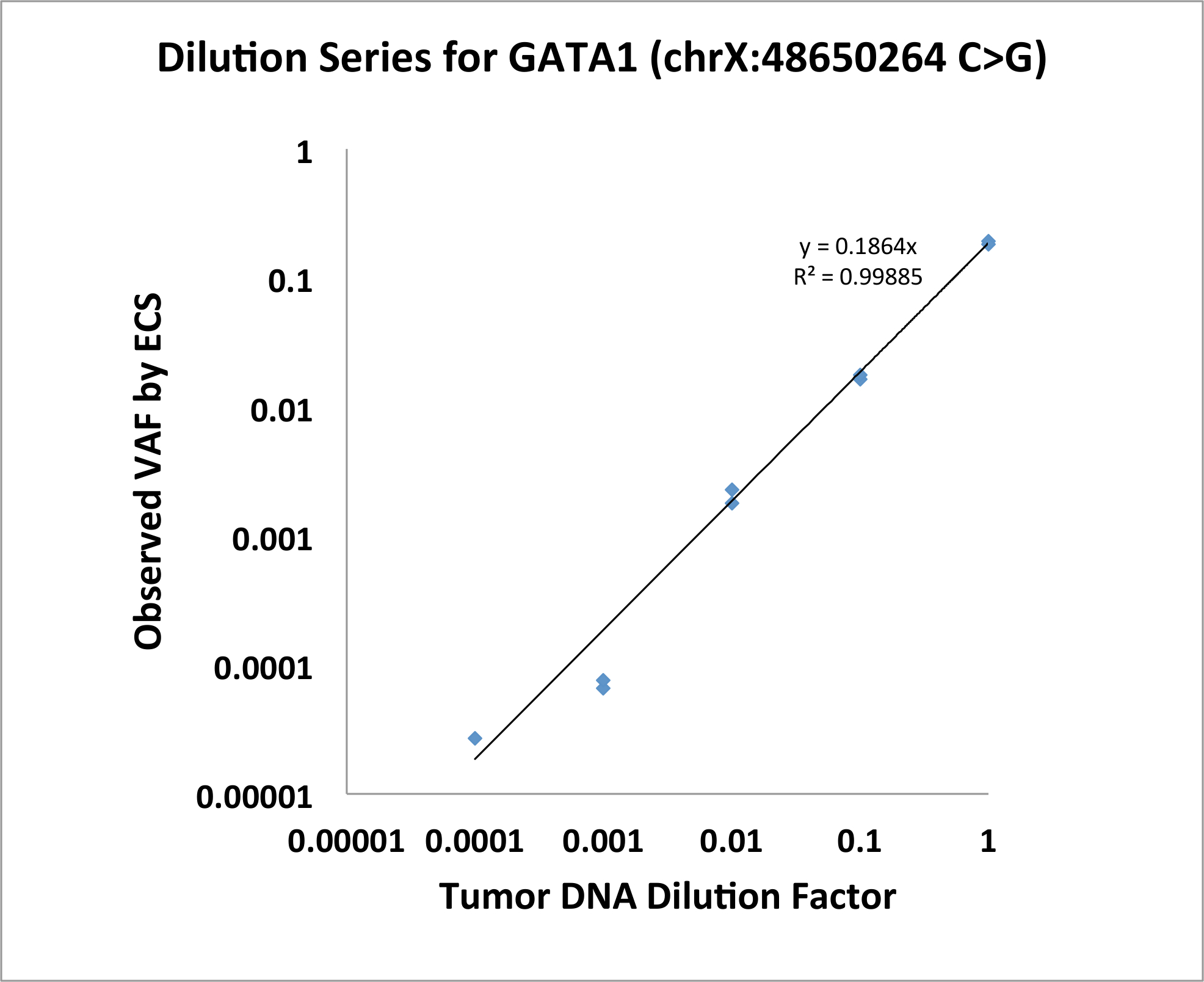
Targeted Error-Corrected dna 시퀀싱, 함께 우리는 돌연변이 환자 상업 genomic DNA에 DNA를 diluting 원리 실험의 증거를 수행 했습니다. 환자 GATA1 돌연변이 했다 (chrX:48650264, C > G) 0.19의 원래 VAF와. ECS 단일 뉴클레오티드 변종 1:10,000의 수준으로 양적은 그림 1 에서 설명 합니다.

