
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
TIRF 현미경에 의한 시험관 내에서 가교 및 단일 미세 소 관 의 역학의 동시 시각화
요약
여기서, TIRF 현미경 기반 시험관내 재구성 분석법이 제시되어 두 미세소관 집단의 역학을 동시에 정량화하고 비교한다. 가교된 미세소관 다발 및 단일 미세소관 상에서 다수의 미세소관-관련 단백질의 집단 활성을 동시에 보는 방법이 기술된다.
초록
미세소관은 세포에서 별개의 구조로 조직되는 αβ-튜불린 이종이량체의 중합체이다. 미세소관 기반 아키텍처 및 네트워크에는 종종 동적 특성이 다른 미세소관 어레이의 하위 집합이 포함되어 있습니다. 예를 들어, 분열 세포에서, 가교결합된 미세소관의 안정한 다발은 동적 비가교된 미세소관에 근접하여 공존한다. TIRF-현미경-기반 시험관 내 재구성 연구는 이들 상이한 미세소관 어레이의 역학의 동역학의 동시 시각화를 가능하게 한다. 이 분석에서, 이미징 챔버는 단일 필라멘트로 존재하거나 가교된 다발로 조직되는 표면 고정화된 미세소관으로 조립된다. 튜불린, 뉴클레오티드 및 단백질 조절제의 도입은 관련 단백질 및 단일 및 가교된 미세소관의 동적 특성의 직접적인 시각화를 가능하게 한다. 또한, 동적 단일 미세소관이 번들로 구성될 때 발생하는 변화를 실시간으로 모니터링할 수 있습니다. 여기에 설명 된 방법은 개별 단백질의 활성 및 국소화에 대한 체계적인 평가뿐만 아니라 동일한 실험 조건 하에서 두 개의 서로 다른 미세 소관 하위 집합에 대한 단백질 조절제의 상승 효과를 허용하여 다른 방법으로는 접근 할 수없는 기계론적 통찰력을 제공합니다.
서문
미세소관은 세포 내 수송 및 소기관 위치에서부터 세포 분열 및 신장에 이르기까지 여러 세포 과정에 필수적인 구조적 스캐폴드를 형성하는 바이오 폴리머입니다. 이러한 다양한 기능을 실행하기 위해 개별 미세 소관은 유사분열 스핀들, 섬모 축삭, 뉴런 번들, 상 간 배열 및 식물 피질 배열과 같은 미크론 크기의 배열로 구성됩니다. 이 구조에서 발견되는 유비쿼터스 건축 모티프는 길이를 따라 가교 된 미세 소관 묶음입니다1. 몇몇 미세소관-기반 구조의 흥미로운 특징은 번들링된 미세소관과 비가교된 단일 미세소관이 가까운 공간적 근접성에서 공존한다는 것이다. 이러한 미세소관 하위집단은 적절한 기능을 위해 필요에 따라 서로 뚜렷하게 다른 중합 역학을 나타낼 수 있습니다2,3,4,5. 예를 들어, 유사분열 스핀들 내에서, 안정한 가교결합된 다발과 동적 단일 미세소관은 세포 중심6에서 미크론 스케일 영역 내에 존재한다. 따라서 공존하는 미세 소관 집단의 동적 특성이 어떻게 지정되는지를 연구하는 것은 미세 소관 기반 구조물의 조립과 기능을 이해하는 데 핵심입니다.
미세소관은 중합과 탈중합의 단계 사이를 순환하는 역동적인 중합체로, 재앙과 구조7로 알려진 사건에서 두 단계 사이를 전환합니다. 세포 미세 소관의 역학은 미세 소관 중합 및 탈중합 속도와 재앙 및 구조 사건의 빈도를 조절하는 무수한 미세 소관 관련 단백질 (MAPs)에 의해 조절됩니다. 세포의 공간적으로 근위 배열에 대한 MAP의 활성을 조사하는 것은 어려운 일이며, 특히 미세 소관 밀도가 높은 영역에서 광 현미경 검사에서 공간 분해능의 한계 때문입니다. 또한, 동일한 세포 영역에 여러 MAP가 존재하면 세포 생물학적 연구의 해석을 방해합니다. TIRF(Total Internal Reflection Fluorescence) 현미경과 함께 수행되는 시험관내 재구성 분석은 MAP의 특정 하위 집합이 근위 세포 미세소관 어레이의 역학을 조절하는 검사 메커니즘의 과제를 회피합니다. 여기서, 시험관내에서 조립된 미세소관의 역학은 조절된 조건하에서 하나 이상의 재조합 MAPs의 존재 하에 검사된다8,9,10. 그러나, 종래의 재구성 분석은 전형적으로 단일 미세소관 또는 한 유형의 어레이에서 수행되어, 공존하는 집단의 시각화를 배제한다.
