
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Dissecção e Cultura de Neurônios da Medula Espinhal comissural embrionárias
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Este vídeo demonstra um método para dissecar e cultura de neurônios comissurais E13 rato medula espinhal dorsal. Dissociada neurônios comissurais são úteis para estudar os mecanismos celulares e moleculares do crescimento do axônio e orientação.
Resumo
Neurônios comissurais têm sido amplamente utilizados para investigar os mecanismos subjacentes a orientação dos axônios durante o desenvolvimento embrionário medula espinhal. Os corpos celulares desses neurônios estão localizados na medula espinhal dorsal e seus axônios seguem trajetórias estereotipados durante o desenvolvimento embrionário. Axônios comissurais inicialmente para o projeto ventralmente floorplate. Depois de cruzar a linha média, esses axônios vez anteriormente e projeto para o cérebro. Cada uma dessas etapas é regulado pela ação de várias pistas de orientação. Culturas altamente enriquecido em neurônios comissurais são ideais para muitas experiências abordando os mecanismos de pathfinding axônio, incluindo ensaios de torneamento, imunoquímica e bioquímica. Aqui, nós descrevemos um método para dissecar e cultura de neurônios comissurais E13 rato medula espinhal dorsal. Em primeiro lugar, a medula espinhal é isolado e tiras dorsal são dissecados. O tecido é então dissociado dorsal em uma suspensão de células por tripsinização e perturbação mecânica. Neurônios são banhados em lamínulas poli-L-lisina-revestido de vidro ou de cultura de tecidos de pratos. Após 30 horas
Protocolo
1. Dissecção da medula espinhal dorsal embrionárias de rato
Recomendações gerais
Mantenha L-15 médio no gelo e muitas vezes mudar o meio no prato dissecção para manter os embriões frescos. Isso ajuda a preservar a integridade dos tecidos. Todas as etapas são realizadas com duas pinças Dumont # 5 a menos que indicado. Para evitar a contaminação, spray todas as ferramentas e superfícies de trabalho com etanol 70% e manter a garrafa meio dissecção fechado. A transferência de embriões entre pratos, use uma pipeta de plástico recortado ou uma colher perfurada. É fundamental para não danificar a medula espinhal (nicking, alongamento) para completar com sucesso a dissecção.
Preparação
- Frio L-15 de médio
- 50 mL de L-15 + inativado pelo calor de 10% soro de cavalo (HiHS). Manter no gelo.
Dissecção da medula espinhal
- Euthanize um E13 gravidez encenado rato (E0 = primeiro dia dia seguinte acasalamento) com uma câmara de CO 2 de acordo com as diretrizes institucionais.
- Spray de etanol a 70% sobre o abdome. Beliscar e puxar para cima a pele da região abdominal inferior com uma pinça e cortado com tesoura cirúrgica. Repita a operação para cortar o músculo e as camadas peritoneal para alcançar a cavidade abdominal. Criar uma incisão em forma de V, cortando o tecido ao longo dos lados do abdômen, até o tórax. Levante e puxe o tecido para expor a cavidade abdominal.
- O útero é anexado em três locais: no abdômen inferior central, e em ambos os cantos superior lateral. Levante o útero, agarrando no tecido entre sacos embrionárias. Corte o tecido conjuntivo para remover o útero e coloque em um prato de Petri cheia de L-15 no gelo.
- Os próximos passos são feitos sob um microscópio de dissecação. Para separar um embrião a partir de tecidos extra-embrionário e as membranas embrionárias, pegue o tecido entre sacos uterina com um par de fórceps,. Com o outro par, aperte o lado mais transparente do saco para cortar através das membranas superficial (o lado mais escuro é a placenta). Esprema o embrião pressionando suavemente o lado placenta do saco. Remova todos os embriões e coloque em um prato de Petri cheia de L-15 no gelo.