그림 1: GATA1 SNV 보여주는 ECS 1:10,000의 수준으로 양적은 희석 시리즈. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
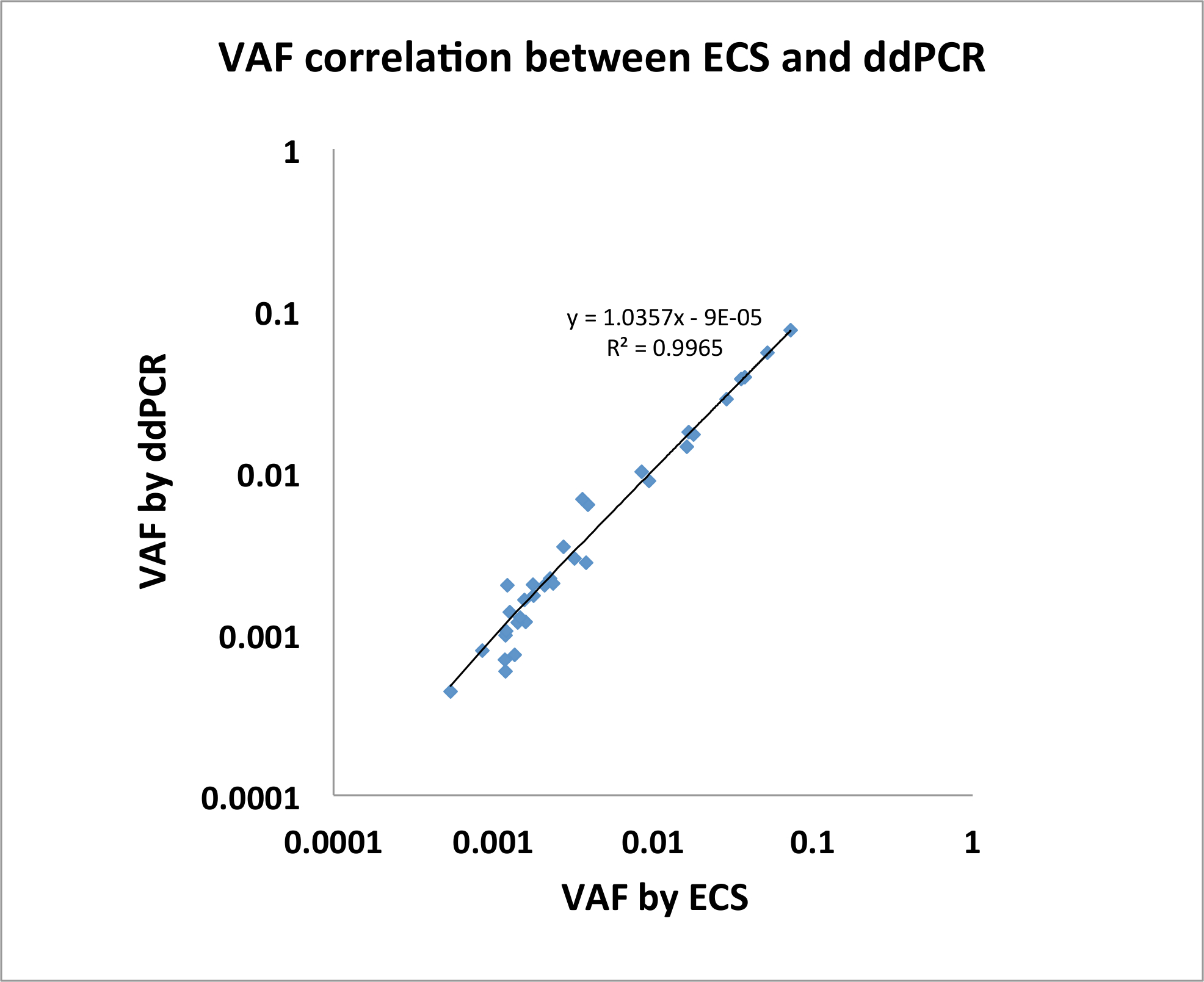
우리는 또한 ECS DNA 안정적으로 성인 급성 골수성 백혈병 (AML) 건강 한 노인 개인4에 recurrently 유전자에 드문 클론 변이 감지 하는지 보여준다. 우리는 간호사의 건강 연구 틀 약 떨어져 ~ 10 년에서에서 20 건강 한 개인에서 버 피 코트 샘플을 얻었다. 우리는이 샘플에 ECS-DNA 패널 프로토콜을 적용. 이 실험에 대 한 우리 Illumina TruSight 골수성 시퀀싱 패널 568 amplicons (자세한 내용은 https://www.illumina.com/products/by-type/clinical-research-products/trusight-myeloid.html에 유전자 목록에) 구성 된 적응 및 시퀀싱 20 개인에서 80 라이브러리 (다른 시간 지점에서 2 컬렉션, 시간 당 개인 당 2 복제 지점) 47.7 백만 쌍 간 읽기의 평균 및 오류를 수정 하는 3.4 백만의 평균을 생성 Illumina NextSeq 플랫폼을 사용 하 여 도서관4당 합의 시퀀스입니다. 라이브러리 당 평균 뉴클레오티드 커버리지 (3.4 수백만 568로 나눈 값) x 6000 대략 이었다. 각 샘플에 대 한 우리는 동일한 샘플에서 되지 않은 시퀀스 된 라이브러리를 사용 하 여 위치 특정 오류 프로필을 건설 한다. 우리는 하나 이상의 컬렉션 시간 지점의 두 복제에 109 클론 체세포 돌연변이 발견. 