여기에서, 우리는 동일한 용액 조건 하에서 두 개의 미세소관 집단의 동시 시각화를 가능하게 하는 시험관내 재구성 검정을 제시한다11. 우리는 단일 미세 소관과 유사분열 스핀들 관련 단백질 PRC1에 의해 가교 된 미세 소관 다발에서 여러 MAP의 집단 활동을 동시에 보는 방법을 설명합니다. 단백질 PRC1은 항평행 미세소관 사이의 중첩에서 우선적으로 결합하고, 이들을 가교결합시킨다9. 간략하게, 이 프로토콜은 (i) 스톡 용액 및 시약의 제조, (ii) 현미경 실험용 이미징 챔버를 만드는 데 사용되는 커버슬립의 세척 및 표면 처리, (iii) 실험 중에 중합이 개시되는 안정적인 미세소관 "종자"의 제조, (iv) 미세소관 역학을 시각화하기 위한 TIRF 현미경 설정의 사양, (v) 미세소관 종자의 고정화 및 가교된 미세소관 번들의 생성 이미징 챔버에서, 및 (vi) TIRF 현미경을 통해 이미징 챔버 내의 미세소관 역학의 시각화는, 가용성 튜불린, MAPs, 및 뉴클레오티드의 첨가시. 이러한 분석은 MAP 국소화의 정성적 평가 및 정량적 검사와 두 개의 미세 소관 집단의 역학에 미치는 영향을 가능하게합니다. 또한, 그들은 광범위한 실험 조건에 걸쳐 이러한 미세 소관 집단에 대한 여러 MAP의 상승 효과의 평가를 용이하게합니다.
프로토콜
1. 시약 준비
- 표 1 및 표 2에 요약된 바와 같이 완충제 및 시약을 준비 한다. 실험 중에 달리 명시되지 않는 한 모든 용액을 얼음 위에 보관하십시오.
| 용액 | 구성 요소 | 권장 저장 기간 | 노트 | ||
| 5X BRB80 | 400 mM K-파이프, 5 mM MgCl2, 5 mM EGTA, KOH로 pH 6.8, 필터 멸균 | 최대 2년 | 4 °C에서 보관 | ||
| 1X BRB80 | 80 mM K-파이프, 1 mM MgCl2, 1 mM EGTA, pH 6.8 | 최대 2년 | 4 °C에서 보관 | ||
| BRB80-DTT | 1X BRB80, 1 mM DTT | 최대 2일 | |||
| 분석 버퍼 | 80 mM K-파이프, 3 mM MgCl2, 1 mM EGTA, pH 6.8, 5% 수크로오스 (또는 1X BRB80, 5% 수크로스, 2 mM MgCl2) | 최대 1년 | 4 °C에서 보관 | ||
| 마스터 버퍼(MB) | 분석 버퍼, 5mM TCEP | 1주일 | 실험 당일에 준비하십시오. 두 개의 튜브로 분리하십시오 : 실온에서 MB-따뜻하고 얼음 위에서 MB - 차가운; 형광 염료를 사용하는 경우 1 mM DTT를 포함하십시오. | ||
| 메틸셀룰로오스를 사용한 마스터 버퍼 (MBMC) | 1X BRB80, 0.8% 메틸셀룰로오스, 5 mM TCEP, 5 mM MgCl2 | 1주일 | 실험 당일에 준비하십시오. 형광 염료를 사용하는 경우 1 mM DTT를 포함하십시오. | ||
| 단백질 희석 완충액 (DB) | MB, 1 mg/mL 소 혈청 알부민 (BSA), 1 μM ATP | 1 일, 얼음 위에 | 실험 당일에 준비하십시오. 형광 염료를 사용하는 경우 1 mM DTT를 포함하십시오. | ||
| 산소 제거 믹스 (OSM) | MB, 389 μg/mL 카탈라아제, 4.44 mg/mL 글루코스 옥시다제, 15.9 mM 2-메르캅토에탄올(BME) | 1 일, 얼음 위에 | 실험 당일 준비 | ||
| 산소 제거 결승 (OSF) | MB, 350 μg/mL 카탈라아제, 4mg/mL 글루코스 옥시다제, 14.3 mM BME, 15 mg/mL 글루코스 | 30 분 이내에 사용 | 9 μL의 OSM에 1 μL의 포도당을 첨가하여 사용 직전에 준비하십시오. | ||
표 1: 이 프로토콜에 사용된 버퍼 및 해당 구성 요소 목록. 각 버퍼를 얼마나 미리 준비할 수 있는지에 대한 지침은 "권장 저장 기간" 열을 참조하십시오.