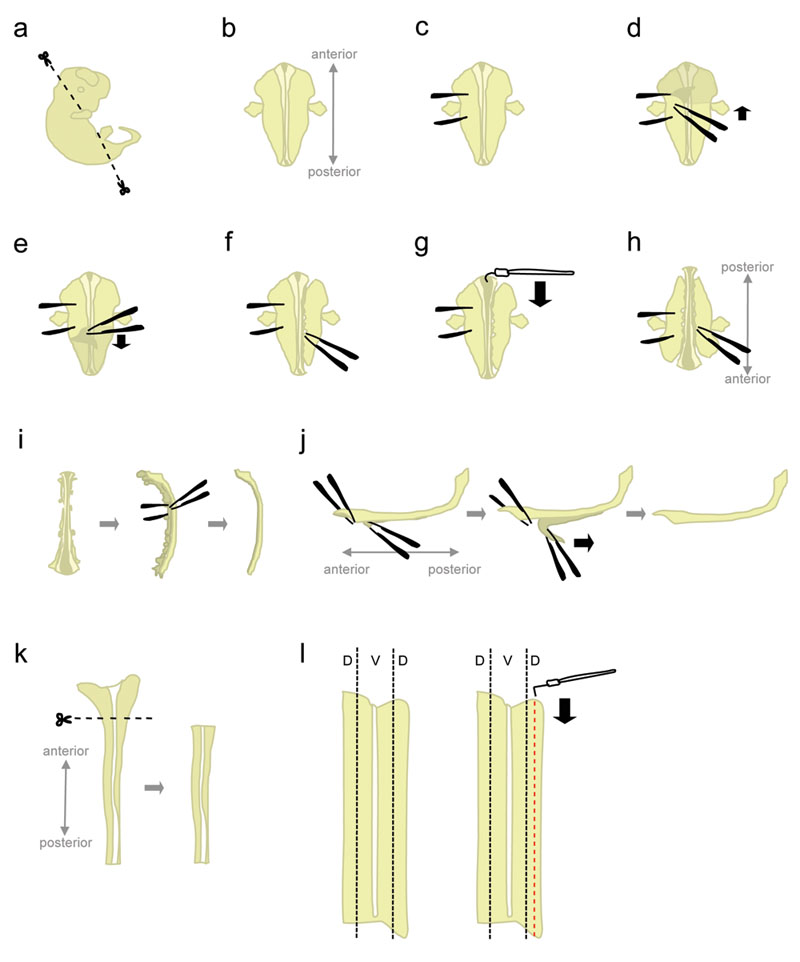
- Embrião único lugar a 10 centímetros prato cheio de Petri gelada L-15. Usando microtesoura, cortar a cabeça e partes posterior em ângulos mostrados na figura 1a.
Corte nestes ângulos irá ajudar a posicionar o embrião quando colocado "face ventral para baixo". - Posição do embrião "face ventral para baixo" (anterior apontando para fora, posterior apontando para o experimentador) (fig. 1b).
- Segure firmemente o embrião de um lado, usando a pinça para "fechar" o embrião (fig. 1c). Não mova a fórceps, uma vez que irá rasgar o tecido. Com o outro par de fórceps, retire a pele que cobre a parte traseira do embrião a partir da região entre a "holding" fórceps (fig 1d-e). Isto irá expor a medula espinhal que, neste momento, é envolto por membranas (as meninges).
Agarre a pele dos lados em vez de acima da medula espinhal para evitar nicking da medula espinhal. - Parcialmente retirar o tecido para o lado direito da medula espinhal (fig. 1f). Começando ao nível do fórceps segurando, picar o tecido com uma pinça fechada, o mais próximo possível para a medula espinhal. Então, abra lentamente a pinça para rasgar o tecido. Isso deve retirar os gânglios da raiz dorsal (DRGs) da medula espinhal, e também deve atrapalhar os órgãos ventral. Deixe um pouco de tecido ligado nas extremidades anterior e posterior. Tecido Deixando anexado adiciona peso ao embrião, impedindo-o de ser puxado para cima durante a próxima etapa. Além disso, o tecido que é usado para segurar o embrião em etapas posteriores.
- A partir do final anterior, use a agulha de tungstênio em forma de gancho para cortar as meninges e abra a medula espinhal ao longo do roofplate (fig. 1g).
- Gire o embrião de 180 graus (posterior apontando para fora, apontando para o experimentador anterior) (fig. 1h).