이러한 돌연변이 VAF 0.0003-0.1451에서 배열 있다. 우리가 알려진된 우주 표현으로 21 돌연변이 선택 하 고 하나 또는 두 개의 컬렉션 시간 포인트 ddPCR를 사용 하 여 모든 21 돌연변이 확인 (n = 34, 그림 2, 젊은 외. 20164에서 적응).

그림 2: ECS로 식별 하는 돌연변이 매우 조화 된 VAFs와 ddPCR를 통해 확인 했다. (n = 34, 젊은 외. 20164수정). 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
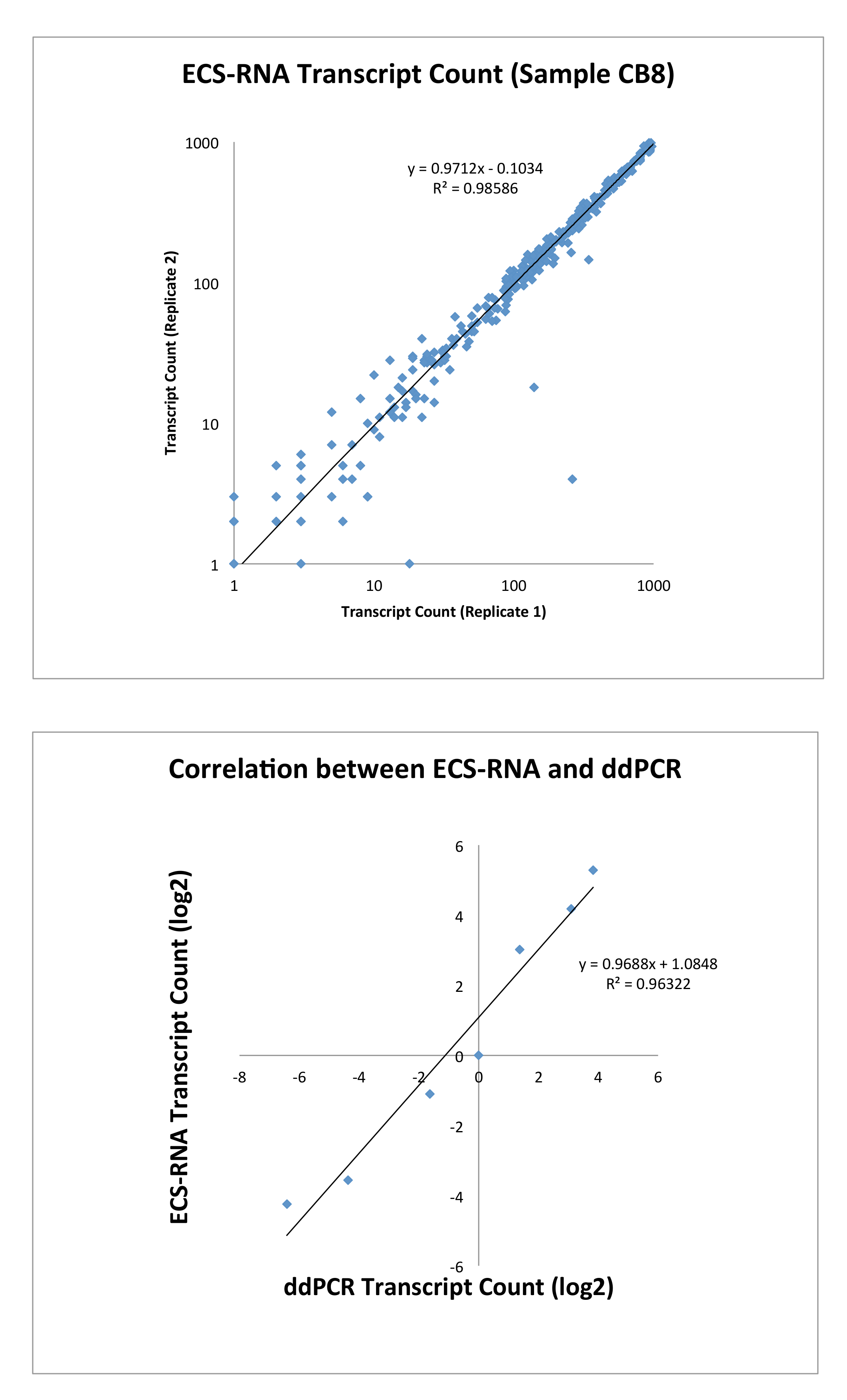
ECS RNA 프로토콜을 사용 하 여 오류 수정 식 수준에 관하여 우리 다양 한 암 (QIAseq 인간의 암 Transcriptome 패널에서 적응), 그리고 우리와 관련 된 것으로 알려진 416 유전자의 구성 된 QIAseq 화학을 사용 하 여 유전자 패널 사용자 정의 ( 보충 자료1에서 유전자 목록) 주어진된 유전자의 가장 일반적으로 표현한 exon 증폭. 우리 쌍 간 형식 라이브러리, 당 8.3 백만 읽기의 평균을 준 Illumina MiSeq 플랫폼을 사용 하 여 라이브러리를 시퀀싱 그리고 우리 0.417 백만 오류 수정 합의 시퀀스의 평균을 잡으려고 관리. 우리 식 수준 낮은 풍부한 사본 보여주었다 (< 50에서 1000 사본 수 총 RNA의 ng) 복제 사이 높은 재현성은 (데이터 요소 n = 300, 그림 3). DdPCR (식의 다양 한 정도의 6 선택 된 유전자)에 의해 유효성 검사 유전자의 식 수준 올바르게 정규화에 대 한 필요 없이 ECS 프로토콜에 의해 점령 되었습니다 했다 설명 했다.