| 시약 | 저장 집중 | 저장 용매 | 보관 온도 | 작업 집중 | 최종 집중 | 권장 저장 기간 | 노트 | |||||||||
| 뉴트라비딘 (NA) | 5 밀리그램/mL | 1X BRB80 | -80°C | 0.2 밀리그램/mL | 0.2 밀리그램/mL | 최대 1년 | 비오틴-뉴트라비딘-비오틴 결합을 통해 미세소관을 고정화하는데 사용; 작은 분취량으로 보관하십시오. | |||||||||
| 카파 카제인 (KC) | 5 밀리그램/mL | 1X BRB80 | -80°C | 0.5 밀리그램/mL | 0.5 밀리그램/mL | 최대 2년 | 이미징 챔버 표면을 차단하기 위해 사용; 작은 분취량에 보관하십시오. 실험 당일에는 실온에서 작은 부피를 따로 두십시오. | |||||||||
| 소 혈청 알부민 (BSA) | 50 밀리그램/mL | 1X BRB80 | -20°C | 1 mg/mL (DB에서) | 해당 없음 | 최대 2년 | 작은 분취량으로 보관하십시오. | |||||||||
| 카탈라아제 | 3.5 밀리그램/mL | 1X BRB80 | -80°C | 350 μg/mL (OSF 기준) | 35 μg/mL | 최대 2년 | 산소 소거제 혼합의 성분; 작은 분취량으로 보관하십시오. | |||||||||
| 글루코스 옥시다제 | 40 밀리그램/mL | 1X BRB80 | -80°C | 4 mg/mL (OSF 중) | 0.4 밀리그램/mL | 최대 2년 | 산소 소거제 혼합의 성분; 작은 분취량으로 보관하십시오. | |||||||||
| 투불린 | 동결건조된 | 해당 없음 | 4°C | 10 밀리그램/mL | 2.12 mg / mL (튜불린 믹스에서) | 최대 1년 | 튜불린이 용액에 들어가면 중합을 피하기 위해 차갑게 유지하십시오. | |||||||||
| 아데노신 트리포스페이트 (ATP) | 100 밀리미터 | 초순수 | -20°C | 10 밀리지미터 | 1 밀리지미터 | 6개월 | 필터 멸균 된 물에 용액을 준비하고 pH를 ~ 7.0으로 조정하고 작은 분취량으로 동결하십시오. | |||||||||
| 구아노신 트리포스페이트 (GTP) | 100 밀리미터 | 초순수 | -20°C | 10 밀리지미터 | 1.29 mM (튜불린 믹스에서) | 6개월 | 필터 멸균 된 물에 용액을 준비하고 pH를 ~ 7.0으로 조정하고 작은 분취량으로 동결하십시오. | |||||||||
| 구아노신-5'-[(α,β)-메틸레노]트리포스페이트 (GMPCPP) | 10 밀리지미터 | 초순수 | -20°C | 10 μM | 0.5 μM | 6개월 | ||||||||||
| 디티오트레이톨 (DTT) | 1 엠 | 멸균수 | -20°C | 1 밀리지미터 | 해당 없음 | 최대 2년 | ||||||||||
| 트리스(2-카르복시에틸)포스핀 (TCEP) | 0.5 엠 | 필터 멸균수 | 실내 온도 | 5 밀리지미터 | 해당 없음 | 최대 2년 | ||||||||||
| 메틸셀룰로오스 | 1% | 멸균수 | 실내 온도 | 0.8% (MBMC에서) | 0.21 % (튜불린 믹스에서) | 최대 1년 | 메틸셀룰로오스를 거의 끓는 물에 천천히 첨가하여 녹입니다. 지속적으로 교반하면서 식히십시오. | |||||||||
| 베타-메르캅토에탄올 (BME) | 143 밀리지미터 | 멸균수 | 실내 온도 | 14.3 mM (OSF에서) | 1.43 밀리지미터 | 최대 5년 | 143 mM은 스톡 BME의 1:100 희석액이다. | |||||||||
| 포도당 | 150 밀리그램/mL | 1X BRB80 | -80°C | 15 mg/mL (OSF 중) | 1.5 밀리그램/mL | 최대 2년 | 사용 직전에 OSM에 추가 | |||||||||
| (±)-6-하이드록시-2,5,7,8-테트라메틸크로만-2-카복실산(트롤록스) | 10mM | 1X BRB80 | -80°C | 10 밀리지미터 | 1 밀리지미터 | 최대 1년 | 완전히 용해되지 않습니다. NaOH를 넣고 ~ 4 시간 동안 저어주고 사용 전에 필터를 멸균하십시오. | |||||||||
| mPEG-숙신이미딜 발레레이트, MW 5,000 | 가루 | 해당 없음 | -20°C | 333 밀리그램/mL (0.1 M 중탄산나트륨에서) | 324 밀리그램/mL (0.1 M 중탄산나트륨에서) | 6개월 | ~ 34mg 분취량을 준비하여 각 튜브에 정확한 중량의 분말을 표시하십시오. 고체 위에 질소 가스를 통과시키고 파라 필름으로 튜브를 밀봉하고 건조제가있는 용기에 -20 ° C에서 보관하십시오. | |||||||||
| 비오틴-페그-스바, MW 5,000 | 가루 | 해당 없음 | -20°C | 111 밀리그램/mL (0.1 M 중탄산나트륨에서) | 3.24 mg/mL (0.1 M 중탄산나트륨 중) | 6개월 | ~ 3mg 분취량을 준비하여 각 튜브에 정확한 중량의 분말을 표시하십시오. 고체 위에 질소 가스를 통과시키고 파라 필름으로 튜브를 밀봉하고 건조제가있는 용기에 -20 ° C에서 보관하십시오. | |||||||||
표 2: 이 프로토콜에 사용된 시약 목록. 권장 보관 조건 및 농도, 실험 중에 사용되는 스톡 용액의 작동 농도 및 이미징 챔버의 최종 농도가 포함됩니다. 추가 메모는 맨 오른쪽 열에 나와 있습니다.
2. 비오틴-PEG 슬라이드 준비
참고 : 이미징 챔버를 가능한 한 실험 시작에 가깝고 2 주 전에 준비하십시오.
- 깨끗한 커버슬립
- 동일한 수의 24 x 60mm 및 18 x 18mm #1.5 커버슬립(그림 1F)을 슬라이드 염색 항아리 및 슬라이드 세척 랙에 각각 배치합니다(그림 1A,B). 18 x 18mm 커버슬립이 들어있는 슬라이드 세척 랙을 100mL 비이커에 넣습니다.