Isso permite que o pesquisador de utilizar a mesma mão para retirar o tecido do outro lado do embrião. - Segure o embrião agarrando-se ao lado anteriormente individual, e perturbar o tecido do lado restante usando o mesmo método (ver 1.8.). Quando terminar, retire completamente o tecido em ambos os lados da medula espinhal.
Alternativa: separar completamente o tecido remanescente no lado esquerdo, mas deixar algum tecido em anexo no final anterior e posterior do lado direito. Isto às vezes pode ajudar a manter a coluna vertebral na posição no passo 1.12. - Coloque a medula espinhal do seu lado e remover a maioria do tecido remanescente mesenquimais e DRGs (fig. 1-I).
- Nesta etapa, as meninges ea medula espinhal são duas "folhas" de tecido apposed para o outro. Pino para baixo o maior, porção ântero-a maior parte da medula espinhal, e descolar um pequeno segmento de meninges da cor da coluna vertebrald (fig. 1-J, à esquerda). Agora, segure os dois segmentos separados por agarrar todo com uma pinça, e descolar as meninges com um movimento suave e constante (fig. 1-J).
Desigual "peeling" levará à ruptura da medula espinhal e / ou a camada de meninges. Geralmente não é possível recuperar a parte da medula espinhal ainda ligado ao meninges. - Usando uma pipeta, transferir o cabo isolado espinhal para uma placa de Petri contendo HiHS L-15 + 10% e deixar no gelo. Tipicamente, medula espinhal de todos os embriões são coletados antes de dissecar as porções dorsal.
Dissecção medula espinhal dorsal
- Medula espinhal um lugar numa placa de Petri contendo L-15 HiHS +10%, deitado em um "livro aberto" de configuração. Cortar a maior parte, anterior (que inclui parte do cérebro posterior) (fig. 1k).
- O tecido dorsal está localizado na parte mais lateral da medula espinhal (fig 1l, esquerda). Enquanto pinagem do cabo com uma agulha de tungstênio em linha reta, use a agulha de tungstênio em forma de L para cortar uma tira que é a largura 1/5th de metade da medula espinhal. (Fig. 1l, direita). Coloque as tiras dorsal em um tubo de plástico contendo 15 mL L-15 + HiHS 10% sobre o gelo.
Dorsal seções do tubo neural de embriões E13 ~ 12 ~ renderá 3,5-4.000.000 células para revestimento após a dissociação. Mais células podem ser obtidas através da realização de uma dissecção grossa do cordão espinhal dorsal (corte mais largo tiras dorsal), mas a pureza de neurônios comissurais será reduzida.
2. Comissural cultura neurônio
Recomendações gerais
Todos os passos devem ser realizados em condições estéreis em uma capa de cultura de tecidos, salvo indicação contrária. Use meio fresco e suplementos recém-descongelado e reagentes. A dissociação e as etapas de trituração são realizados em Ca 2 + / Mg 2 + livre HBSS para minimizar Ca 2 + / Mg 2 + dependente de adesão.
Preparação
- lamínulas revestido (uso de vidro Desag alemão) ou placas de cultura de tecidos (ver procedimento de revestimento abaixo).
- quente Mídia Plating Neurobasal (veja abaixo), em pratos de cultura de tecidos e CO 2 equilibrado na cultura de tecidos incubadora de pelo menos 1,5 h antes do plaqueamento.
- Tripsina 2,5% no banho-maria 37 ° C
- 2 garrafas de HBSS Ca 2 + / Mg 2 + livre, um de 4 ° C, uma temperatura de 37 ° C.
- dois-fogo de vidro polido pipetas Pasteur uma com um diâmetro de metade do tamanho normal, e um com um diâmetro ligeiramente menor que a metade. Use pipetas esterilizadas Pasteur. Ao fogo polonês as pipetas, use um bico de Bunsen (o topo da chama azul claro) para derreter a ponta para diminuir ligeiramente o diâmetro. Porque esta etapa é realizada fora do capô de cultura de tecido, spray as pipetas com etanol 70% antes de colocar sob o capô cultura de tecidos. Para uma recuperação mais eficiente dos neurônios, o revestimento do pipetas Pasteur com a mídia contendo soro antes do início da dissociação ou apenas antes da etapa de trituração (preencher pipetas com a mídia e manter por 30 segundos). Isso vai evitar que as células de degola para o interior da pipeta Pasteur durante a trituração.