그림 3: 상단, 대 본의 상관 관계 계산 ECS RNA에 의해 동일한 샘플의 복제 사이 (n = 300). 하단, ECS로 식별 하는 건의 했다 ddPCR에 의해 확인 하는 사본 (n = 6). 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
{kind=link}
토론
다른 질병에 낮은 VAFs와 돌연변이 연구를 쉽게 구현할 수 있는 오류 수정 시퀀싱 프로토콜의 집합을 설명 합니다. 그들은 원시 읽기 오류 보정을 가능 하 게 가장 중요 한 요소 시퀀싱 전에 각 분자 Umi의 법인입니다. 여기에 설명 된 방법을 상용 유전자 패널을 자체 설계 유전자 특정 oligos 사용자 지정된 Umi를 통합 하는 연구를 허용 합니다.
표준 NGS 프로토콜 precludes 시퀀싱 오류 비율 때문 2% 아래 VAF와 변이의 탐지 하 고이 희소 한 이체의 탐지는 중요 한 연구에서 NGS의 응용 프로그램을 제한 합니다. 우회 표준 NGS 오류율, ECS이 원시 이체의 민감한 감지 수 있습니다. 예를 들어, 이러한 돌연변이 처음 (따라서 데 낮은 VAF) 발생 때 병원 성 변이의 탐지 병14,15의 조기 개입에 게 필수적입니다. 백혈병 연구, 최소한의 잔류의 검출에에서 질병 (잔여 leukemic 세포 후 처리) 위험 계층을 알립니다 및 이진 흐름 cytometric 평가 수 없습니다 하는 방식으로 치료 옵션을 통보 하는 데 사용할 수. 또한, ECS 순환 종양 핵 산을 검출 하 고 단단한 종양 환자에서 전이성 잠재력을 평가 하는 존재/부재로의 기본 특성은 특정 돌연변이의 변형 부담을 평가 하 여 적용 됩니다. 종양16.