- 모든 커버슬립을 초순수(18.2MΩ-cm 저항율)로 5-6배 헹구고 진공관에 부착된 피펫 팁으로 각각 헹구어 낸 후 과량의 액체를 제거합니다(그림 1C).
- 커버슬립이 들어있는 비커와 슬라이드 염색 항아리를 초순수로 채우고, 파라필름으로 밀봉하고, 10분 동안 초음파 처리하십시오.
- 두 개의 150 mL 비커를 200 프루프 에탄올로 채 웁니다. 핀셋을 사용하여 각 커버슬립을 에탄올로 채워진 비이커 하나에 담근 다음 다른 비커에 담그십시오.
- 핀셋을 사용하여 커버슬립을 슬라이드 건조 랙으로 옮기고(그림 1D), 질소 기류 하에서 건조시킨 다음 완전히 건조될 때까지(~15분) 37°C에서 배양합니다.
- 건조 된 커버 슬립을 플라즈마 클리너 내부의 단일 층에 놓습니다. 진공 밀봉을 형성한 다음 플라즈마 청소기의 무선 주파수(RF) 레벨을 ~8MHz로 설정합니다.
- 플라즈마가 생성되면 커버슬립을 플라즈마 클리너에 5분 동안 두십시오. 플라즈마 청소기를 끄고 진공을 천천히 해제하십시오.
- 진공 씰이 풀리면 커버슬립을 뒤집고 커버슬립의 다른 쪽에 대해 5분 동안 플라즈마 청소를 반복합니다.
- 플라즈마 세척에 대한 대안 : 2.1.2-2.1.3 단계 대신에 초음파 처리 덮개를 2 % 세제 (초순수)의 따뜻한 용액에서 10 분 동안 미끄러 뜨립니다. 그런 다음 커버 슬립을 초순수로 철저히 씻고 초순수에서 2-3 회 (각각 10 분) 초음파 처리하십시오. 다음으로, 에탄올로 세척하고 단계 2.1.4-2.1.5에서와 같이 건조시킨다. 2.1.6-2.1.8단계를 건너뜁니다.
- 비오틴-PEG 처리
- 사용 직전에 400 μL의 3-아미노프로필트리에톡시실란 40 mL를 아세톤에 녹인다. 핀셋을 사용하여 플라즈마로 청소한 커버슬립을 슬라이드 세척 랙과 슬라이드 염색 항아리로 옮깁니다. 커버슬립을 3-아미노프로필트리에톡시실란 용액에 침수시키고 5분 동안 인큐베이션한다12,13.
- 모든 커버 슬립을 초순수로 5-6 번 씻으십시오.
- 커버슬립을 슬라이드 건조 랙으로 옮기고 질소 기류 하에서 건조시키고 완전히 건조될 때까지(~20분) 37°C에서 인큐베이션한다.
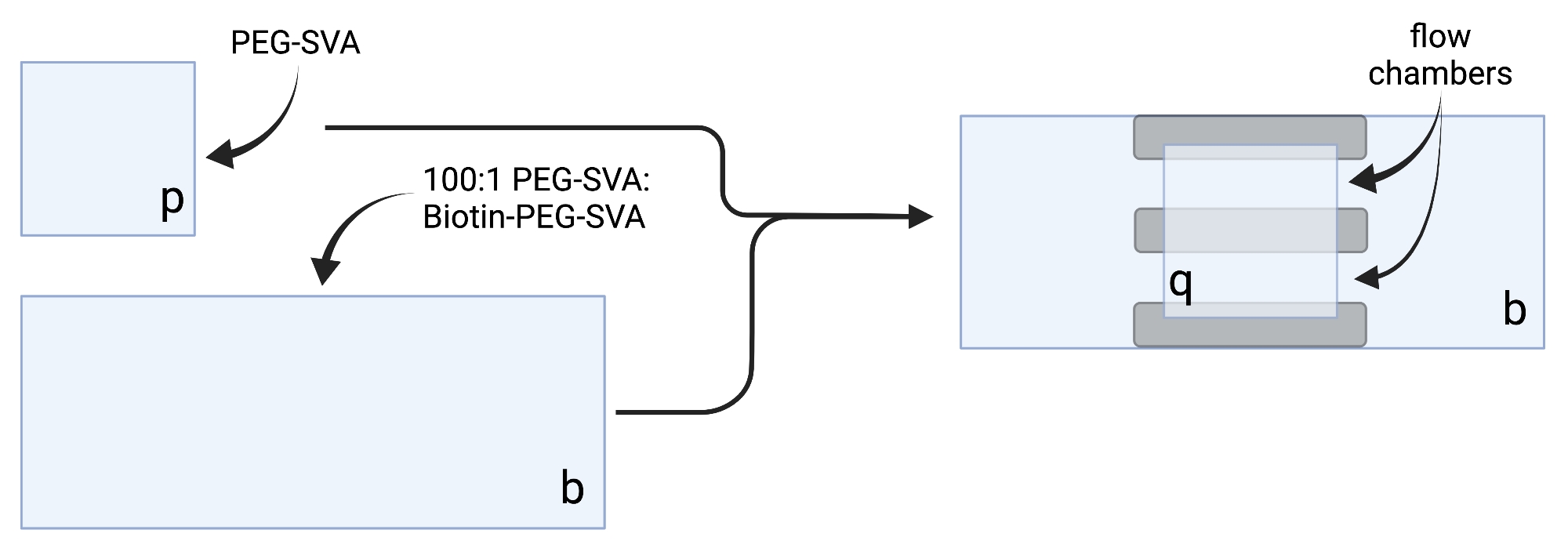
- 건조된 커버슬립을 섬세한 작업 물티슈에 놓고 각 커버슬립을 한 모서리에 라벨을 붙입니다(예: 각 18 x 18mm 커버슬립에 'p', 각 24 x 60mm 커버슬립에 'b'가 표시됩니다( 그림 2 참조).