- água MilliQ estéril para lavar PLL revestido pratos ou lamelas
- 12,5% MgSO 4 em solução HBSS
Poli-L-lisina revestimento
Se estiver usando lamínulas de vidro, ácido lavagem por 24 horas e esterilizar antes de chapeamento (ver Kaech e Banker, 2006). Use lamínulas de vidro alemão Desag.
Para o revestimento de poli-L-lisina em lamínulas de vidro ou placas de cultura de tecido de plástico:
- sob uma capa de cultura de tecidos, cobrir superfícies com uma pequena cúpula de 100 ug / ml solução PLL para 1,75-2 horas.
- lavar duas vezes com água MilliQ, pelo menos 5 minutos por lavagem (pode ser realizada durante a etapa de dissociação de células abaixo).
- armazenamento em água até o uso. Não deixe que a superfície PLL revestido seco.
Para reduzir o desperdício de PLL para o revestimento de lamelas, coloque as lamelas em estéril pratos bacteriana Petri. Brasão e lavar as lamelas, colocando uma cúpula de líquido no lamínulas. Os pratos bacteriana Petri são hidrofóbicos, de modo que o líquido deve permanecer no lamínulas de vidro. Transferir as lamelas para placas de cultura de tecidos logo antes do plaqueamento das células.
Para os neurônios semeadas em placas de cultura de tecido de plástico, a adesão é normalmente mais elevada, alongamento, assim, neurite poderiam ser diminuídos.
Dissociação e chapeamento
- Verifique se as tiras dorsal ter resolvido até o fundo do tubo. Remover a maioria dos HiHS L-15 +10% com uma pipeta Pasteur. Rapidamente lavar a dorsal tiras do tubo neural, uma vez adicionando 3 ml de frio (4 ° C) HBSS.
- Deixe o dorsal tiras do tubo neural se contentar com dois minutos, depois remover o HBSS com uma pipeta Pasteur.
- Adicionar quente (37 ° C) HBSS a um volume de 4,7 ml. Tgalinha adicionar 0,3 ml de tripsina 2,5% para dar uma concentração final de tripsina 0,15%.
- Incubar a 37 ° C em banho-maria por 7 min. Misture delicadamente uma vez a meio da incubação.
- Adicionar 30 mL DNAse (25 000 U / mL) para uma concentração final de 150 U / mL. Adicionar 60 mL de MgSO 4 e misture rapidamente, para uma concentração final de 0,15%.
Para o tecido de dissecção grossa, incubar por um minuto extra de 1 a 37 ° C em banho-maria.
Nesta fase, o dorsal seções de tubo neural devem ser fragmentado. Se o dorsal seções do tubo neural não ter começado a se fragmentar, isso normalmente significa que o estoque de tripsina 2,5% é velho, e novas ações devem ser descongeladas, ou a lavagem com HBSS frio não retirar o HiHS da amostra. - Centrifugar os fragmentos de tecido em 200 g por 4 min.
- Remover o sobrenadante com uma pipeta Pasteur, deixando ~ 5-10 mL de líquido no fundo do tubo.
- Flick suavemente o tubo para soltar a pelota, em seguida, lave as células, adicionando 5 ml de HBSS quente. Vamos resolver em temperatura ambiente por 2 min.
- Centrifugar a 200 g por 5 min.
- Remover o sobrenadante com uma pipeta Pasteur, deixando ~ 5-10 mL de líquido no fundo do tubo.
- Flick suavemente o tubo para soltar a pelota e parcialmente ressuspender as células. Em seguida, adicione 2 ml de HBSS quente.
- Use o pequeno (diâmetro meia)-fogo de vidro polido pipeta Pasteur para dissociar as células por pipetagem lentamente para cima e para baixo 4-6 vezes. Evitar fazer bolhas, e pipetar o líquido contra a parede do tubo. Não excesso de triturar.
- Use o menor-fogo de vidro polido pipeta Pasteur para continuar a dissociar as células por pipetagem lentamente para cima e para baixo 3-4 vezes. Evitar fazer bolhas, e pipetar o líquido contra a parede do tubo. Não excesso de triturar.