표 1에서처럼 변종 전화 위치 특정 오류 이항 분포-기반 모델을 사용 하 여 전원 시퀀싱 오류 모델을 구축 하는 데 사용의 깊이 뿐만 아니라 시퀀스 라이브러리의 수에 따라 달라 집니다. 오류 모델의 견고성 샘플 및 시퀀싱 깊이 더 높은 수가 증가합니다. 각 샘플에 대 한 오류 프로필 범위의 오류 수정 읽기 샘플 당 3000 x의 평균 최소한 10 시퀀스 샘플을 사용 하는 것이 좋습니다. 위치-특정 방식은 비슷한 MAGERI, 하지만 모든 6 다른 대체 종류에 대 한 집계 오류 비율을 사용 하는 대신 (A > C/T > G, A > G/T > C, A > T/T > A, C > A/G > T, C > G/G > C C > T/G > A)13, 우리 모든 위치에 독립적으로 각 대체 모델. 예를 들어, C의 오류 비율 > T는 genomic 위치는 다른 위치에서 다른. 우리의 접근 방법 또한 걸립니다 계정에 연속 배치 효과 관찰 한 시퀀싱 실행 기본 대체 속도에서 실행 하는 다른 다를 수 있습니다. 따라서 그것은 특히 다른 시퀀싱 실행에서 샘플 모델 구축 풀링되지 때 모든 대체 형식에 대 한 각 위치를 모델링 하는 것이 중요.
ECS 실험을 설계할 때 중요 한 고려 사항은 원하는 검출 임계값입니다. NGS 연구의 아름다움은 그 그들은 쉽게 확장할 수 유전자/대상 관심, 검출 임계값 (시퀀싱의 깊이 의해 결정), 및 쿼리 하는 개인의 수의 점에서. 예를 들어 연구원은 0.0001의 감지 임계값을 갖는 2 개의 amplicons에 드문 돌연변이 찾을 관심이 있다면, 그들은 최대 15 백만 읽기 출력 MiSeq V2 화학을 사용 하 여 실행 한 시퀀싱에 극대로 75 샘플 수영장 수 (2 amplicons * 10000 분자 * 10 오류 수정에 대 한 읽기 * 75 샘플 15 백만 시퀀싱 읽기 =). 연구원은 시퀀싱 또는 단일 시퀀스 검출 임계값 조정 실행에 풀링된 샘플 수로가 하는 분자의 수를 변화할 수 있다. 우리의 연구에서 우리 0.0001 VAF (1:10, 000)의 감지 임계값으로 돌연변이 찾을 목적 Illumina 유전자 패널을 사용 하 여. 우리가 일상적으로 250를 사용 하 여 상기 검출 임계값을 달성 하기 위해 충분 한 분자 캡처되는지 확인에 DNA 시작의 ng. 연구팀은 DNA의 낮은 금액으로 시작 하도록 선택할 수 있습니다 (50 ng는 것이 좋습니다) 원하는 검색 제한이 > 0.001 VAF.
Umi i5 인덱스에 추가 됩니다, 시퀀싱 설정을 적절 하 게 수정 되어야 할. 예를 들어 16 N Umi, 사용 하 고 시퀀싱 설정 된 2 x 144 짝된 끝 읽기, 인덱스 1의 8 주기 및 인덱스 2의 일반적인 8 사이클 반대로 인덱스 2의 16 주기. 인덱스 2 주기 증가 사이클 읽기에 할당 된의 총 수에 있는 감소에 의해 보상 된다. 연구원은 12N Umi10,17를 사용 하기로 선택한 경우 인덱스 2의 12 주기를 설정은 변경 되어야 합니다.
이 우미 기반 시퀀싱 방법은 시퀀싱 오류 수정 최적화 됩니다. 모든 확대 기반 방법에 대 한 문제는 차선의 PCR jackpotting 처리에 남아 있습니다. 라운드 후 시퀀싱 및 게시물-생물 정보학 유효성 검사 ddPCR를 사용 하 여 수행 하 고 거의 PCR jackpotting 때문에 어떤 잘못 된 반응 감지. 그럼에도 불구 하 고, 연구원은 낮은 증폭 오류를 고화질 중 합 효소를 사용 하 여 실험을 실시 하는 것이 좋습니다.