- 실험 당일, 0.84 mg의 NaHCO3를 초순수 10 mL에 용해시켜 신선한 0.1 M 중탄산나트륨 용액을 제조하였다.
- mPEG-Succinimidyl Valerate (PEG-SVA) 및 Biotin-PEG-SVA의 분취량을 사용 직전에 실온으로 가져 오십시오. 표 2의 폴리에틸렌 글리콜(PEG) 분취량 제제에 대한 참고를 참조한다.
참고: 숙신이미딜발레레이트(SVA) 잔기의 가수분해 반감기가 ~30분이기 때문에 신속하게 작업하십시오. - 34mg의 PEG-SVA에 102μL의 0.1 M NaHCO3를 첨가하고, 20초 동안 2,656 x g의 벤치탑 마이크로원심분리기에서 회전시킨 다음, 위아래로 피펫팅하여 혼합한다. 3 mg의 비오틴-PEG-SVA를 27 μL의 0.1 M NaHCO3에 상하로 피펫팅하여 용해시킨다. 튜브 상에 기록된 PEG의 정확한 중량에 따라 희석 부피를 조정한다(표 2 참조).
- 100:1 w/w PEG:비오틴-PEG 혼합물을 20개의 커버슬립에 PEG-SVA 용액 75μL와 비오틴-PEG-SVA 용액 2.25μL, 30개의 커버슬립에 대해 100μL 및 3μL, 또는 40개의 커버슬립에 대해 125μL 및 3.75μL를 조합하여 준비합니다.
- 젖은 종이 타월을 빈 10μL 팁 상자 하단의 팁 랙 아래에 배치하여 수화 챔버를 구성합니다(그림 1E). 이것은 PEG 용액의 증발을 방지할 것이다.
- 6 μL의 100:1 PEG-SVA:비오틴-PEG 혼합물을 표지된 측에 1개의 24 x 60 mm 커버슬립의 중심 상에 피펫. 첫 번째 커버슬립 위에 또 다른 24 x 60mm 커버슬립을 올려 쌍이 X 자형을 형성하고 'b'라고 표시된 면이 서로 마주보도록 합니다. 쌍을 수화 챔버의 빈 팁 랙에 놓고 나머지 24 x 60mm 커버슬립에 대해 반복하십시오.
- 6 μL의 PEG-SVA를 표지된 쪽에 18 x 18 mm 커버슬립의 중심 상에 피펫한다. 첫 번째 커버슬립 위에 또 다른 18mm x 18mm 커버슬립을 놓고 'p'라고 표시된 면이 서로 마주보고 있습니다. 수화 챔버의 빈 팁 랙에 쌍을 놓고 나머지 18 x 18mm 커버슬립에 대해 반복하십시오.
- 수화 챔버를 닫고 3시간 또는 하룻밤 동안 인큐베이션한다.
- 커버 슬립 쌍을 분리하고 초순수로 헹구십시오.
- 커버슬립을 질소 스트림으로 건조시키고 이를 37°C 인큐베이터에 넣어 완전히 건조시킨다.
- 이미징 챔버를 구성하려면 'b'라고 표시된 측면의 24 x 60mm 커버슬립에 양면 테이프 세 스트립을 붙입니다. 테이프 스트립의 반대편에 18 x 18mm 커버슬립을 부착하고 면에는 'p'라고 표시된 면이 더 큰 커버슬립을 향하도록 부착합니다. 이것은 현미경 실험을위한 두 개의 유동 챔버를 형성하며, 처리 된 표면은 서로 마주 봅니다 (그림 2 및 그림 1G).

그림 1: 커버슬립 처리 및 이미징 챔버 준비용 장비. (A) 24 x 60mm 커버슬립용 슬라이드 염색 항아리, (B) 18mm 커버슬립용 슬라이드 세척 랙, (C) 진공 셋업, (D) 슬라이드 건조 랙, (E) 수화 챔버, (F) 커버슬립, (G) 이미징 챔버, (H) 슬라이드 홀더. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2: 양면 테이프(회색) 및 PEG/비오틴-PEG 처리 커버슬립을 사용한 이미징 챔버 제조 회로도. BioRender.com 로 만들었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
3. 미세소관 중합
- GMPCPP 씨앗 준비
참고: GMPCPP 씨앗을 냉장실에서 준비하여 모든 시약, 팁 및 튜브를 4°C로 유지하십시오. GMPCPP 씨앗은 미리 준비하고 -80°C에서 최대 1년 동안 보관할 수 있습니다. 1 mL 원심분리 튜브를 함유하는 고정각 초원심분리 로터를 초원심분리기에 넣고 온도를 4°C로 설정한다.- 동결건조된 튜불린(표 2)을 사용 직전에 1X BRB80에 ~10mg/mL로 재현탁한다.