Para o tecido de dissecção grossa, adicionar um ml um extra de HBSS quente para o tubo no final da dissociação.
Ao dissociar as células, não é necessário dissociar todos os aglomerados de células e agregados. Parar pipetando para cima e para baixo ou mudar para uma pipeta Pasteur com um diâmetro menor, se você não vê nenhuma redução ainda maior no tamanho dos agregados de células com mais de pipetagem. - Que qualquer fragmento de tecido restantes se estabelecer no tubo durante 1 min. Não é necessário transferir as células para um novo tubo.
- Tome 20 l da suspensão celular e adicionar 5 mL de azul de tripano. Contagem de células em um hemocitómetro.
Neurônios deve ser ≥ 95% viáveis por exclusão do azul tripan. - Placa de neurônios no Media Plating Neurobasal (veja receitas abaixo).
- Sugeridos para a obtenção de densidades plating neurônios isolados (culturas de baixa densidade para evitar a aglomeração de neurônios ou sobrepostos uns aos outros):
- 120 000 - 180 000 células / poço de uma placa de 6 bem
- 60 000 - 75 000 células / poço de uma placa de 12 poços
- 16-18 horas mais tarde, mudar a mídia para mídia Crescimento Neurobasal (veja receitas abaixo)
Não use uma bomba de vácuo para aspirar a mídia da placa de cultura quando se muda de mídia; delicadamente use uma pipeta. Isto evita o desprendimento de neurônios.
Resultados representativos:
Quatro horas após o plaqueamento, os neurônios devem ter aderido à poli-L-lisina (PLL) revestido de superfície. Sob iluminação de contraste de fase, corpos celulares aderidos são tipicamente relativamente plana e de formato oval (fig. 2a). Células que não têm bem aderidas aparecem como esferas que se movem levemente quando o prato é muito gentilmente bateu na lateral. Muitos fatores podem potencialmente impedir a adesão de células (ver discussão).
Após 30 horas in vitro, a maioria dos neurônios estenderam um axônio com um cone de crescimento visível (fig. 2c, d). Se o crescimento axonal pobres é observado, verifique se o meio de crescimento Neurobasal foi feito com meio fresco e suplementos. Neurônios permanecem saudáveis por pelo menos seis dias nestas condições. Este procedimento tem se mostrado confiável rendimento preparações altamente enriquecido em neurônios comissurais, com 90% de neurônios expressando DCC (Yam et al. 2009). A largura da faixa de medula espinhal dorsal que é usado para preparar a suspensão de células irá afetar a pureza da cultura, com maior pureza, quando tiras mais finas são utilizadas. Um exemplo de aplicação é mostrado na figura 3 (imunofluorescência). Ver o artigo de Yam et al. (2009) para mais exemplos.

Figura 1. Schematics de passos dissecção da medula espinhal. D = dorsais, V = Ventral. Clique aqui para ver uma figura maior.
{kind=link}

Figura 2. Resultado Representante da comissural isoladoneurônios semeadas em uma lamela de vidro PLL-revestido. a, b) 4 horas após o plaqueamento, os neurônios têm aderido à superfície. Bar = 20 mM. c, d) 30 horas após o plaqueamento, a maioria dos neurônios ter estendido um axônio com um cone de crescimento visível. Bar = 20 mM.

Figura 3. Um neurônio comissural histoquímica para gama-tubulina (verde), com F-actina marcados por phalloidin (F-actina, vermelho) eo núcleo por DAPI (azul).
Receitas e comentários
Neurobasal Mídia Plating
- Neurobasal
- 10% inativadas pelo calor FBS (HiFBS)
- 2 mM L-glutamina (de 200 mM solução estoque)
Opcional: antibióticos penicilina / estreptomicina (use metade da concentração normal)
Neurobasal Mídia Crescimento
- Neurobasal
- B27 (1 / 50 de diluição do estoque)
- 2 mM L-glutamina (de 200 mM solução estoque)
- Opcional: antibióticos penicilina / estreptomicina (use metade da concentração normal)
Uma vez que a mídia é feita, ela pode ser mantido a 4 ° C por duas semanas. Para equilibrar a temperatura eo pH dos meios de comunicação antes do plaqueamento das células, a mídia local em pratos de cultura de tecidos Petri e coloque em uma cultura de tecidos incubadora de pelo menos 1,5 horas.