공개
저자는 공개 없다.
감사의 말
우리 아 이들의 종양학 그룹 AAML1531 연구 및 환자 샘플의 형태로 그들의 공헌에 대 한 간호사 건강 연구에서 참가자를 감사합니다. 이 작품은 건강의 국가 학회 (UM1 CA186107, RO1 CA49449 RO1 CA149445), 아 이들의 발견 연구소의 워싱턴 대학교 세인트 루이스 어린이 병원 (엠씨-II-2015-461)와 일 라이 세스 매튜 스 백혈병 재단에 의해 투자 되었다.
자료
| Name | Company | Catalog Number | Comments |
| Q5 High Fidelity Hot Start Master Mix | New England BioLabs | M0492S | |
| Agencourt AMPure XP | Beckman Coulter | A63880 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| SYBR Safe DNA Gel Stain | Thermo Fisher Scientific | S33102 | |
| Truseq Custom Amplicon Index Kit | Illumina | FC-130-1003 | |
| UMI i5 adapter sequences | Integrated DNA Technologies | - | |
| NEBNext Ultra End Repair/dA-Tailing Module | New England BioLabs | E7442S | |
| NEBNext Ultra II Ligation Module | New England BioLabs | E7595S | |
| QX200 ddPCR EvaGreen Supermix | Bio-Rad | 1864034 | |
| QX200 Droplet Generation Oil for EvaGreen | Bio-Rad | 1864005 | |
| QX200 Droplet Digital PCR System | Bio-Rad | 1864001 | |
| ddPCR 96-Well Plates | Bio-Rad | 12001925 | |
| DG8 Cartridges for QX200/QX100 Droplet Generator | Bio-Rad | 1864008 | |
| DG8 Gaskets for QX200/QX100 Droplet Generator | Bio-Rad | 1863009 | |
| Bioanalyzer | Agilent Genomics | G2939BA | |
| TapeStation | Agilent Genomics | G2991AA | |
| TruSight Myeloid Sequencing Panel | Illumina | FC-130-1010 | |
| Bowtie 2 | Johns Hopkins University | - | |
| Customized QIAseq Targeted RNA Panel | Qiagen | - | |
| Rneasy Plus Mini Kit (50) | Qiagen | 74134 |
참고문헌
- Hoang, M. L., et al. Genome-wide quantification of rare somatic mutations in normal tissues using massively parallel sequencing. Proceedings of the National Academy of Sciences USA. 113, 9846-9851 (2016).
- O'Roak, B. J., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 485, 246-250 (2012).
- Young, A. L., et al. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia. 29 (7), 1608-1611 (2015).
- Young, A. L., Challen, G. A., Birmann, B. M., Druley, T. E. Clonal hematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. NatureCommunications. 7, 12484(2016).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New England Journal of Medicine. 366, 1079-1089 (2012).
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Kohlmann, A., et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia. 28, 129-137 (2014).
- Luthra, R., et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 99, 465-473 (2014).
- Kinde, I., Wu, J., Papadopoulos, N., Kinzler, K. W., Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences USA. 108 (23), 9530-9535 (2011).
- Schmitt, M., et al. Detection of ultra-rare mutations by next-generation sequencing. Proceedings of the National Academy of Sciences USA. 109 (36), 14508-14513 (2012).
- Vander Heiden, J. A., et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 30 (13), 1930-1932 (2014).
- Newman, A. M., et al. Integrated digital error suppression for improved detection of circulating tumor DNA. NatureBiotechnology. 34, 547-555 (2016).
- Shugay, M., et al. MAGERI: Computational pipeline for molecular-barcoded targeted resequencing. PLOSComputationalBiology. 13 (5), e1005480(2017).
- Wong, T. N., et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 518, 552-555 (2014).
- Krimmel, J. D., et al. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proceedings of the National Academy of Sciences USA. 113 (21), 6005-6010 (2016).
- Phallen, J., et al. Direct detection of early-stage cancers using circulating tumor DNA. ScienceTranslationalMedicine. 9, eaan2415(2017).
- Egorov, E. S., et al. Quantitative profiling of immune repertoires for minor lymphocyte counts using unique molecular identifiers. The Journal of Immunology. 194 (12), 6155-6163 (2015).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유