- 표 3에 기재된 바와 같이 GMPCPP 종자의 성분을 혼합한다.
참고: 가용성 튜불린의 중합을 최소화하기 위해 모든 튜불린 성분을 얼음 위에 가능한 한 많이 보관하십시오. - 4°C에서 5분 동안 352,700 x g 의 고정각 초원심분리 로터에서 혼합을 명확히 한다.
- 상청액을 5 μL 분취량으로 분리하고, 이를 액체 질소에 스냅하여 동결시키고, 이를 -80°C에서 보관한다.
- 실험 당일에 씨앗을 중합하십시오.
- 1-2 mL의 BRB80-DTT (표 1)를 37°C로 가온한다.
- 단계 3.1.4로부터의 GMPCPP 종자 (-80°C)의 5 μL 분취량을 얼음 상에 놓고 즉시 20 μL의 따뜻한 BRB80-DTT에 용해시킨다. 실온에서 5초 동안 2,000 x g 로 회전하고 탭하여 혼합합니다.
참고: 초기 희석 부피는 13 μL와 21 μL 사이에서 변할 수 있으며 종자의 각 배치에 대해 경험적으로 결정된다. 종자가 중합되지 않으면 초기 희석 완충액 (단계 3.2.2)을 0.5 μM GMPPP로 보충하여 문제를 해결하십시오. - 빛으로부터 보호하고 37°C에서 30-45분 동안 인큐베이션한다.
참고 : 미세 소관의 길이는 배양 기간에 따라 다릅니다. 짧은 미세 소관의 경우, 배양 시간은 15 분만큼 짧을 수 있습니다. 긴 미세 소관의 경우, 배양 시간은 2 시간만큼 길어질 수 있습니다. 비오티닐화 미세소관은 비오티닐화 미세소관보다 더 긴 배양 시간을 필요로 하는 경향이 있다. - 500 μL 원심분리 튜브를 함유하는 고정각 초원심분리 로터를 초원심분리기에 넣고 30°C로 미리 가온한다.
- 인큐베이션 후, 50 μL의 따뜻한 BRB80-DTT (단계 3.2.1)를 중합된 GMPCPP 종자에 첨가하고, 혼합물을 500 μL 원심분리 튜브로 옮긴다. GMPCPP 종자가 들어있는 빈 튜브를 또 다른 50 μL의 따뜻한 BRB80-DTT로 세척하고, 피펫을 상하로 세척하고, 이 완충액을 혼합물이 들어있는 500 μL 원심분리 튜브에 첨가한다.
- 회전하기 전에 원심 분리 튜브의 테두리를 표시하여 펠렛이 어디에 있는지 표시하십시오 (펠릿이 너무 작아서 볼 수 없습니다). 30°C12에서 244,900 x g 에서 10분 동안 회전시킨다.
- 조심스럽게 상층액을 피펫하고 버리십시오. 펠렛을 100 μL의 따뜻한 BRB80-DTT에 재현탁시킨다. 탭하여 혼합합니다.
- 30°C에서 244,900 x g 에서 10분 동안 회전시키고, 동일한 장소에서 펠릿에 로터로 마킹을 정렬한다.
- 상청액을 제거하고 펠렛을 16 μL의 따뜻한 BRB80-DTT에 재현탁시켰다. 미세소관 용액을 깨끗한 0.6 mL 마이크로원심분리 튜브로 옮긴다. 빛으로부터 보호하고 실온 이상으로 유지하십시오.
참고 : 중합 후 미세 소관을 실온 이상으로 유지하십시오. 그들이 추워지면 탈중합 될 것입니다. 추가 안정성을 위해 28°C에서 인큐베이션한다.
- TIRF 현미경을 통해 미세 소관 확인
- 4.5 μL의 BRB80-DTT와 1 μL의 미세소관 용액(단계 3.2.9)의 혼합물을 현미경 슬라이드 상에 피펫팅한다. 18 x 18mm 커버슬립으로 덮고 가장자리를 투명한 매니큐어 또는 실온에서 고체이고 95°C에서 액체인 바셀린, 라놀린 및 파라핀(valap 실란트)의 1:1:1 혼합물로 밀봉합니다.
- TIRF 목표를 커버슬립 아래에 위치시키고(권장 현미경 설정은 4단계 참조) 브라이트 믹스에서 형광 표지된 튜불린에 적합한 파장으로 새로 중합된 미세소관을 시각화하고(표 3), 다가오는 실험에서 사용할 미세소관의 희석액을 결정한다.
| 시약 | 밝은 믹스 (μL) | 추가 순서 | 밝은 믹스 + 비오틴 (μL) | 추가 순서 |
| 형광 튜불린, 10 mg/mL | 2 | 6 | 2 | 7 |
| 비오틴-튜불린, 10 mg/mL | 0 | 해당 없음 | 2 | 6 |
| 라벨이 부착되지 않은 튜불린, 10 mg/mL | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| DTT, 0.2 엠 | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| 멸균수 | 52.9 | 1 | 52.9 | 1 |
| 총 부피 (μL) | 132 | 132 |
표 3: GMPCPP 종자 혼합. GMPCPP 미세소관 종자의 성분, 부피 및 첨가 순서를 포함한다. 5 μL 분취량을 제조하고 -80°C에서 최대 1년 동안 보관한다.
4. 현미경 설정
- 온도: 현미경 온도를 28°C로 설정하여 동적 미세소관을 볼 수 있습니다.
- 필터: 이미징할 형광 채널에 따라 필터 큐브와 방출 필터의 최상의 조합을 사용합니다. 동일한 실험에서 488nm, 560nm 및 647nm 파장을 시각화하려면 지정된 파장에 대한 방출 필터와 결합된 405/488/560/647nm 레이저 쿼드 밴드 세트를 사용합니다.
- 레이저 정렬: 실험에 사용된 레이저 빔이 정렬되었는지 확인합니다. 실험에 대한 레이저 강도를 경험적으로 결정하여 모든 형광 단백질을 가능한 가장 높은 신호 대 잡음비로 이미징 할 수 있지만 실험 과정에서 상당한 광표백을 겪지 않도록하십시오.
- 목표: 렌즈 용지를 사용하여 70% 에탄올로 100x 대물렌즈를 청소합니다. 이미징하기 전에 현미경 침지 오일 한 방울을 목표에 추가하십시오.
- 이미징 시퀀스 설정
- 647 nm 형광단-표지된 비오티닐화 미세소관, 560 nm 형광단-표지된 비-비오티닐화 미세소관 및 가용성 튜불린, 및 GFP 표지된 관심있는 단백질에 대한 실험을 위해, 20분 동안 이미지화하였다. 10초마다 560nm 및 488nm 채널을 이미지화하고 647nm 채널을 30초마다 이미징합니다.
- 가용성 튜불린과 MAP를 첨가하기 전에 번들의 참조 이미지를 캡처하려면 560nm 및 647nm 파장에서 각각 하나의 이미지로 시퀀스를 설정하십시오.
5. 표면 고정화 된 미세 소관 번들 생성
주: 다음 단계의 경우, 필터 용지를 다른 쪽에 배치하면서 한쪽 열린 면으로 피펫팅하여 모든 용액을 유동 챔버로 흐르게 하십시오. 이미징 챔버를 빛으로부터 보호하여 형광 표지된 단백질의 광표백을 줄입니다. 준비된 이미징 챔버를 슬라이드 홀더에 테이프로 붙입니다(그림 1G,H). 프로토콜 steps 5.2-6.4에 해당하는 표 4의 단계를 따르십시오.
- 표 5에 따라 가용성 튜불린 믹스를 준비하고 얼음 위에 보관하십시오.
참고: 가용성 튜불린은 중합을 방지하기 위해 항상 얼음 위에 놓아야 합니다. 신선한 가용성 튜불린 믹스를 대략 매 2 시간마다, 또는 미세 소관이 더 이상 중합하지 않을 때 준비하십시오. - 비오틴-뉴트라비딘-비오틴 결합을 통해 미세소관을 고정화시키기 위해, 먼저 챔버가 채워질 때까지 뉴트라비딘(NA) 용액으로 유동시키고(∼7.5 μL) 5분 동안 인큐베이션한다.
- 10 μL의 MB-콜드로 세척하십시오.
- 블로킹 단백질 κ-카제인(KC)의 7.5 μL를 유동시키고 2분 동안 인큐베이션한다.
- 10 μL의 MB-warm으로 세척하여 미세소관의 도입을 위한 챔버를 제조하였다.
- 비오티닐화 미세소관의 스톡을 1X BRB80-DTT에 희석하고(단계 3.3.2의 관찰에 따라) 이 희석액의 1 μL를 MB-warm의 9 μL에 첨가한다. 혼합물을 챔버 내로 유동시키고 10분 동안 인큐베이션한다. 더 많은 번들을 위해 더 높은 농도의 미세 소관을 사용하십시오.
- 고정화되지 않은 미세소관을 10 μL의 MB-warm으로 씻어내십시오.
- 7.5 μL의 따뜻한 KC를 챔버 내로 유동시키고 2분 동안 인큐베이션한다.
- 인큐베이션 동안, 따뜻한 KC에서 가교결합 단백질 PRC1의 2 nM 용액을 준비한다. 이 용액 10 μL를 유동 챔버에 넣고 5분 동안 인큐베이션한다.
참고: 재조합 PRC1은 앞서 기술된 바와 같이 박테리아 세포로부터 발현되고 정제된다13. - 다발을 만들기 위해, 10 μL의 비오티닐화 미세소관을 챔버 내로 흘려보내고 10분 동안 인큐베이션한다. PRC1은 비오티닐화되고 고정화된 비오티닐화 미세소관15,16을 가교결합시킨다(도 3).
참고: 인큐베이션 시간 동안 어세이 믹스를 제조하기 위한 단계 6.1을 참조한다. - 챔버를 10 μL의 MB-warm으로 두 번 세척한다. 부착 된 미세 소관은이 시점으로부터 약 20 분 동안 안정적입니다.
| 걸음 | 시약 | 부피 (μL) | 인큐베이션 시간(분) |
| 1 | 뉴트라비딘 | 7.5 | 5 |
| 2 | MB-콜드 | 10 | - |
| 3 | κ-카제인 | 7.5 | 2 |
| 4 | MB 따뜻한 | 10 | - |
| 5 | 비오티닐화 된 미세 소관 (MB 온난으로 희석) | 10 | 10 |
| 6 | MB 따뜻한 | 10 | - |
| 7 | 따뜻한 κ 카제인 | 7.5 | 2 |
| 8 | κ-카제인으로 희석된 2 nM PRC1 | 10 | 5 |
| 9 | 비-비오티닐화 미세소관 | 10 | 10 |
| 10 | MB 따뜻한 x 2 | 10 | - |
| 11 | 분석 믹스 | 10 | - |
| 부착 된 씨앗은이 시점에서 약 20 분 동안 안정적입니다. | |||
표 4: 분석 단계. 세척(-) 또는 인큐베이션 시간의 표시와 함께 이미징 챔버에 첨가된 시약 목록.
| 시약 | 부피 (μL) |
| 재활용 튜불린, 10 mg/mL | 10 |
| MB-콜드 | 10.3 |
| 증권 시세 표시기 | 13.7 |
| BRB80-DTT | 3.4 |
| GTP, 10 밀리지미터 | 6.7 |
| ATP, 10 밀리지멘트 (키네신을 사용하는 경우) | 6.7 |
| 형광 표지된 튜불린, 10 밀리그램/mL | 1 (차가운 BRB80-DTT에서 동결건조된 표지된 튜불린을 재현탁) |
표 5: 가용성 튜불린 혼합 성분. 실험 시작시 혼합하고 얼음 위에 보관하십시오.

도 3: 형광 표지된 다발과 단일 미세소관을 만들고 이미지화하기 위한 분석 성분의 첨가의 개략도. 비오티닐화 종자는 청색, 비오티닐화 종자 및 가용성 튜불린은 적색, PRC1은 검정, 및 시안에 관심있는 단백질로 표시된다. 그림의 단계 번호는 표 4의 단계 번호와 일치합니다. 단계 9에 대응하는 패널은 미리 형성된 다발(좌측 아래)을 나타내고; 단계 11은 새로 형성된 번들(좌측 상단)을 도시한다. BioRender.com 로 만들었습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
6. 이미지 미세 소관 역학
- 단계 5.10에서 10분 인큐베이션 시간 동안, 표 6에 따라 관심있는 단백질, 가용성 튜불린, 뉴클레오티드, 산소 제거제14, 및 항산화제를 함유하는 10 μL의 분석 믹스를 제조하였다. 믹스를 얼음 위에 보관하십시오.
- 준비된 이미징 챔버를 슬라이드 홀더에 테이프로 붙인 100x TIRF 목표에 로드합니다. 560nm 및 647nm 채널을 사용하여 단일 미세소관 및 번들의 최적 수와 밀도를 포함하는 시야를 찾으십시오.
참고: 비오티닐화 및 비오티닐화 미세소관이 모두 동일한 형광단으로 표지되는 경우, 형광 강도에 대한 라인 스캔 분석은 단일 미세소관과 다발을 구별할 수 있습니다. - 시야각이 식별되면 참조 이미지를 가져옵니다.
- 이미징 챔버를 방해하지 않고 어세이 믹스를 조심스럽게 유동시킨다.
- 챔버의 열린 끝을 valap 실란트로 밀봉하십시오.
- 단계 4.5.1에 설명된 대로 이미징 시퀀스를 시작하십시오.
| 시약 | 부피 (μL) |
| 가용성 튜불린 믹스 | 4 |
| 증권 시세 표시기 | 1 |
| Trolox (쉽게 광표백 형광단으로 표지 된 미세 소관을 사용하는 경우) | 1 |
| ATP, 10 밀리지멘트 (키네신을 사용하는 경우) | 1 |
| PRC1 (또는 선택의 가교제) | 1 |
| 관심있는 단백질 | X |
| MB-콜드 | 2-X |
표 6: 분석 혼합 성분. 혼합, 이미징 챔버로 흐르고, 30 분 이내에 미세 소관 역학을 이미지화하십시오.
결과
상기 기재된 실험은 647 nm 형광단-표지된 비오티닐화 마이크로소관, 560 nm 형광단-표지된 비오티닐화 마이크로소관, 및 560 nm 형광단-표지된 가용성 튜불린 혼합물을 사용하여 수행되었다. 미세소관은 가교결합 단백질 PRC1 (GFP 표지)에 의해 가교결합되었다. 표면-고정화된 다발 및 단일 미세소관이 생성된 후(단계 5.11), 이미징 챔버를 TIRF 100X 1.49 NA 오일 대물 상에 장착하고 560 nm 및 647 nm 형광 채널에?...
토론
여기에 설명 된 실험은 전통적으로 단일 미세 소관 또는 한 유형의 어레이에서 수행되는 기존의 미세 소관 재구성 분석의 범위와 복잡성을 크게 확장합니다. 현재의 분석은 두 집단, 즉 단일 미세소관 및 가교결합된 다발에 대한 조절 MAP 활성을 동시에 정량화하고 비교하는 방법을 제공한다. 또한,이 분석은 두 가지 유형의 번들을 검사 할 수 있습니다 : 역학이 시작되기 전에 안정적인 씨앗으로 ?...
공개
저자는 경쟁 이익이 없다고 선언합니다.
감사의 말
이 사업은 NIH (No. 1DP2GM126894-01)의 보조금과 퓨 자선 신탁 및 스미스 가족 재단의 기금으로 R.S.에 지원되었습니다. 저자들은 프로토콜의 개발 및 최적화에 기여한 Shuo Jiang 박사에게 감사드립니다.
자료
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
참고문헌
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595 (2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880 (2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119 (2022).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유