Neurobasal
Depois de uma garrafa de meio Neurobasal foi aberto, ele pode ser mantido por um mês a 4 ° C no escuro. Dispor de Neurobasal que foi aberta há mais de um mês, a sobrevivência da célula será menor.
Inativado pelo calor soro fetal bovino (HiFBS) ou soro de cavalo (HiHS)
Para aquecer-inativar calor FBS ou HS, a 56 ° C em banho-maria por 30 minutos. Agitar o frasco aproximadamente a cada 10 minutos ou assim. (Para maior precisão utilizar um frasco de tamanho semelhante cheio de água. Coloque um termômetro na garrafa de água para ver quando 56 ° C seja atingida. Inicie o tempo neste momento.) Inativadas pelo calor FBS pode precisar de ser centrifugadas para eliminar precipitados e pode ser aliquotado e re-congeladas a -20 ° C.
L-Glutamina
Descongelar sempre uma nova porção da L-glutamina para cada experimento.
B27
Alíquotas de B27 pode ser mantida a 20 ° C para o armazenamento a longo prazo, ou a 4 ° C por até um mês.
Discussão
Nós descrevemos um método para dissecar e cultura neurônios comissurais do cordão espinhal de ratos embrionárias. Este procedimento tem sido utilizado rotineiramente em nosso laboratório para preparar os neurônios de forma confiável para estudar os mecanismos celulares e moleculares de orientação axônio. Para a biologia celular e experimentos imunoquímica, a dissecção de uma ninhada produz neurônios suficientes. Quando as células são mais necessários, como em muitos experimentos de bioquímica, dissec?...
Agradecimentos
Este trabalho foi financiado por concessões do Canadian Institutes of Health Research (CIHR), o Peter Lougheed Medical Research Foundation, o Programa de McGill em Neuroengenharia, o Fonds de Recherche en Santé du Québec (FRSQ), ea Fundação Canadense para Inovação (CFI ). Sébastien D. Langlois foi apoiada pelo Prêmio de Mestre de Formação do Fonds de la Recherche en santé du Québec (FRSQ) e Frederick Banting por Charles e Prêmio de Melhor Canadá de Pós-Graduação Mestrado Bolsas de Estudo a partir do Canadian Institutes of Health Research (CIHR). Somos gratos a Jessica MT Pham de assistência com os números.
Materiais
Dissecção da medula espinhal dorsal embrionárias de rato (ver também Quadro I)
- E13 gravidez encenado rato
- Etanol 70%
- Tesoura cirúrgica, Ferramentas Ciência Belas
- Fórceps, Dumont # 5, Ferramentas Ciência Belas
- Placas de Petri
- L-15 de médio
- Microscópio de dissecção, Leica
- Plástico pipetas de transferência
- Microtesoura, Ferramentas Ciência Belas
- Agulhas de tungstênio e retentores de pinos (uma agulha em forma de gancho, uma agulha L-formas, uma agulha), Ferramentas da Ciência Belas
- Inativado pelo calor soro de cavalo (HiHS)
- 15 ml de tubos de plástico
Comissural neurônio cultura (ver também Quadro I)
- Tecido incubadoras cultura (37 ° C, umidade 5% CO 2, controlada)
- Alemão Desag lamínulas de vidro e / ou placas de cultura de tecido de plástico
- Poli-L-lisina, Sigma
- Estéril MilliQ H 2 O
- Neurobasal, Invitrogen
- Inativado pelo calor soro fetal bovino (HiFBS)
- L-Glutamina
- B27, Invitrogen
- Penicilina / estreptomicina antibióticos
- Banho-maria 37 ° C
- Pasteur estéril Pipettes
- Bunsen queimador de gás
- HBSS, Ca 2 + / Mg 2 + livre, Invitrogen
- DNAse, Worthington
- MgSO 4
- Centrifugador
- Hemocitômetro
- Solução Azul Trypan
Referências
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados