
Method Article
Adsorção molecular induzida por luz de proteínas usando o sistema PRIMO para Micropadronização para estudar as respostas celulares às proteínas da matriz extracelular
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Nosso objetivo geral é entender como as células sentem sinais extracelulares que levam ao crescimento axonal direcionado. Aqui, nós descrevemos a metodologia da adsorção molecular Light-induced das proteínas, usadas para produzir micropatterns definidos de componentes da matriz extracelular a fim estudar os eventos específicos que governam o conseqüência do AXON e o pathfinding.
Resumo
As células sentem uma variedade de sinais extracelulares, incluindo a composição e geometria da matriz extracelular, que é sintetizado e remodelado pelas próprias células. Aqui, apresentamos o método de adsorção molecular induzida por luz de proteínas (LIMAP) utilizando o sistema PRIMO como técnica de padronização para a produção de substratos de matriz extracelular (ECM) micropadronizada usando uma única ou combinação de proteínas. O método permite a impressão de padrões ECM em resolução mícron com excelente reprodutibilidade. Nós fornecemos um protocolo passo-a-passo e demonstrar como isso pode ser aplicado para estudar os processos de pathfinding neuronal. O LIMAP tem vantagens significativas sobre os métodos de microimpressão existentes em termos de facilidade de padronização de mais de um componente e a capacidade de gerar um padrão com qualquer geometria ou gradiente. O protocolo pode ser facilmente adaptado para estudar a contribuição de quase qualquer componente químico para o destino celular e comportamento celular. Finalmente, discutimos questões comuns que podem surgir e como elas podem ser evitadas.
Introdução
Nos últimos anos, as ciências biológicas têm feito cada vez mais uso dos avanços prestados pelas ciências materiais. Um exemplo proeminente é a micropadronização de substratos, que pode ser usado para estudar respostas celulares, como a proliferação celular1,2Diferenciação3,4,5,6, migração de células7,8,9e pathfinding10,11. Há uma série de técnicas disponíveis que permitem a micropadronização de substratos, tais como fotoquímica animado multifóton12, Nanolitografia AFM DIP-Pen13, pino e jato de tinta impressão direta14, litografia por feixe de elétrons15ou microfluínicos16. No entanto, duas técnicas que são amplamente utilizadas no campo biológico são a impressão de microcontato17,18,19ou padronização assistida por laser3(Figura 1). A padronização assistida por laser é considerada para fornecer resultados mais confiáveis em termos de estabilidade de proteínas e PEG e confinamento de células nos padrões, em comparação com a impressão de microcontato20. Uma aproximação mais nova para o micropatterning descrito aqui é o uso da adsorção molecular Light-induced das proteínas21AEUFigura 1 D) utilizando um sistema disponível comercialmente (PRIMO,Tabela de materiais). Cada um dos métodos tem vantagens e limitações que são brevemente descritas abaixo. A impressão de microcontato usa moldes PDMS (selos) com microcaracterísticas desejadas que são geradas a partir de mestres litografados. Os selos são incubados com uma proteína escolhida que é então transferida (carimbada) para o substrato de cultura celular18(Figura 1 Um). O padronização laser-ajudado usa a luz UV para Cleave uma película antiincrustante22,23,24,25, expondo regiões que posteriormente podem ser revestidas com a proteína de interesse (Figura 1 B). Quando a definição conseguida com as aproximações Photo-patterning estiver na escala do mícron25,26, a maioria dessas técnicas requer uma máscara fotográfica, seja em contato com a amostra, ou situada no plano de objeto do objetivo do microscópio23,27,28. As exigências para máscaras na impressão do microcontact e no Photo-patterning podem ser uma limitação; máscaras específicas são necessárias para cada padrão geométrico e tamanho, que pode ser caro e demorado para gerar. Em contraste com essas técnicas, o LIMAP não requer uma máscara (Figura 1 D). O uso do sistema PRIMO para LIMAP pode ser de custo intensivo no início, pois requer a compra de equipamentos. No entanto, o software de código aberto é usado para projetar padrões de qualquer geometria desejada, dando muito mais liberdade e permitindo experimentos mais complexos, incluindo o uso de gradientes de concentração proteica. O laser PRIMO é controlado e dirigido por um dispositivo Micromirror (DMD) controlado digitalmente para criar padrões em qualquer número de geometrias definidas pelo usuário. O LIMAP requer que a superfície da cultura seja revestida com moléculas que impedem a fixação da célula. O polietileno glicol (PEG) é o mais comumente usado como tal um "antifouling" reagente; forma uma película antiadesiva densa na superfície de vidro ou plástica29. Subseqüentemente, um Photo-Initiator é adicionado que permite que a película do PEG seja removida com a elevada precisão através de um mecanismo do photoscission30pela exposição local à luz UV o controle do DMD. Estas regiões Peg-livres podem ser revestidas com as proteínas que adsorver à superfície laser-gravada, gerando um micro-teste padrão. Variando a potência do laser, as quantidades diferentes de PEG podem ser removidas da superfície permitindo que o usuário gere gradientes da proteína. A remoção do PEG e o procedimento do revestimento podem ser repetidos para criar testes padrões com duas ou mais proteínas distintas no mesmo micro-poço21. Os micropadrões gerados fornecem superfícies adesivas para as células, permitindo o estudo do comportamento celular. Em nossos estudos, nós usamos a micropadronização para estudar o pathfinding do neurite ou do AXON de uma linha celular neuronal (CAD (Cathecholaminergic-um diferenciado) pilhas31) ou neurônios primários do gânglio dorsal-raiz do rato (DRG), respectivamente. Aqui, nós esboçamos um protocolo passo a passo para LIMAP (A Figura 2) utilizando o sistema PRIMO disponível comercialmente e acompanhando o software Leonardo. Nós demonstramos como ele pode ser usado para a geração de padrões com geometrias definidas e múltiplas proteínas, que usamos para estudar pathfinding axonal. Discutimos questões comuns que podem surgir e como elas podem ser evitadas.
Protocolo
1. projeto de moldes do teste padrão
Nota: os modelos para padronização são gerados com o software de desenho digital (tabela de materiais). Desenhar em diferentes níveis de cinza determinará intensidades de laser. Usar o software para projetar modelos padrão permite a geração rápida de padrões com qualquer geometria desejada e gradientes (Figura 3).
- Desenhe digitalmente o modelo de padrão desejado usando o software de desenho. Selecione um tamanho de imagem de 1824 pixels de comprimento e 1140 pixels de largura (que neste estudo corresponde a 415 μm de comprimento e 260 μm de largura). Salve o modelo de padrão como um arquivo TIFF de 8 bits.
Nota: um protocolo passo a passo para gerar modelos está disponível mediante solicitação à marca que comercializa o equipamento de micropadronização (tabela de materiais).
2. limpeza do plasma
Nota: os resultados óptimos exigem a limpeza do plasma das superfícies antes do patterning, que removerá toda a matéria orgânica e ativará a superfície. No presente caso, o ar ambiente é suficiente para a ativação da superfície. Um limpador de plasma (tabela de materiais) foi usado com uma pressão de processo de 1000-1300 mTorr e uma potência de 29,6 W para 1-5 min.
- Use o prato inferior de vidro/es com um tamanho bem interno de 20 milímetros e uma espessura de vidro de 0.16-0.19 milímetros. Para testar várias condições, use um prato inferior de vidro de 6 poços. Se não, use um único prato inferior de vidrodo poço (tabela dos materiais).
Nota: um protocolo passo a passo para a limpeza de plasma está disponível mediante solicitação à marca que comercializa o equipamento de limpeza de plasma (tabela de materiais).
3. passivação
Nota: esta etapa gera uma película antifouling que impeça a adsorção da proteína à superfície de vidro. O PEG oferece a resistência elevada à adsorção29 da proteína como um agente antifouling. LIMAP usa um Photo-Initiator para remover localmente PEG através de photoscission UV. A proteína/s de interesse, em seguida, adsorverá a estas superfícies PEG-Free21, gerando micro-padrões.
- Passivação com PLL-PEG
- Em condições estéreis, corte os estênceis PDMS (ver Figura 4a, B e tabela de materiais) para garantir que eles cabem na parte inferior do vidro interior bem, e remover o interior micro-bem enchimentos com fórceps estéril. Estêncis da vara no poço de vidro usando o fórceps.
- Assegure-se de que os estênceis furam firmemente no vidro bem, impedindo a formação de bolhas de ar que podem causar fugas durante o processo da passivação.
Nota: os stencils de PDMS podem igualmente ser fabricados em casa usando protocolos publicados18,32. - Prepare a solução PLL-PEG (tabela de materiais, 0,1 mg/ml) em soro fisiológico tamponado com fosfato (PBS). Adicionar 20 μL de solução de PLL-PEG a cada micropoço e incubar à temperatura ambiente, durante 1 h.
- Retire 15 μL de PLL-PEG dos micropoços e lave-os cinco vezes com 20 μL de PBS (tabela de materiais) sem deixá-los secar.
Nota: deixar sempre cerca de 5 μL de PBS entre as lavas. Dado o seu pequeno volume, os micropoços são particularmente suscetíveis à secagem. A secagem resultará em micropadrões de má qualidade. - Ou manter o prato de cultura em PBS (3 mL por poço) a 4 ° c por até 3 dias ou continuar com o próximo passo (passo 3.1.5).
- Remova 18 μL de PBS de um único micro-poço (por exemplo, o micro-poço esquerdo superior, veja Figura 4D), adicione 5 μL de Photo-Initiator (plpp, tabela de materiais) e deixe 20 μL de PBS nos micro poços restantes. Este micropoço com PLPP será usado para criar um padrão de referência (veja a Figura 4D, E) durante a etapa de calibração do sistema (etapa 4). Mantenha PLPP protegido da luz.
- Passivação com PEG-SVA duradouro
Nota: para gerar a superfície antiadesiva uma pode usar-se: 1) um PEG lig ao poly-L-lysine (PLL-PEG, etapa 3,1) ou 2) um PEG-Succinate N-hydroxisuccinimide (PEG-SVA). A decisão de escolher uma ou outra opção de passivação depende das opções de armazenamento (consulte a etapa 10). Os pratos da cultura incubados com Peg-SVA exigem a quantidade dobro de passivação e de tempo photopatterning.- Prepare stencils conforme descrito na etapa 3.1.1.
- Adicionar 20 μL de 0, 1% de poli-L-lisina (PLL, tabela de materiais) a cada micropoço e incubar à temperatura ambiente durante 30 min para pré-revestimento com PLL.
- Retire 15 μL de PLL dos micropoços e lave-os três vezes com 20 μL de tampão HEPES de 1 M (tabela de materiais) sem deixar secar os poços.
Nota: deixar sempre cerca de 5 μL de HEPES ou PBS entre as lavas. Dado o seu pequeno volume, os micropoços são particularmente suscetíveis à secagem. A secagem resultará em micropadrões de má qualidade. - Prepare a solução PEG-SVA. A solução de PEG-SVA (50 mg/mL no tampão de HEPES 1M) deve ser preparada fresca cada vez imediatamente antes de usar. Prepare 20 μL de PEG-SVA por micropoços.
- O tampão de HEPES deve ter um pH entre 8-8.5. Teste o pH de HEPES antes de preparar a solução de PEG-SVA com papel do pH. Pesar PEG-SVA em um tubo de centrífuga usando uma balança de precisão. Adicionar tampão HEPES e Vortex 30 s até dissolver. PEG-SVA é completamente dissolvido quando a solução é transparente.
Nota: o SVA é o éster que permite a ligação de PEG ao PLL previamente revestido. Uma vez que o tampão de HEPES é adicionado ao PEG-SVA, o SVA tem uma meia-vida de 15 minutos e deve ser usado imediatamente. - Adicionar 20 μL de solução de PEG-SVA a cada micropoço e incubar à temperatura ambiente, durante 1 h. Retire 15 μL de PEG-SVA dos micropoços e lave cinco vezes com 20 μL de PBS (tabela de materiais) sem deixar secar os poços.
- Prepare o prato de cultura para armazenamento de longo prazo (até 1 mês, consulte a etapa 10,2) ou prossiga para a próxima etapa (etapa 3.2.8).
- Remova 18 μL de PBS de um único micro-poço (por exemplo, o micro-poço esquerdo superior, veja Figura 4D), adicione 5 μL de plpp (tabela de materiais) e deixe 20 μL de PBS nos micro poços restantes. Este micropoço será utilizado para criar um padrão de referência (ver Fi Gure 4D, E). Assegure-se de que PLPP seja homogêneo em toda a superfície do micropoço. Mantenha PLPP protegido da luz.
4. calibração do sistema
Nota: nestas etapas, o foco do laser será ajustado ao tipo particular de prato da cultura (etapa 4,1). Um padrão de referência será gerado em apenas um micropoço (etapa 4,2) seguido de incubação com uma solução proteica (etapa 4,3) para garantir as condições ideais de foco do laser (passo 4,4), necessário para obter padrões afiados e definidos.
- Calibração do laser
Nota: durante o processo de calibração, uma imagem a laser de calibração será projetada para uma superfície de vidro com destaque de fluorescentamente (calibração bem, marcada com um marcador fluorescente, Figura 4C), que precisa ser colocada em foco na Microscópio.- Use um marcador fluorescente (tabela de materiais) para marcar um poço de vidro interno vazio.
Nota: a calibração bem deve ter a mesma espessura de vidro (0.16-0.19 mm) como o prato de cultura em que os micropadrões serão gerados. Se um prato inferior de vidro de 6 poços está sendo usado, um poço vazio pode ser usado para a calibração e deve ser marcado com o Highlighter fluorescente circunstâncias estéreis. - Ligue o microscópio, o palco e o computador. Ligue o equipamento micro-padronização PRIMO, abra o micro-Manager e o software Leonardo. Leonardo software é operado através de micro-gerente plugins. Verifique as marcas/números de catálogo de equipamentos e softwares na tabela de materiais.
- No menu inicial de Leonardo, selecione calibrar. Verifique se o cubo de filtro PRIMO dedicado está na posição correta (caminho óptico) na torre do filtro; Selecione o objetivo 20X (0,75 DIC S Plan fluor, sem anel de fase) tanto no microscópio quanto no software Leonardo.
Nota: Este protocolo é ajustado a Leonardo versão 4,4. O protocolo pode precisar de ajuste para outras versões. - Posicione bem o poço de calibração realçado anteriormente (Figura 4C) acima do objetivo. Selecione o caminho da câmera. Ajuste o foco objetivo até que a projeção do laser do logotipo primo e da linha de tag cuide de suas células estejam em foco.
- Deixe o tempo de exposição da câmera padrão em 25 MS. Ajuste a intensidade do laser para ver a letra I da projeção do logotipo primo em cor cinzenta e o resto das letras em branco.
- Registre a posição Z do plano focal, (altura do objetivo para a amostra) mais tarde referida como Z-posição de calibração. Esta será uma aproximação ao foco ideal obtido após a geração do teste padrão de referência (veja a etapa 4.2-4.4).
- Use um marcador fluorescente (tabela de materiais) para marcar um poço de vidro interno vazio.
- Padrão de referência
- Posicione o prato de cultura com o micropoço que contém o Photo-Initiator (padrão de referência micro-bem, Figura 4D) acima do objetivo e selecione o padrão agora no software Leonardo.
- Visualize a borda do micropoço com luz transmitida através da câmera e escolha o símbolo de ROI no menu à direita. Defina o diâmetro do círculo de ROI para 4000 μm e alinhe a borda do ROI digital com a borda do micropoço atual.
- Assegure-se de que haja uma sobreposição exata entre o ROI digital e o micro-poço atual movendo o estágio em torno das bordas do micro-poço. A posição do ROI será acoplada aos movimentos do estágio.
- Selecione Bloquear no software Leonardo para bloquear o ROI na posição desejada. Desligue a luz transmitida.
- Selecione primo para carregar o modelo de padrão desejado, que será projetado no ROI como uma unidade de design (consulte a Figura 5). Os padrões serão exibidos em uma lista suspensa denominada ações e mostradas no menu Ações no software.
Observação: modelos de padrão precisam ser projetados anteriormente (consulte a etapa 1) e salvos como um arquivo TIFF de 8 bits antes de carregar o modelo no software. - Apenas um pequeno padrão é necessário para o padrão de referência; por exemplo, 3 linhas, 1 coluna (ver Figura 4e e Figura 5B). No menu de replicação , defina o número desejado de colunas e linhas (linhas no software Leonardo). Clique em Atualizar para observar uma visualização digital do design do padrão.
- Defina a dose de laser no menu de replicação . A dose óptima do laser neste ajuste e usando PLL-PEG é 1390 mJ/mm2.
Nota: a potência do laser pode variar entre 5-7,5 mW/mm2. Neste caso, é 7,5 mW/mm2, que leva aproximadamente 30 s para padronizar cada unidade de projeto, usando uma dose de laser de 1390 MJ/mm2. As doses mais elevadas do laser podem ser exigidas se a superfície do prato da cultura é passivated com PEG-SVA (dose aproximadamente dobro do laser comparada ao PLL-PEG). Isso precisa ser testado de antemão. - Navegue até uma região periférica do micropoço, (por exemplo, parte superior) longe da região principal de interesse para a geração de padrões (região central) do micropoço (consulte a Figura 4e) eselecione Bloquear. Aguarde até que o padrão seja exibido.
- Ajuste o foco para a posição Z da calibração (consulte a etapa 4.1.6).
Nota: é recomendável fazer uma etapa de calibração do sistema adicional se outro usuário estiver usando o mesmo microscópio entre as rodadas de fotopadronização. - Selecione o símbolo de reprodução para iniciar a padronização. Assegure-se de que o laser esteja ligado no software. Aguarde até que o processo de padronização seja concluído. A duração de padronização será exibida no painel tempo estimado . Na versão do software Leonardo 4,4, a padronização é concluída quando todas as ações aparecem em azul no menu Visualização.
- Incubação de proteínas no padrão de referência
- Em condições estéreis, lave o padrão de referência micro-poço três vezes com 20 μL de PBS para remover o PLPP.
- Adicionar 20 μL de proteína de ECM com rótulo fluorescentamente (10 μg/mL de laminina, fibronectina ou fibrinogênio em PBS, ver tabela de materiais e passo 6) para o padrão de referência micro-bem. Incubar à temperatura ambiente durante 10-20 min (dependendo da proteína) protegida da luz (embrulhe o prato em folha de alumínio).
- Após a incubação, retire 18 μL de solução proteica e lave três vezes com 20 μL de PBS; manter os micropoços que não são utilizados para criar um padrão de referência (ver Figura 4D, E) em 20 μL de PBS.
- Visualização e definição do foco laser ideal
- Visualize o padrão de referência usando um microscópio de epifluorescência, objetivo 20X e software adequado (verifique o software utilizado neste estudo na tabela de materiais). O padrão de referência deve ser visível na região periférica (por exemplo, região de poço superior), na qual o padrão de referência foi gerado (ver Figura 4E).
- Ajuste o foco nas bordas do padrão através do caminho da câmera. Registre a posição Z de acordo com o melhor foco no padrão de referência. Esta posição Z ajustada será o foco laser ideal usado para a padronização subsequente.
5. software de set-up e photopatterning
Nota: uma vez que a calibração do sistema foi alcançada (etapa 4), o usuário carregará os moldes desejados do teste padrão (configuração do molde, Figura 5) para o photopatterning, com a opção para gerar testes padrões para uma ou proteínas múltiplas em cada micro-bem. O processo de micropadronização envolve a fotopadronização e as etapas de incubação de proteínas (ver Figura 2).
- Em condições estéreis, retire 18 μL de PBS de todos os micropoços e adicione 5 μL de PLPP a cada micropoço. Assegure-se de que o PLPP seja homogêneo em toda a superfície dos micropoços.
- Posicione o prato de cultura com o padrão de referência micro-bem (Figura 4D, e) acima do objetivo e selecione padrão agora no software Leonardo.
- Visualize o micropoço com luz transmitida através do caminho da câmera e escolha o símbolo de ROI. Defina o diâmetro do ROI para 4.000 μm e sobreponha a borda do ROI digital à borda do micropoço atual. Selecione Bloquear.
Nota: a forma e o diâmetro do ROI dependem do design e do tamanho do estêncil PDMS que está sendo usado. Por exemplo, se estiver usando 5.000 x 5.000 μm PDMS quadrados estênceis, use um 5.000 x 5.000 μm ao quadrado ROI. - Repita a etapa de sobreposição (etapa 5,3) para cada micropoço do prato. Após a conclusão, desligue a luz transmitida.
- Navegue até o centro do padrão de referência micro-bem, longe da região de padrão de referência e selecione primo para carregar o modelo de padrão desejado, que será projetado no ROI como uma unidade de design (consulte a Figura 5). Os padrões serão exibidos em uma lista suspensa denominada ações e mostradas no menu Ações no software.
Observação: modelos de padrão precisam ser projetados antes do experimento e salvos como um arquivo TIFF de 8 bits antes de carregar o modelo no software. - A fim criar um teste padrão através do micro-poço inteiro, a unidade de projeto precisa de ser replicada. Uma unidade de design abrange cerca de 0,1 mm2 da área de micropoços. No menu Replication , defina o número desejado de colunas e linhas (linhas no software) (consulte a Figura 5).
- Para gerar um padrão contínuo, ajuste o espaçamento entre colunas e linhas; Neste estudo, padrões de listras contínuas são obtidos usando-20 a-35 μm espaçamento (espaçamento negativo) entre as colunas. Esse espaçamento negativo cria uma sobreposição entre as unidades de design (Figura 5B, C).
- Defina a dose de laser no menu de replicação . A dose óptima do laser neste ajuste e usando PLL-PEG é 1390 mJ/mm2. A duração de padronização será exibida no painel tempo estimado .
Nota: neste caso, a potência do laser é de 7,5 mW/mm2, tendo aproximadamente 30 s para padronizar cada unidade de projeto, usando uma dose de 1390 MJ/mm2. Por exemplo, uma unidade de design (0,1 mm2) replicada em 4 colunas e 4 linhas (cerca de 1,6 mm2), levaria 8 min para ser padronizada. As doses mais elevadas do laser podem ser exigidas se a superfície do prato da cultura é passivated com PEG-SVA (dose aproximadamente dobro do laser comparada ao PLL-PEG). - Selecione Bloquear e aguarde até que o padrão seja exibido virtualmente.
- Para atualizar os parâmetros de um padrão, clique na açãorelacionada e, em seguida, desbloqueie e atualize os parâmetros. Selecione Bloquear novamente quando a atualização de padrão for concluída.
- A padronização de múltiplas proteínas no mesmo micropoço (rodadas seqüenciais de micropadronização) requer um alinhamento preciso dos padrões. Para alcançar esse alinhamento, carregue todos os conjuntos de modelos de padrão desejados simultaneamente (padrões de primeira e segunda rodadas de photopatterning).
- Defina os parâmetros de replicação e dose dos modelos de padrão. Depois que os parâmetros são definidos, os padrões serão exibidos como ações na lista de ações . Guarde esta configuração de modelo como um ficheiro no software (consulte a Figura 5D).
- Na lista de ações , selecione apenas as ações específicas a serem padronizadas durante a primeira rodada de padronização e desmarque as ações que serão padronizadas durante a segunda rodada de padronização (consulte a Figura 5D).
- Navegue até a área onde o padrão de referência foi produzido na etapa 4,2 (por exemplo, a região superior do padrão de referência micro-bem, consulte a Figura 4E). Ajuste o foco para a posição Z ideal (obtida na etapa 4,4).
Nota: é altamente recomendável selecionar o sistema de focagem perfeito no microscópio que está sendo usado (se disponível), o que garante que a posição Z ideal para padronização será mantida ao longo de todo o processo de fotopadronização. - Selecione o símbolo de reprodução para iniciar a padronização. A duração de padronização será exibida no painel tempo estimado . Na versão do software Leonardo 4,4 a padronização é concluída quando todas as ações aparecem em azul no menu de Visualização .
6. incubação de proteínas
Nota: os micropoços são incubados com proteínas ECM (preferencialmente rotuladas com fluorescentamente). Estes só se ligam às áreas onde PEG foi clivada através do processo de fotopadronização descrito no passo 5. Cada poço contem um estêncil de PDMS com os 4 micro-poços, que permitirão testar 4 circunstâncias diferentes simultaneamente, por exemplo, incubação de uma proteína diferente em cada micro-poço (veja Figura 4D).
- Utilize proteínas rotuladas com fluorescentamente (por exemplo, laminina, fibronectina ou fibrinogênio conjugadas a fluoróforos vermelhos ou verdes) para visualizar os micropadrões (ver passo 6,7 e 9,4). Alternativamente, as proteínas sem rótulo adsorveu podem ser visualizadas em estágios mais atrasados usando a imunofluorescência.
Nota: as proteínas de ECM (por exemplo, fibronectina) podem ser rotuladas usando protocolos existentes33 e kits de rotulagem de fluorescência comercialmente disponíveis (ou seja, kit de rotulagem Alexa 488), ou comprados prontamente rotulados (por exemplo, fibrinogênio-488 conjugados ou laminina-vermelho fluorescente RHODAMINE, ver tabela de materiais). - Em condições estéreis, prepare a concentração desejada de proteínas de ECM (10 μg/mL de laminina, fibronectina ou fibrinogênio em PBS, ver tabela de materiais e etapa 4,3).
Nota: as proteínas de ECM rotuladas Fluorescently são sensíveis à luz e devem ser protegidas da luz (prato do envoltório na folha de alumínio) e devem ser mantidas no gelo em todas as vezes. - Em condições estéreis, lave os micropoços três vezes com 20 μL de PBS para remover o PLPP.
- Retire 18 μL de PBS de todos os micropoços e adicione 20 μL de solução de proteína ECM a cada micropoço. Incubar à temperatura ambiente, por 20-30 min e proteger da luz.
Nota: os tempos de incubação de revestimento ideais podem variar consoante o tipo e a concentração de proteínas. - Após a incubação, retire 15 μL de solução proteica de ECM e lave os micropoços três vezes com 20 μL de PBS.
Nota: deixar sempre cerca de 5 μL de PBS entre as lavas. - Se a padronização com apenas uma proteína (uma rodada de micropadronização), proceder tanto para o armazenamento do prato de cultura (passo 10) ou para o chapeamento de células (passo 11, e ver Figura 2). Se executar uma segunda rodada de micropadronização com uma proteína diferente no mesmo micro-bem, proceder tanto para o armazenamento do prato de cultura (etapa 10) ou para bloquear sites de ligação inespecíficos (etapa 7, e veja a Figura 2).
- Etapa opcional do controle da qualidade: visualize e testes padrões impressos imagem usando um microscópio da epifluorescência antes das pilhas do chapeamento. Selecione os canais fluorescentes apropriados e ajuste os tempos de exposição de acordo.
Nota: para comparar a intensidade de fluorescência dos padrões entre experimentos, é essencial usar os mesmos tempos de exposição para as mesmas proteínas.
7. bloqueando locais de ligação inespecíficos (apenas para múltiplos padrões proteicos)
Nota: a micropadronização com múltiplas proteínas no mesmo micropoço envolve etapas de padronização seqüenciais (veja a Figura 2). Um agente de bloqueio (PLL-PEG ou BSA) é adicionado aos micropoços para evitar a ligação cruzada, que ocorre quando a segunda proteína incubada (etapa 9) se liga à primeira proteína incubada (etapa 6), evitando assim uma mistura de proteínas dentro dos padrões.
- Em condições estéreis, adicione 20 μL de PLL-PEG (0,1 mg/mL em PBS) ou BSA (1% BSA em PBS) como um passo de bloqueio para evitar a ligação cruzada.
Nota: a eficiência de bloqueio pode variar dependendo da natureza das proteínas que estão sendo usadas e da afinidade entre eles. Recomenda-se testar de antemão PLL-PEG e BSA como agentes de bloqueio (Figura 10D-I). - Incubar o agente de bloqueio à temperatura ambiente durante 1 h e proteja-o da luz. Remova 15 μL de agente de bloqueio e lave todos os micropoços três vezes com 20 μL de PBS. Deixe sempre cerca de 5 μL de PBS entre as lavas.
- Retire 18 μL de PBS de todos os micropoços e adicione 5 μL de PLPP a cada micropoço, assegurando que o PLPP seja homogêneo em toda a superfície dos micropoços.
8. segunda rodada de photopatterning (apenas para vários padrões de proteínas)
Nota: após a primeira rodada de fotopadronização e incubação de proteínas, um micropadrão é gerado. Durante a segunda rodada de fotopadronização, o padrão para a segunda proteína será gerado no mesmo micropoço (ver Figura 2C e Figura 5D). No software, selecione os modelos de padrão corretos (ações), que serão padronizados durante esta rodada (veja a Figura 5D).
- Navegue até o primeiro micropoço (Figura 4D). Assegure a sobreposição apropriada entre o ROI digital e o micro-poço.
- Carregue a configuração do modelo salva anteriormente (etapa 5,12) e selecione as ações a serem padronizadas durante a segunda rodada de fotopadronização (desmarque as ações da primeira rodada, consulte a Figura 5D).
- Selecione o símbolo de reprodução para iniciar a padronização. Assegure-se de que o laser esteja ligado no software.
9. segunda rodada de incubação de proteínas (apenas para múltiplos padrões proteicos)
Nota: nesta parte do protocolo, a proteína/s com rótulo fluorescently será incubada no prato de cultura após a segunda rodada de fotopadronização.
- Em condições estéreis, lave o micropoço três vezes com 20 μL de PBS para remover o PLPP. Retire 18 μL de PBS e adicione 20 μL de solução de proteína ECM a cada micropoço. Incubar à temperatura ambiente durante 20-30 min e proteja-se da luz.
Nota: os tempos de incubação de revestimento ideais podem variar consoante o tipo e a concentração de proteínas. - Após a incubação, retire 15 μL de solução proteica de ECM e lave os micropoços três vezes com 20 μL de PBS. Deixe sempre cerca de 5 μL de PBS entre as lavas.
- Ou proceder ao armazenamento de prato de cultura (passo 10) ou ao chapeamento de células (passo 11, e ver Figura 2).
- Etapa opcional do controle da qualidade: usando um microscópio da epifluorescência, visualize e testes padrões impressos imagem antes de pilhas do chapeamento. Selecione os canais fluorescentes apropriados e ajuste o tempo de exposição.
Nota: para comparar a intensidade de fluorescência dos padrões entre experimentos, é essencial usar os mesmos tempos de exposição para as mesmas proteínas.
10. armazenamento de micro-padrões
Nota: os Micropadrões com proteínas adsorvedas podem ser armazenados durante diferentes etapas do protocolo (ver Figura 2). Se micropadronização com múltiplas proteínas, os micropadrões podem ser armazenados após a primeira rodada de micropadronização ou após a conclusão das duas rodadas seqüenciais de micropadronização (ver Figura 2B).
- Quando passivated com PLL-PEG, armazene os micro-testes padrões em PBS (3 mL por o poço) em 4 ° c por até 3 dias.
- Se o prato de cultura foi passivated com PEG-SVA, os micro-testes padrões podem ser armazenados por até 1 mês. A fim fazer assim, enxague os micro-testes padrões intensamente com água deionizada destilada dobro e seque com uma pistola de ar estéril do argônio ou do nitrogênio, embora o ar normal possa igualmente ser usado. Após a secagem, os micropadrões podem ser armazenados a 4 ° c por até 1 mês (equipe de suporte do sistema PRIMO, comunicação pessoal).
11. células de chapeamento
Nota: durante os próximos passos, as células serão chapeadas no preparado micro-padronizado cultura prato/es. Nestes estudos, é utilizada uma linha celular neuronal (células CAD)31. Entretanto este protocolo pode ser ajustado para estudar outros tipos da pilha de interesse (ajuste o protocolo do chapeamento de pilha como exigido).
- Diferenciar células CAD para 48 h usando meio de diferenciação (DMEM suplementado com 1% de glutamina, sem soro, e com 1% Pen/strep, ver tabela de materiais).
- Placa 1 mL de meio com células no interior de vidro bem, cobrindo todos os micropoços e coloque o prato de cultura em um 37 ° c, 5% CO2 incubadora.
Resultados
Seguindo o protocolo acima resulta em superfícies micromodeladas, revestidas com proteína ECM/s de interesse. Nós estamos usando estes testes padrões para seguir o pathfinding neuronal.
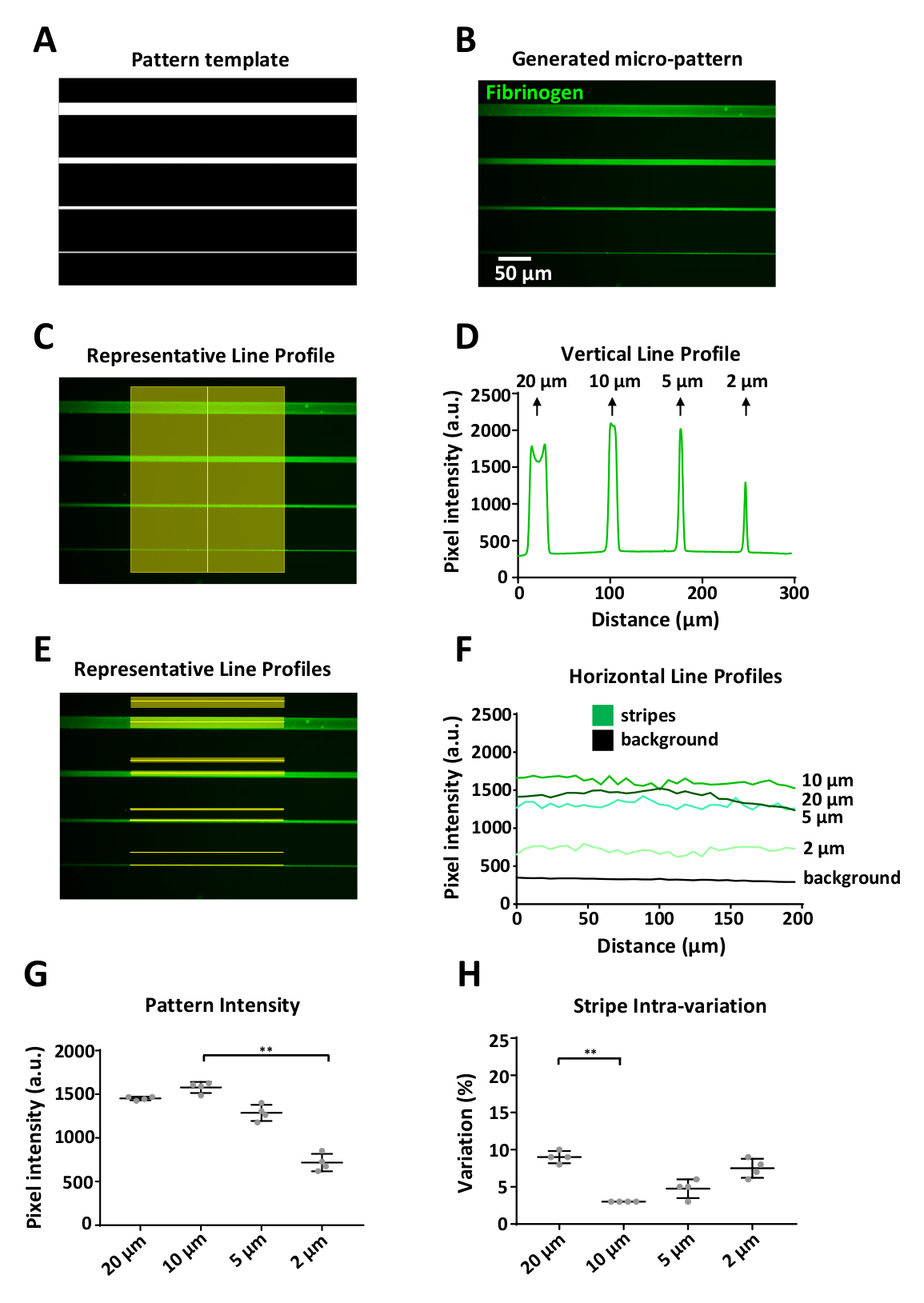
Os padrões gerados devem ser uma representação precisa do modelo. Um exemplo é mostrado na Figura 6 , onde um modelo de padrão digital (Figura 6a) representando uma unidade de projeto (Figura 5B), resultou em micropadrões definidos, variando de 20 a 2 μm de largura, revestido com rotulado Fibrinogênio (Figura 6B). Usando ImageJ, as medições de intensidade de fluorescência foram obtidas verticalmente (Figura 6C) e horizontalmente (Figura 6e) ao longo da listra e de uma região de fundo correspondente 15 μm acima de cada faixa. As medições de fundo foram subtraída das medições de padrão para cada largura da listra.
Uma limitação do sistema é que um efeito de aresta pode ser observado (Figura 6B, listra superior) ao imprimir características ≥ 20 μm, com um sinal de maior intensidade nas bordas do padrão em comparação com o centro (Figura 6D, primeiro pico de Perfil de intensidade fluorescente). Em nossos experimentos o limite de resolução foi de aproximadamente 2 μm; nessa largura observamos uma diminuição significativa (por aproximadamente 50%) na intensidade da fluorescência do fibrinogênio em comparação com a intensidade das listras mais largas (Figura 6F, G). A padronização com o sistema PRIMO e o protocolo aqui delineado produziram padrões reprodutíveis, com o maior desvio padrão da intensidade média fluorescente medida para a largura de 2 μm de quatro unidades de projeto replicadas individuais (Figura 6 G). a variação dentro das listras modeladas foi encontrada igualmente para ser baixa; o coeficiente de variação variou de 3 a 10%, com as listras de 20 μm e 2 μm com a maior variação interna. Isso provavelmente será resultado do efeito de aresta e do limite de resolução do sistema, respectivamente. Note-se que para estas medições só medimos a intensidade no centro das listras, para evitar a iluminação desigual resultante do objetivo utilizado para adquirir essas imagens (vinheta, Figura 6E).
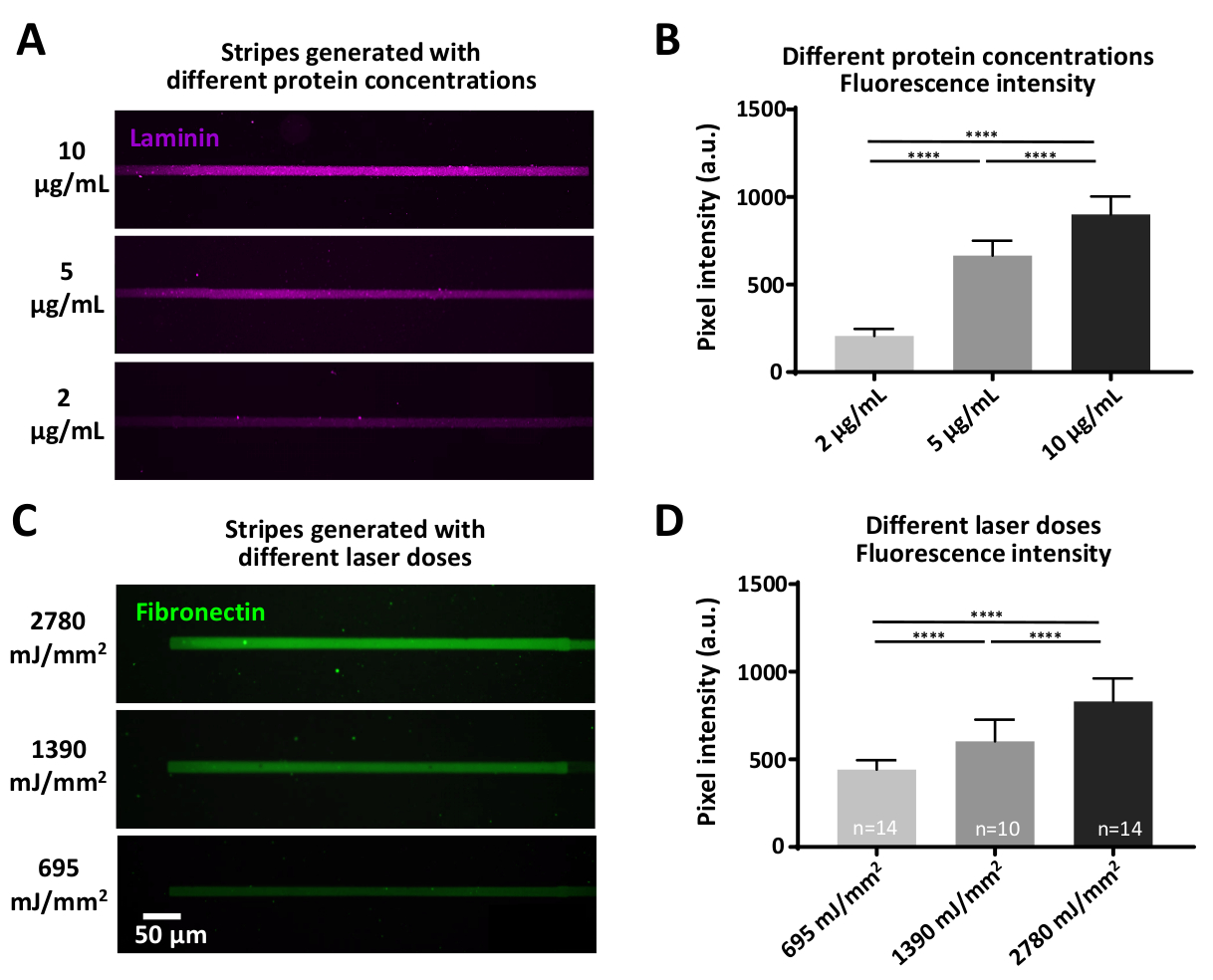
Alguns experimentos podem envolver questões que exijam concentrações de proteínas definidas, que podem ser alcançadas de duas maneiras: 1) variando a concentração proteica (Figura 7a, B). A incubação com diferentes concentrações de laminina, resulta em intensidades de fluorescência significativamente diferentes, aumentando com maiores concentrações proteicas (Figura 7B). 2) a dose do laser que é usada para Cleave a película antiadesiva (PEG) pode ser variada. Doses mais elevadas de laser removerão o filme antifouling em maior medida, gerando locais mais vinculativos para as proteínas de interesse (Figura 7C, D) resultando em intensidades de fluorescência significativamente diferentes, aumentando com maior doses (Figura 7D).
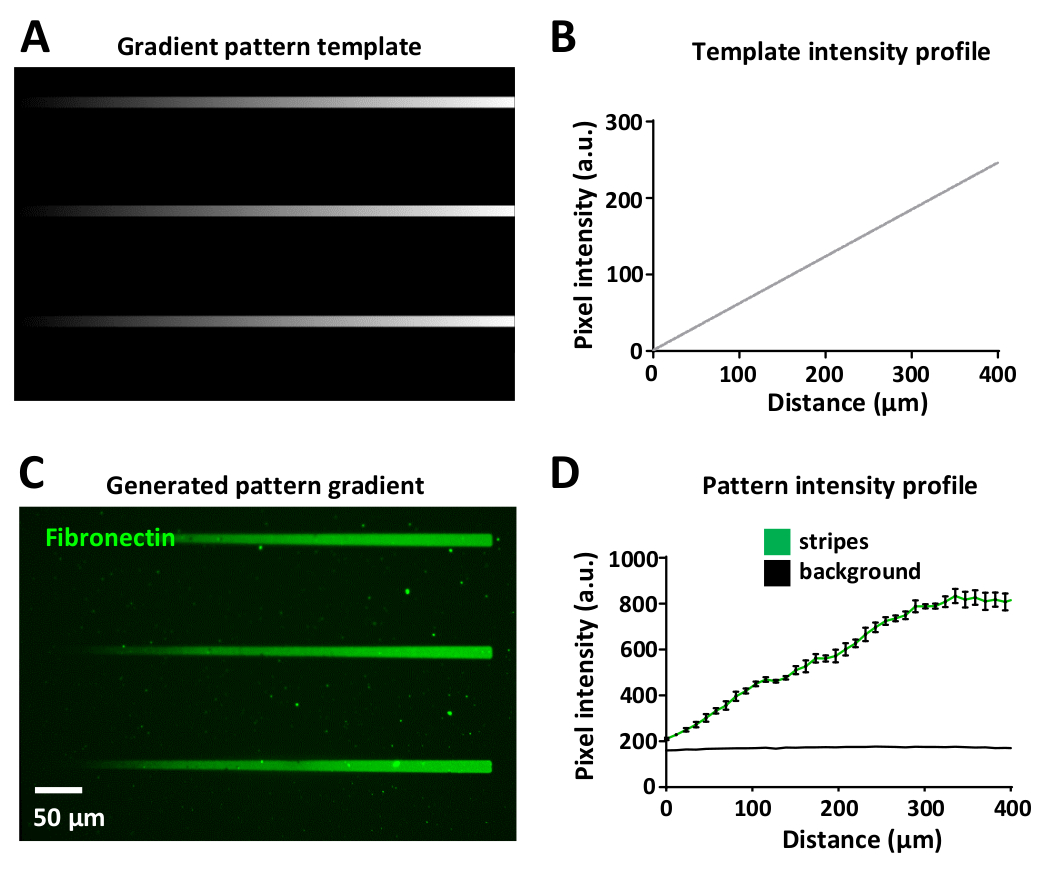
Variando a dose de laser permite a geração de gradientes de proteínas dentro do mesmo padrão. Isso é exibido na Figura 8a, onde um modelo de gradiente foi projetado usando diferentes níveis de escala de cinzentos, de preto (sem potência do laser) para branco (potência máxima do laser).
A intensidade do laser é proporcional ao nível de escala de cinzentos do modelo (variando de 0 a 255 em uma imagem de 8 bits), gerando gradientes de iluminação UV. A medida do perfil de intensidade de fluorescência ao longo da listra de gradiente é linear no modelo de padrão (Figura 8B) e no padrão de gradiente gerado (Figura 8C, D). Isso é reprodutível entre todas as listras de gradiente dentro do mesmo padrão de modelo e gradiente (Figura 8B, D). A geração de tais gradientes é extremamente útil e ajuda a imitar ambientes in vivo onde as células frequentemente respondem a gradientes de proteínas bioativas34,35,36,37.
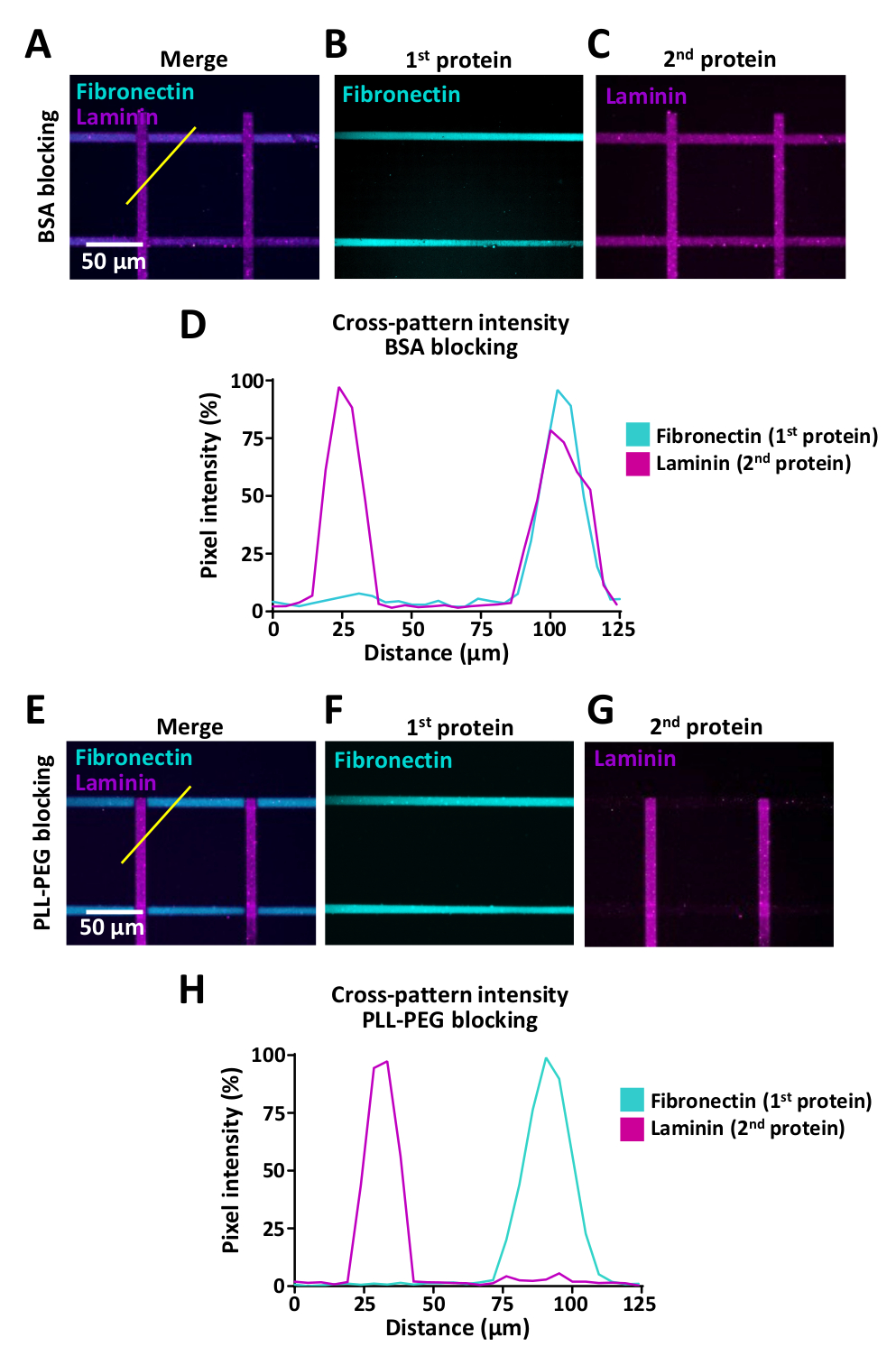
As células sentem a mudança de ambientes extracelulares, mas os ensaios que permitem o estudo do comportamento celular quando as células encontram tais alterações são limitadas. LIMAP pode ser usado para micro-padrão com múltiplas proteínas no mesmo micro-bem. Exemplos são mostrados na Figura 9 onde os padrões cruzados foram gerados com listras de fibronectina (horizontal) e laminina (vertical). Ao criar padrões com múltiplas proteínas, é crucial usar um passo de bloqueio entre a primeira e segunda incubação de proteínas, para evitar a ligação cruzada de proteínas (ver passo 7). A eficiência de bloqueio pode variar dependendo das características bioquímicas das proteínas que são usadas para o revestimento e aconselhamos testar vários buffers de bloqueio, incluindo PLL-PEG (0,1 mg/mL) e BSA (1%). Para avaliar este efeito de ligação cruzada, realizamos medições de intensidade de fluorescência utilizando ImageJ (Figura 9) e mostramos que a ligação cruzada pode ser reduzida dramaticamente, utilizando tampão PLL-PEG (0,1 mg/ml) para fibronectina e laminina padrões cruzados (Figura 9D, H).
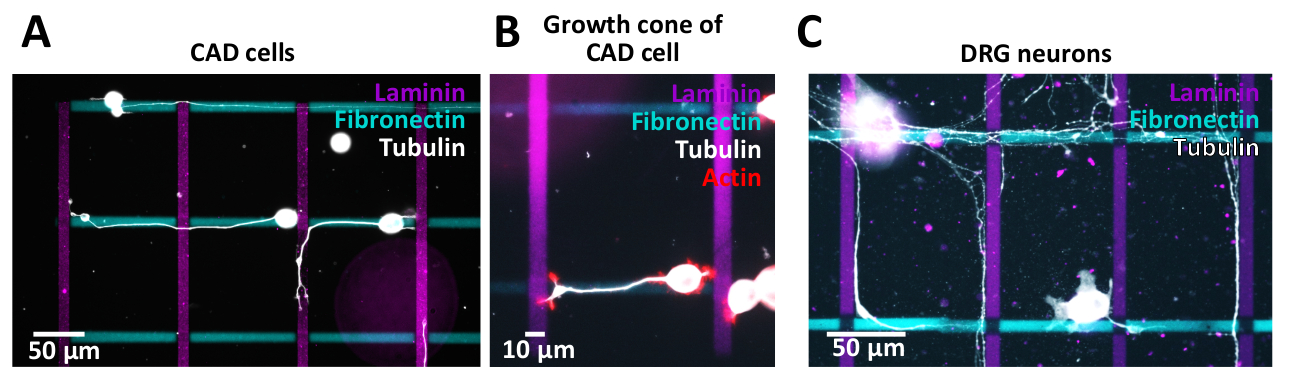
Os padrões cruzados gerados foram utilizados para ensaios celulares com células CAD (Figura 10A, B) ou neurônios do gânglio da raiz dorsal do rato (DRG) (Figura 10C). Seus neuritos (CAD) e axônios (DRG) crescem ao longo de diferentes linhas. Células CAD são usadas como um modelo neuronal, uma vez que mostram um perfil de expressão de integrina semelhante em comparação com os neurônios primários e eles ainda exibem cones de crescimento ricos em actina após 48 h na cultura (Figura 10B), tornando-os adequados para pathfinding Estudos.
A fim investigar os efeitos citotóxica possíveis dos micro-testes padrões gerados para neurônios preliminares, os neurônios de DRG foram isolados e cultivados em micro-testes padrões que seguem um protocolo previamente publicado38. Os resultados demonstram que os neurônios primários toleram o ambiente de micropadrões (Figura 10C). Nós estamos estudando atualmente como uma variedade de proteínas do ECM influenciam o pathfinding axonal (neurite). A comprovação preliminar de conceitos encontrados em células CAD será ainda mais investigada usando neurônios DRG. A fim validar a qualidade de micro-testes padrões gerados, é desejável aos testes padrões da imagem pela microscopia de fluorescência para assegurar-se de que as bordas do teste padrão estejam bem definidas antes de prosseguir ao chapeamento de pilha. Durante o processo de imagem, é importante garantir o ajuste óptico entre o microscópio e a câmera para evitar o efeito de escurecimento periférico (vinheta) que afeta a análise posterior e a interpretação dos dados. Além disso, adquira uma imagem de uma região sem padrão usando os mesmos tempos de exposição que serão usados para fazer a imagem dos padrões e subtrair essa imagem da imagem padrão.
Em síntese, para a geração de micropadrões de boa qualidade, é aconselhável avaliar a concentração proteica (Figura 7A, B), doses de laser (figura7C, D), níveis de fundo proteico (Figura 6e, F, H) e um etapa de bloqueio eficiente (Figura 9) ao usar múltiplas proteínas. De forma conclusiva, a qualidade dos micropadrões gerados com o LIMAP é essencial para obter dados confiáveis e reprodutíveis de ensaios celulares.

Figura 1: esquema de técnicas de micro padronização: impressão de microcontato e padronização assistida por laser. (A) a impressão de microcontato usa um mestre litografado com microcaracterísticas definidas para gerar um carimbo PDMS que é incubado com a proteína de interesse. Esta proteína é então transferida (carimbada) para uma superfície de vidro, gerando micropadrões proteicos. (B) técnicas de padronização assistida por laser incluem fotopadronização e padronização direta do laser. (C) a maioria das abordagens de fotopadronização usam uma fonte de luz UV e uma máscara fotográfica (seja em contato com a superfície do substrato ou no plano focal do objetivo) com geometrias desejadas, a fim de clicar a superfície ANTIFOULING Peg em posições específicas, Criando um padrão definido. Uma etapa subseqüente da incubação da proteína conduz à adsorção da proteína somente às regiões laser-cleaved. (D) o limap é uma técnica de fotopadronização que não requer uma Fotomáscara em contacto com o substrato (ou seja, uma abordagem sem contacto com o maskless). Limap usa um Photo-Initiator, que é ativado por baixas doses de um laser, clivagem regiões expostas à luz de Peg. Isto cria locais do acessório para a adsorção seqüencial da proteína. (E) o laser direto que padronização usa a luz de alta energia para gravar diretamente a película do Peg, permitindo a ligação da proteína naquelas regiões gravadas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

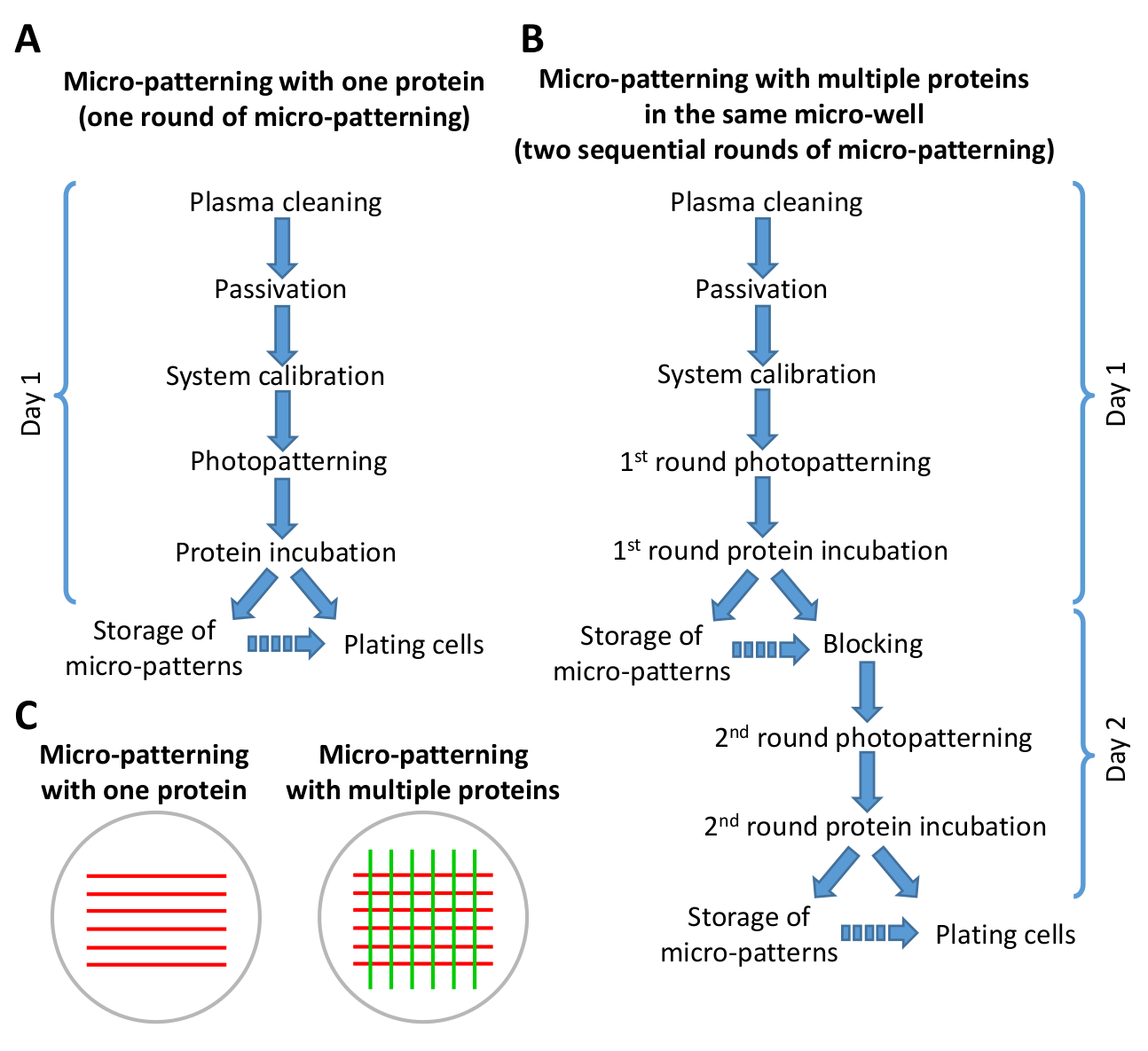
Figura 2: esquema mostrando um resumo das etapas no protocolo de micropadronização. (A) micro-padronização com uma proteína envolve apenas uma rodada de micropadronização (fotopadronização e incubação de proteínas) e pode ser realizada em menos de 8 h. (B) micro-padronização com múltiplas proteínas requer duas rodadas sequenciais de micro-padronização e pode ser concluída em 1-2 dias, dependendo do número de micro-padrões que estão sendo preparados. É possível percorrer a versão B do protocolo em 1 dia de trabalho. As setas contínuas indicam o fluxo direto das etapas no protocolo. As setas descontínuas indicam que há um intervalo de tempo significativo entre uma etapa e o outro (veja etapa 6,6 e 9,3). (C) visão esquemática dos padrões de exemplo obtidos após uma rodada de micropadronização (listras vermelhas) ou duas rodadas sequenciais de micro padronização (listras vermelhas e verdes). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: a geração do modelo de padrão é versátil com o LIMAP. (A, B) Exemplos de modelos de padrões projetados com ImageJ (um crossbows, letras B). As formas desenhadas no branco foram projetadas na potência máxima do laser e as formas desenhadas no preto não foram projetadas. (C, D) Micro-padrões obtidos com LIMAP a partir de modelos após incubação com 10 μg/mL de fibrinogênio (verde). (C) os crossbows são 50 μm de largura e 50 μm de altura espaçadas por 75 μm horizontalmente e 50 μm verticalmente. (D) as letras são 80 μm de largura e 85 μm de altura. As barras de escala em C e D representam 50 μm. por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: materiais essenciais para o protocolo LIMAP. (A) stencils utilizados neste protocolo são de 20 mm de diâmetro, finas peças de forma circular pdms (250 μm de espessura) contendo 4 micropoços (4 mm de diâmetro cada). Os volumes utilizados nos micropoços variam de 5 a 20 μL, reduzindo consideravelmente a quantidade de reagentes e proteínas necessárias para cada experimento. (B) prato inferior de vidro de 6 poços onde os stencils foram colocados já em cada poço. Os micro-poços contêm 20 μL de PBS para torná-los visíveis. (C) prato de calibração em que o poço de vidro interior foi marcado com um marcador verde, que será usado para calibrar o foco do laser. (D) vista esquemática do canto superior esquerdo bem do prato inferior de vidro de 6 poços em B (esboçado com círculo vermelho Tracejado). O poço de fundo de vidro interior é representado em branco e o estêncil é mostrado em cinza. O estêncil contem 4 micropoços (numerados 1-4), para o teste de 4 circunstâncias experimentais diferentes (por exemplo, concentrações diferentes da proteína, geometrias do teste padrão, combinações de proteínas, etc.). O asterisco representa o micropoço que contém o padrão de referência. (E) vista esquemática do micropoço onde um padrão de referência foi gerado na parte superior (seta com ponta de flecha preenchida). Este padrão de referência é necessário para obter o foco laser ideal para Padronização (consulte a etapa 4). A seta com ponta de flecha vazia indica a área central do micropoço, que será usada para a padronização subsequente após a calibração do sistema. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

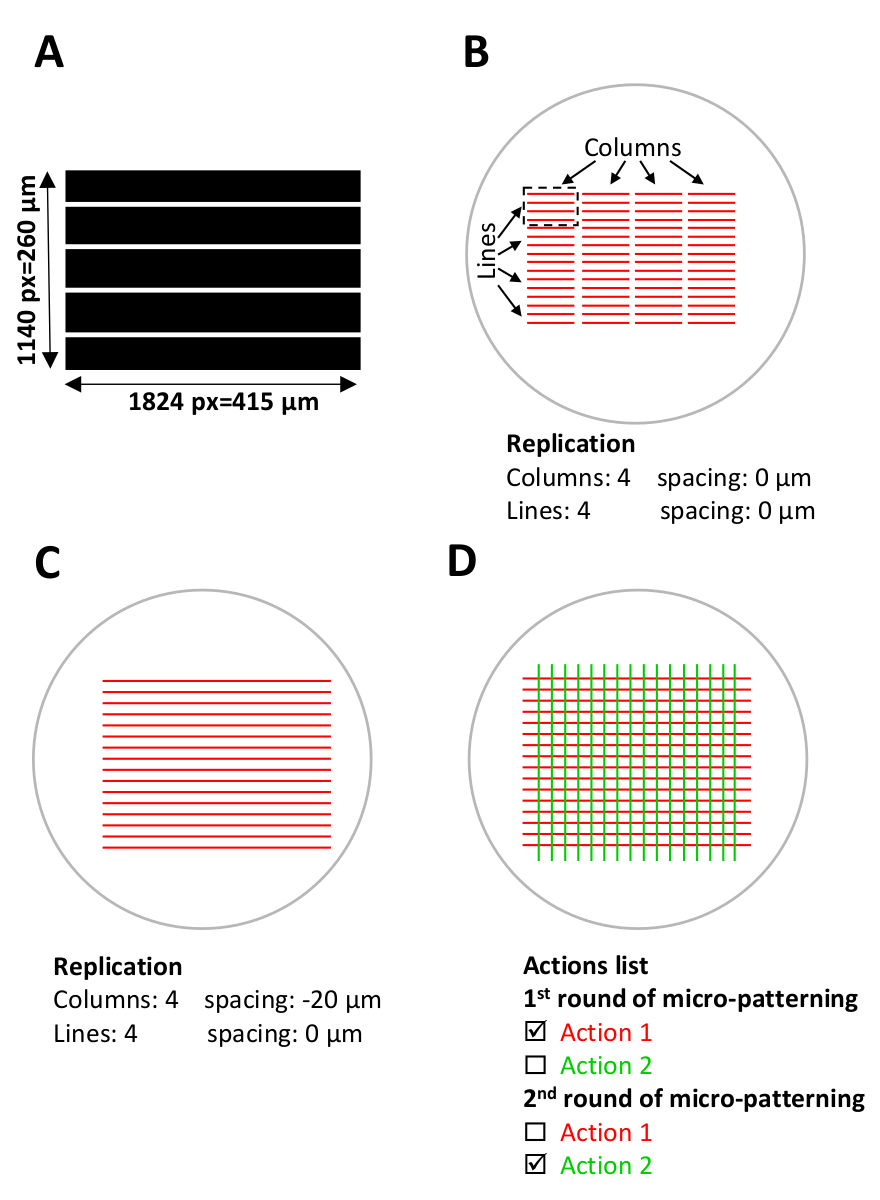
Figura 5: set-up de software para micropadronização. (A) modelo padrão com listras paralelas projetadas com ImageJ e salvas como um arquivo TIFF de 8 bits. (B-D) Visão esquemática de ROIs digitais (regiões de interesse) que se sobrepõem com os micropoços atuais onde os micropadrões serão gerados. (B) o modelo de padrão a ser usado (comprimento 1824 pixel = 415 μm, largura 1140 pixel = 260 μm) é selecionado em Leonardo e é projetada no ROI como uma unidade de projeto (listras vermelhas no retângulo tracejado preto), que cobrirá aproximadamente 0,1 mm2 do área de micropoços. A unidade de design é replicada em 4 colunas e 4 linhas no menu de replicação (configuração de modelo), criando um padrão em todo o micro-bem. Anote o espaço entre as colunas. (C) para listras contínuas padrão, o espaçamento entre as colunas deve ser ajustado. Nesse caso, para obter uma sobreposição entre as unidades de design, o espaçamento entre colunas é definido no menu de replicação como espaçamento negativo,-20 μm. (D) para padronizar múltiplas proteínas no mesmo micro-poço, um alinhamento preciso dos padrões é Necessário. Durante a etapa de set-up do software (etapa 5), carregue todos os moldes desejados do teste padrão simultaneamente. Na lista de ações , selecione apenas as ações específicas a serem padronizadas durante cada rodada de padronização e desmarque o restante das ações (etapa 5,12, 5,13 e 8,2). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: análise da variabilidade do padrão usando LIMAP. (A) modelo padrão projetado com ImageJ usado para micro-padrão de quatro listras de largura variável (20, 10, 5, 2 μm, de cima para baixo). (B) micropadrão obtido após incubação com 10 μg/ml de fibrinogênio com rótulo fluorescentamente (verde). (C) medições de intensidade ao longo de uma linha vertical cruzando as listras do micropadrão. D) perfil de intensidade de fluorescência vertical obtido a partir da medição em (C). NOTE que em larguras maiores (20 μm) há uma variação no perfil vertical causada pelo acúmulo de proteínas nas bordas da listra, resultando em dois picos de intensidade de fluorescência distintos (efeito de aresta). Este efeito só é observado em larguras de listra ≥ 20 μm. (e) medições de intensidade ao longo das linhas horizontais representadas (fluorescência e fundo). F) perfis de intensidade de fluorescência horizontais obtidos a partir de medições em (E). (G) gráfico mostrando a intensidade média para cada largura da faixa, medida a partir de quatro unidades de projeto replicadas individuais (variação entre padrões). Anote a adsorção reduzida da proteína aos testes padrões da largura da listra de 2 μm. (H) a variação dentro das listras padronizadas (coeficiente de variação) foi baixa para todas as larguras da listra, variando de 3 a 10%. Os dados em G e H mostraram-se como média ± DP. a análise estatística em G e H foi realizada utilizando-se ANOVA One-Way (Kruskal-Wallis) teste não paramétrico com comparações múltiplas. Valor de P é < 0,001 para * * significância. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: o efeito das variações no poder do laser e na concentração da proteína para a eficiência da adsorção da proteína. (A) asuperfície do PLL-Peg era laser-cleaved com uma dose constante do laser (1390 MJ/mm2) e incubadas com as concentrações indicadas do laminina fluorescently-etiquetado (magenta). (B) quantificação da intensidade de fluorescência das listras de laminina em (a). (C) as diferentes doses de laser indicadas foram aplicadas seguidas de incubação com a mesma concentração (10 μg/ml) de fibronectina fluorescentamente rotulada (verde). (D) quantificação da intensidade de fluorescência das listras de fibronectina em (C) mostrando que doses mais elevadas de laser se correlacionam com níveis mais elevados de proteínas adsorvedas. Todas as medições são de fundo subtraída. Os números de amostra são indicados na parte inferior das colunas; os dados são mostrados como média ± SEM. a análise estatística foi realizada por meio do teste não paramétrico de Mann-Whitney com cálculo bicaudal. Valor de P é < 0,0001 para * * * * significância. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 8: geração de um gradiente de concentração proteica dentro de um micropadrão. (A) modelo de padrão de gradiente em escala de cinzentos. (B) perfil de intensidade de fluorescência medido a partir de (a). (C) padrão obtido com limap do modelo de padrão em (a) após incubação com 10 μg/ml de fibronectina com rótulo fluorescentamente (verde). (D) perfil de intensidade de fluorescência de n = 3 listras e fundo representado como média ± MEV, mostrando o aumento linear na intensidade do gradiente proteico. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 9: o efeito de ligação cruzada ao padronar várias proteínas sequencialmente. (A-C, E-G) Padrões cruzados com listras de 10 μm de fibronectina com rótulo fluorescentamente (ciano, horizontal) e laminina com rótulo fluorescentamente (magenta, vertical). (A-C) Amostras tratadas com BSA bloqueando buffer. (E-F) Amostras tratadas com PLL-PEG para bloquear locais de ligação inespecíficos (passo 7). (A, E) Canais de fluorescência mesclados mostrando tanto fibronectina e laminina. (B, F) Imagem mostrando apenas fibronectina. (C, G) Imagem que mostra o laminina somente. Para C, anote a presença de laminina igualmente em listras positivas horizontais do fibronectina que é devido ao bloqueio ineficaz de locais de ligação desocupados com BSA. Para G, anote que obstruir com PLL-PEG impede eficientemente a ligação do laminina às listras do fibronectina. (D, H) Perfis de intensidade de fluorescência obtidos a partir de medições indicadas (linha amarela diagonal) em A e e, respectivamente. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 10: padrões cruzados para investigar o pathfinding do neurite/AXON. (A-C) Padrões cruzados com listras de 10 μm de fibronectina com rótulo fluorescentamente (ciano, horizontal) e laminina com rótulo fluorescentamente (magenta, vertical). (A, B) Imagens fluorescentes de células CAD com neuritos crescendo ao longo dos micropadrões. Para visualizar os neuritos, as células foram cultivadas por 48 h, fixadas com PFA a 4% e coradas para tubulina (A) ou tubulina e actina (B). (C) neurônios do gânglio dorsal-raiz do rato (DRG) com axônios crescendo ao longo dos micropadrões. Para visualizar axônios, os neurônios DRG foram cultivados por 72 h, fixados com 4% de PFA e corados para tubulina. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 11: exemplos de resultados negativos comuns obtidos ao gerar micropadrões com LIMAP. (A-C) As listras padronizadas sub-optimal de 10 μg/mL fibrinogênio fluorescently-etiquetado (verde) obtidos circunstâncias diferentes. (A) o micro-bem seco durante ageração de padrões. Observe os altos níveis de fluorescência no fundo (seta) e a presença de cristais de PBS (asteriscos). (B) a costura entre as listras não foi ajustada corretamente durante a criação de software, resultando em listras descontínuas com lacunas (seta) entre as unidades de projeto (ver etapa 3.4.7). (C) o foco do laser era secundário-optimal que causa as listras difundas (setas) que não representam as larguras reais da listra do molde do teste padrão, que devem ser 20, 10, 5, 2 μm de de cima para baixo, como em (B). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Vantagens de micro padronização LIMAP (PRIMO) e comparação com a impressão de microcontato
Enquanto a impressão de microcontato é possivelmente a técnica de micro padronização mais comumente usada no campo biológico39, parece haver um número crescente de pesquisadores usando a tecnologia limap40,41,42 ,43,44. Aqui, apresentamos um protocolo utilizando o PRIMO, um sistema comercialmente disponível para o LIMAP. Abaixo nós discutimos brevemente as vantagens e as limitações potenciais da impressão do microcontact e do Photo-patterning de LIMAP.
A impressão de microcontato requer mestres litografados produzidos por spin-Coating uma máscara fotográfica (geralmente SU-8) em uma bolacha de vidro ou silício, que é então gravada a laser com as microcaracterísticas desejadas. Esses mestres são usados como modelos para criar um carimbo PDMS45. O carimbo é incubado com uma proteína escolhida que adsorba a ele, e é então transferido (carimbado) para o prato de cultura celular. O processo de adsorção da proteína para o carimbo PDMS é dependente da concentração proteica, tempo de tampão e incubação. Esses parâmetros precisam ser testados de antemão para obter os melhores resultados46.
Os mestres podem ser usados em um número substancial de experiências, durando por meses ou mesmo anos, se preservado corretamente. No entanto, um fator limitante desta tecnologia é a necessidade de re-projetar novos mestres litografados para cada modificação desejada. Mudanças nos designs experimentais podem resultar na produção demorada de novos mestres (até várias semanas), atrasando experimentos. Em comparação, o photopatterning de LIMAP não exige um mestre físico; Ele usa modelos de padrão gerados por software que podem ser usados para adaptar de forma flexível as geometrias desejadas de micropadrões para alterar as questões de pesquisa. O LIMAP também pode ser usado para gerar gradientes proteicos dentro do mesmo micropadrão (Figura 8), o que é mais difícil de obter de forma reprodutível usando a impressão de microcontato47.
Além disso, a resolução de micropadrão alcançada com o LIMAP, no nosso caso, é de 2 μm (Figura 6B).
Abordar essa resolução aumentou a variabilidade intra e interpadrão. A geração de padrões em torno ou acima de 10 μm de largura foi altamente reprodutível (Figura 6G, H). Pelo contrário, com a impressão de microcontato é difícil obter consistentemente resoluções abaixo de 10 μm e é comum encontrar artefactos ao carimbar pequenos recursos (dados não mostrados).
Nós mostramos que o LIMAP pode ser usado para micro-padrão de proteínas múltiplas (Figura 9) dentro do mesmo micro-bem, permitindo que outros níveis de complexidade sejam adicionados aos experimentos. Embora isso possa ser conseguido com a impressão de microcontato, alinhar diferentes proteínas com alto nível de precisão pode ser tecnicamente bastante exigente. Embora a padronização de múltiplas proteínas usando LIMAP pareça direta, é importante mencionar que a ligação cruzada de proteínas através de procedimentos de revestimento seqüencial pode ser reduzida através do bloqueio de reagentes, mas não totalmente eliminado (Figura 9).
Em relação ao custo de uma ou outra técnica, o LIMAP, conforme descrito aqui, requer a compra de equipamento de micropadronização (PRIMO) que pode ser instalado em microscópios de fluorescência diferentes e requer um estágio motorizado. Embora esse investimento seja inicialmente de custo intensivo, não há compras adicionais que não sejam itens consumíveis (estênceis, PEG e PLPP) a longo prazo associados ao LIMAP. Alternativamente, os stencils de PDMS podem igualmente ser produzidos no laboratório pelo próprio experimentador que segue os protocolos publicados18,32. Os maiores custos para a impressão de microcontato podem estar associados à produção de novos mestres, o que pode se tornar substancial se os experimentos exigirem novos padrões.
Uma desvantagem de LIMAP é a abordagem de taxa de transferência relativamente baixa desta técnica. A impressão de microcontato pode produzir um grande número de micropadrões de forma rápida e eficiente em uma etapa de estampagem simultânea, em comparação com a micropadronização sequencial de laser necessária com o LIMAP. Por exemplo, é possível produzir 6 coberturas de vidro estampadas em cerca de 2 h com impressão de microcontato usando carimbos PDMS (excluindo a preparação do carimbo); padronizar uma área semelhante (prato de 6 poços) com o LIMAP levaria cerca de 4 h, excluindo o procedimento de passivação superficial (considerando a configuração do modelo de padrão descrita na etapa 5,12 e veja a Figura 5B).
Outro fator limitante de taxa da tecnologia LIMAP é o tempo de iluminação longo necessário para padronar grandes áreas (30 s por unidade de design com um laser de 7,5 mW/mm2 ). Nesses casos, a impressão de microcontato pode ser uma opção preferida. Uma foto-iniciador recentemente disponível (gel de PLPP, tabela de materiais) deve consideravelmente reduzir o tempo tomado para o patterning, permitindo a geração de centenas de micro-testes padrões em áreas grandes (até 8 milímetros2) em apenas alguns minutos.
Outro fator importante a ser tido em conta quando a micropadronização de superfícies para cultura celular é a reprodutibilidade dos micropadrões entre diferentes repetições experimentais, em comparação com a variabilidade obtida com a impressão de microcontato. Por exemplo, os gráficos mostrados na Figura 7B, D são dados representativos de três repetições experimentais independentes com resultados muito semelhantes (dados não mostrados). Com base em nossa experiência e publicações anteriores, esse nível de reprodutibilidade é difícil de conseguir com a impressão de microcontato48,49,50,51,52.
Em contraste com outras técnicas Photo-patterning que exigem química dedicada para o engenheiro de materiais fotossensíveis ou o uso de foto-sensibilizadores, que geralmente não são muito biocompatíveis3, o componente fotossensível de limap (plpp ) é biocompatível e bem tolerado pelas células21; em nossas mãos, não experimentamos qualquer citotoxicidade em uma variedade de células, incluindo DAC, neurônios DRG (Figura 10), fibroblastos, células epiteliais e células de melanoma (dados não mostrados). Uma outra vantagem de limap usando o primo comparado a outras técnicas Photo-patterning é que nenhuma Fotomáscara é exigida. Similar à impressão do microcontact, os photomasks novos precisariam de ser projetados e gerados para cada teste padrão desejado.
Todas as limitações mencionadas acima para a impressão do microcontact, referem a aproximação manual da técnica. No entanto, é possível aumentar a taxa de transferência e a reprodutibilidade da impressão de microcontato usando um dispositivo automatizado com carga de carimbo e controle de pressão53.
Principais passos do protocolo e resolução de problemas para LIMAP usando PRIMO
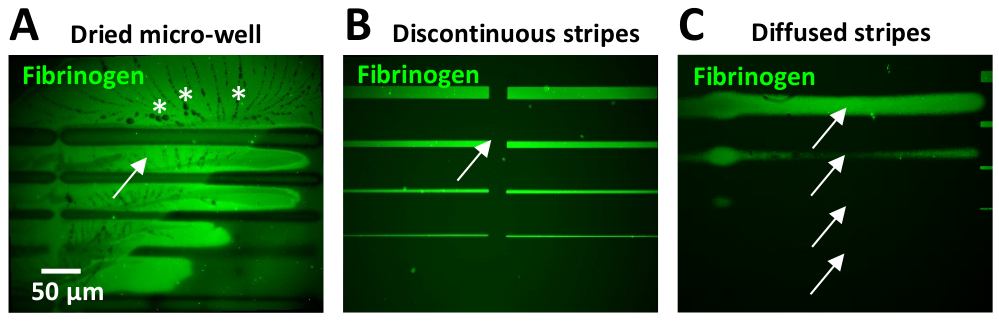
Um dos problemas mais comuns encontrados durante este protocolo é ter altos níveis de fluorescência de fundo dentro dos micropadrões. Isto pode ser devido à secagem de micropoços que muitas vezes ocorre devido ao seu pequeno volume. Quando isso ocorre, os cristais de PBS geralmente aparecem em torno dos padrões de ECM (Figura 11A).
As etapas de lavagem insuficientes ou ineficientes após a incubação de proteínas também podem resultar em altos níveis de fluorescência de fundo. Isso pode ser observado principalmente no uso de concentrações proteicas de 10 μg/mL (Figura 11B) ou superior. O excesso da proteína no fundo pode ser reduzido incluindo etapas de lavagem adicionais com PBS.
A presença de fundo protéico precisa ser mensurada e caracterizada em cada experimento, calculando a intensidade da fluorescência de fundo (Figura 6e) e subtraindo-a da intensidade dos micropadrões (Figura 6F-H e Figura 7B, D). O fundo elevado da proteína pode ter um impacto no acessório e no sprouting de pilhas do CAD, comprometendo a interpretação dos resultados.
Ter lacunas entre as unidades de design é um problema comum quando os usuários têm experiência limitada (Figura 11B), que ocorre como resultado de sobreposição insuficiente entre padrões. Dois parâmetros no software de Leonardo podem ser ajustados para superar isto: 1) um afastamento negativo entre colunas pode ser exigido, dependendo do projeto do teste padrão (etapa 5,7 e ver Figura 5B, C). Alternativamente, 2) Use a opção de gradiente no menu expert para costurar as colunas. Um teste rápido para determinar os parâmetros de espaçamento ideais pode ser realizado usando adesivo UV (tabela de materiais). Uma gota pequena deste adesivo é aplicada a uma corrediça de vidro, que seja coberta então com um COVERSLIP de vidro, fazendo um filme. O adesivo UV incorporado é fotomodelado com o molde do teste padrão do interesse usando uma baixa dose do laser (30 mJ/mm2). As regiões UV-expostas do adesivo incorporado serão curadas, tornando-se visíveis a microscopia do brilhante-campo. Os resultados do teste são visualizados para avaliar o espaçamento obtido dentro do padrão. Em nossos experimentos neuronais, uma lacuna entre as listras pode afetar negativamente o comportamento celular, produzindo variações na dinâmica de crescimento (velocidade reduzida ou abandono do trajeto).
Na última atualização do software Leonardo (no momento da publicação, Leonardo 4,11), é possível fazer upload de modelos de padrão maiores projetados anteriormente que cobrem uma área muito maior (até 8 mm2 usando o objetivo 20x) da superfície de micropoços comparado ao atual 0,1 milímetro2 por a unidade do projeto, eliminando a necessidade de costurar junto as unidades menores do projeto. Bordas indefinidas podem resultar da falta de ajuste do foco do laser durante a geração de padrões (Figura 11C). É conseqüentemente crítico calibrar o laser e executar etapas do teste padrão de referência (veja a etapa 4) antes do patterning. Listras mal definidas resultam em variações na largura da listra, dificultando a correlação entre a dinâmica de crescimento do AXON e a largura da listra. Os axônios também tendem a abandonar listras que têm bordas difuso. Além disso, a variabilidade nas bordas também pode ser encontrada ao imprimir listras de 10-20 μm de largura ou superior, resultando em um maior teor de proteínas nas bordas em comparação com as regiões centrais do padrão (Figura 6B, D). Este efeito de aresta é produzido por uma difusão não homogênea do Photo-Initiator durante o processo de fotopadronização. A reação da photoscission é dependente do oxigênio, que difunde mais nas bordas. Este efeito de aresta pode ser minimizado homogeneizar o Photo-Initiator com uma pipeta no micropoço durante o processo de fotopadronização. Além disso, um novo Photo-iniciador comercializado (PLPP gel), também pode reduzir o efeito de borda (equipe de suporte do sistema PRIMO, comunicação pessoal).
A microimpressão de mais de uma proteína pode resultar em ligação cruzada (Figura 9A-D). Isto pode ser minimizado aumentando a eficiência de bloqueio que é usada para ocupar locais de ligação inespecíficos entre as etapas de incubação para as duas proteínas diferentes. A ligação cruzada das proteínas pode perturbar a reprodutibilidade dos desfechos experimentais e pode levar à má interpretação dos dados, uma vez que é difícil determinar a contribuição de cada proteína para a dinâmica de crescimento do AXON e para outros comportamentos celulares.
Conclusão
Esperamos que o protocolo fornecido utilizando o LIMAP facilite a geração de micropadrões proteicos através do uso do sistema PRIMO. Enquanto nosso protocolo se concentra em como produzir de forma confiável micropadrões em superfícies de vidro 2D, outros mostraram que é possível usar o LIMAP para micropadronização de substratos macios54e superfícies Microestruturadas para culturas 3D42. Esses micropadrões podem ser uma ferramenta versátil para estudar as respostas celulares às mudanças em seu microambiente.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho é apoiado pelo BBSRC, EPSRC, MRC e Wellcome Trust. O laboratório C.B. é parte do centro de confiança Wellcome para a pesquisa Cell-Matrix, Universidade de Manchester, que é apoiado pelo financiamento do núcleo da confiança de Wellcome (concessão número 088785/Z/09/Z). Os autores desejam reconhecer o financiamento prestado pelo Conselho de pesquisa em biotecnologia e ciências biológicas (BBSRC) a K.J. (BB/M020630/1) e P.A. (BB/P000681/1) e pelo Conselho de pesquisa em engenharia e ciências físicas (EPSRC) e Medical Centro de investigação (MRC) para a formação de doutoramento em medicina regenerativa a AK (EP/L014904/1). Os autores agradecem Alvéole por sua correspondência e sua equipe de suporte pós-venda. Os autores agradecem a Peter March e Roger Meadows da BioImaging Facility, da Universidade de Manchester, por sua ajuda com a microscopia. Os microscópios de BioImaging Facility utilizados neste estudo foram adquiridos com subsídios da BBSRC, Wellcome Trust e do fundo estratégico da Universidade de Manchester.
Materiais
| Name | Company | Catalog Number | Comments |
| Alexa 488 protein labeling kit | Invitrogen | A10235 | Working concentration: N.A. |
| Alexa 647 protein labeling kit | Invitrogen | A20173 | Working concentration: N.A. |
| CAD cells | ECACC | 8100805 | Working concentration: N.A. |
| Conjugated fibrinogen-488 | Molecular Probes | F13191 | Working concentration: 10 μg/ml |
| DMEM culture medium | Gibco | 11320033 | Working concentration: N.A. |
| Epifluorescence Microscope** | Nikon | Eclipse Ti inverted | Working concentration: N.A. |
| Fibronectin | Sigma | F4759 | Working concentration: 10 μg/ml (after labelling with Alexa 488 protein labeling kit, see above) (diluted in PBS) |
| Fiji-Image J | www.imagej.nih.gov | Version 2.0.0-rc-54/1.51f | Working concentration: N.A. |
| Fluorescent highlighter | Stabilo | Stabilo Boss Original | Working concentration: N.A. |
| HEPES | Gibco | 15630080 | Working concentration: 1M |
| Inkscape software | Inkscape | Check last update | Working concentration: N.A. |
| Laminin-red fluorescent rhodamine | Cytoskeleton, Inc. | LMN01 | Working concentration: 10 μg/ml (diluted in PBS) |
| Leonardo software | Alvéole | version 4.11 | Working concentration: N.A. |
| L-Glutamine | Sigma | G7513 | Working concentration: 1% |
| Micro-manager software | Open imaging | Check last update | Working concentration: N.A. |
| Motorized x/y stage | PRIOR Scientific | Proscan II | Working concentration: N.A. |
| NIS Elements Software | Nikon | NIS Elements AR 4.60.00 64-bit (With Nikon jobs) | Working concentration: N.A. |
| PBS (without Ca2+, Mg2+) | Sigma | D8537 | Working concentration: 1X |
| PDMS Stencils | Alvéole | visit www.alveolelab.com | Working concentration: N.A. |
| PEG-SVA | Laysan bio, Inc. | MPEG-SVA-5000-1g | Working concentration: 50 mg/ml |
| Phalloidin 405 | Abcam | ab176752 | Working concentration: 1:1000 |
| Photo-initiator (PLPP) | Alvéole | Classic PLPP | Working concentration: 14.5 mg/ml |
| Photo-initiator (PLPP gel) | Alvéole | PLPP gel | Working concentration: 4.76% diluted in ethanol |
| Plasma cleaner | Harrick Plasma | PDC-32G (115V) | PDC-32G-2 (230V) | Working concentration: N.A. |
| PLL-PEG | SuSoS (also distributed by Alvéole) | www.alveolelab.com | Working concentration: 0.1 mg/ml (diluted in PBS) |
| Poly-L-Lysine | Sigma | P4707 | Working concentration: 0.01% |
| Primo equipment | Alvéole | www.alveolelab.com | Working concentration: N.A. |
| Pen/Strep | Thermo Fisher | 15140122 | Working concentration: 1% |
| Tubulin anti-alpha antibody | Abcam | DM1A | Working concentration: 1:1000 CAD cells |
| Tubulin anti-beta 3 antibody | Sigma | T8660 | Working concentration: 1:500 DRG neurons |
| UV adhesive | Norland Products | NOA81 | Working concentration: N.A. |
| 1 well glass bottom dish | Cellvis | D35-20-1.5-N | Working concentration: N.A. |
| 6 well glass bottom dish | Cellvis | P06-20-1.5-N | Working concentration: N.A. |
| 20x objective** | Nikon | no phase ring (check updated catalogue) | Working concentration: N.A. **Epifluorescence microscope: images were acquired and patterns were generated on an Eclipse Ti inverted microscope (Nikon), coupled to PRIMO micro-patterning equipment (Alvéole), using a 20x objective (0.75 S Plan Fluor (nophasering, Nikon). Nikon specific filter sets for GFP, mCherry and Cy5 were used and fluorescent light source was LED (Lumencor) although other fluorescence sources and filter sets can be used. The microscope has an automated x/y stage (PRIOR Scientific) for the printing of multi-field patterning and Nikon Perfect Focus to prevent focus drift. The images were collected using a Retiga R6 (Q-Imaging) camera. |
Referências
- Alamdari, O. G., Seyedjafari, E., Soleimani, M., Ghaemi, N. Micropatterning of ECM Proteins on Glass Substrates to Regulate Cell Attachment and Proliferation. Avicenna Journal of Medical Biotechnology. 5 (4), 234-240 (2013).
- Sunami, H., Yokota, I., Igarashi, Y. Influence of the pattern size of micropatterned scaffolds on cell morphology, proliferation, migration and F-actin expression. Biomaterials Science. 2 (3), 399-409 (2014).
- Thery, M. Micropatterning as a tool to decipher cell morphogenesis and functions. Journal of Cell Science. 123 (Pt 24), 4201-4213 (2010).
- Marino, A., et al. Two-photon polymerization of sub-micrometric patterned surfaces: investigation of cell-substrate interactions and improved differentiation of neuron-like cells. ACS Applied Materials & Interfaces. 5 (24), 13012-13021 (2013).
- Joo, S., et al. Effects of ECM protein micropatterns on the migration and differentiation of adult neural stem cells. Scientific Reports. 5, (2015).
- Morgani, S. M., Metzger, J. J., Nichols, J., Siggia, E. D., Hadjantonakis, A. K. Micropattern differentiation of mouse pluripotent stem cells recapitulates embryo regionalized cell fate patterning. Elife. 7, (2018).
- Javaherian, S., O'Donnell, K. A., McGuigan, A. P. A Fast and Accessible Methodology for Micro-Patterning Cells on Standard Culture Substrates Using Parafilm (TM) Inserts. Plos One. 6 (6), (2011).
- Smirnov, M. S., Cabral, K. A., Geller, H. M., Urbach, J. S. The effects of confinement on neuronal growth cone morphology and velocity. Biomaterials. 35 (25), 6750-6757 (2014).
- Albert, P. J., Schwarz, U. S. Dynamics of Cell Ensembles on Adhesive Micropatterns: Bridging the Gap between Single Cell Spreading and Collective Cell Migration. PLOS Computational Biology. 12 (4), (2016).
- Evans, A. R., et al. Laminin and fibronectin modulate inner ear spiral ganglion neurite outgrowth in an in vitro alternate choice assay. Developmental Neurobiology. 67 (13), 1721-1730 (2007).
- Nichol, R. H., Hagen, K. M., Lumbard, D. C., Dent, E. W., Gomez, T. M. Guidance of Axons by Local Coupling of Retrograde Flow to Point Contact Adhesions. Journal of Neuroscience. 36 (7), 2267-2282 (2016).
- Burdick, J. A., Khademhosseini, A., Langer, R. Fabrication of gradient hydrogels using a microfluidics/photopolymerization process. Langmuir. 20 (13), 5153-5156 (2004).
- Schwartz, P. V. Molecular transport from an atomic force microscope tip: A comparative study of dip-pen nanolithography. Langmuir. 18 (10), 4041-4046 (2002).
- Barbulovic-Nad, I., et al. Bio-microarray fabrication techniques--a review. Critical Reviews in Biotechnology. 26 (4), 237-259 (2006).
- Shafagh, R. Z., Vastesson, A., Guo, W. J., van der Wijngaart, W., Haraldsson, T. E-Beam Nanostructuring and Direct Click Biofunctionalization of Thiol-Ene Resist. Acs Nano. 12 (10), 9940-9946 (2018).
- Kobayashi, J., Yamato, M., Itoga, K., Kikuchi, A., Okano, T. Preparation of microfluidic devices using micropatterning of a photosensitive material by a maskless, liquid-crystal-display projection method. Advanced Materials. 16 (22), (2004).
- Bernard, A., et al. Printing patterns of proteins. Langmuir. 14 (9), 2225-2229 (1998).
- Ruiz, S. A., Chen, C. S. Microcontact printing: A tool to pattern. Soft Matter. 3 (2), 168-177 (2007).
- Qin, D., Xia, Y., Whitesides, G. M. Soft lithography for micro- and nanoscale patterning. Nature Protocols. 5 (3), 491-502 (2010).
- Fink, J., et al. Comparative study and improvement of current cell micro-patterning techniques. Lab Chip. 7 (6), 672-680 (2007).
- Strale, P. O., et al. Multiprotein Printing by Light-Induced Molecular Adsorption. Advanced Materials. 28 (10), 2024-2029 (2016).
- Belisle, J. M., Correia, J. P., Wiseman, P. W., Kennedy, T. E., Costantino, S. Patterning protein concentration using laser-assisted adsorption by photobleaching, LAPAP. Lab Chip. 8 (12), 2164-2167 (2008).
- Belisle, J. M., Kunik, D., Costantino, S. Rapid multicomponent optical protein patterning. Lab Chip. 9 (24), 3580-3585 (2009).
- Heinz, W. F., Hoh, M., Hoh, J. H. Laser inactivation protein patterning of cell culture microenvironments. Lab Chip. 11 (19), 3336-3346 (2011).
- Azioune, A., Carpi, N., Tseng, Q., Thery, M., Piel, M. Protein Micropatterns: A Direct Printing Protocol Using Deep UVs. Microtubules: In Vivo. 97, 133-146 (2010).
- Vignaud, T., Ennomani, H., Thery, M. Polyacrylamide hydrogel micropatterning. Methods in Cell Biology. 120, 93-116 (2014).
- Waldbaur, A., Waterkotte, B., Schmitz, K., Rapp, B. E. Maskless projection lithography for the fast and flexible generation of grayscale protein patterns. Small. 8 (10), 1570-1578 (2012).
- Kang, J., Choi, J. C., Kim, M., Jung, H. R., Doh, J. Photopatterning with a printed transparency mask and a protein-friendly photoresist. Methods in Cell Biology. 119, 55-72 (2014).
- Falconnet, D., Csucs, G., Grandin, H. M., Textor, M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials. 27 (16), 3044-3063 (2006).
- Morlat, S., Gardette, J. L. Phototransformation of water-soluble polymers. Part II: photooxidation of poly(ethylene oxide) in aqueous solution. Polymer. 44 (26), 7891-7897 (2003).
- Qi, Y., Wang, J. K., McMillian, M., Chikaraishi, D. M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. Journal of Neuroscience. 17 (4), 1217-1225 (1997).
- Shrirao, A. B., et al. A Versatile Method of Patterning Proteins and Cells. Journal of Visualized Experiments. (120), (2017).
- Pankov, R., Momchilova, A. Fluorescent labeling techniques for investigation of fibronectin fibrillogenesis (labeling fibronectin fibrillogenesis). Methods in Molecular Biology. 522, 261-274 (2009).
- Dertinger, S. K., Jiang, X., Li, Z., Murthy, V. N., Whitesides, G. M. Gradients of substrate-bound laminin orient axonal specification of neurons. Proceedings of the National Academy of Sciences of the United States of America. 99 (20), 12542-12547 (2002).
- Chelli, B., et al. Neural cell alignment by patterning gradients of the extracellular matrix protein laminin. Interface Focus. 4 (1), (2014).
- Tang, Y., Qiu, Q. F., Zhang, F. L., Xie, M., Huang, W. H. Quantifying orientational regeneration of injured neurons by natural product concentration gradients in a 3D microfluidic device. Lab Chip. 18 (6), 971-978 (2018).
- Srinivasan, P., Zervantonakis, I. K., Kothapalli, C. R. Synergistic effects of 3D ECM and chemogradients on neurite outgrowth and guidance: a simple modeling and microfluidic framework. PLoS One. 9 (6), (2014).
- de Luca, A. C., Faroni, A., Reid, A. J. Dorsal root ganglia neurons and differentiated adipose-derived stem cells: an in vitro co-culture model to study peripheral nerve regeneration. Journal of Visualized Experiments. (96), (2015).
- Khadpekar, A. J., Khan, M., Sose, A., Majumder, A. Low Cost and Lithography-free Stamp fabrication for Microcontact Printing. Scientific Reports. 9 (1), (2019).
- Delepine, C., et al. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Human Molecular Genetics. 25 (1), 146-157 (2016).
- Decock, J., Schlenk, M., Salmon, J. B. In situ photo-patterning of pressure-resistant hydrogel membranes with controlled permeabilities in PEGDA microfluidic channels. Lab Chip. 18 (7), 1075-1083 (2018).
- Stoecklin, C., et al. A New Approach to Design Artificial 3D Microniches with Combined Chemical, Topographical, and Rheological Cues. Advanced Biosystems. 2 (7), (2018).
- Toraille, L., et al. Optical Magnetometry of Single Biocompatible Micromagnets for Quantitative Magnetogenetic and Magnetomechanical Assays. Nano Letters. , (2018).
- Theodoly, O., et al. Live nanoscopic to mesoscopic topography reconstruction with an optical microscope for chemical and biological samples. PLoS One. 13 (12), (2018).
- Ermis, M., Antmen, E., Hasirci, V. Micro and Nanofabrication methods to control cell-substrate interactions and cell behavior: A review from the tissue engineering perspective. Bioactive Materials. 3 (3), 355-369 (2018).
- von Philipsborn, A. C., et al. Microcontact printing of axon guidance molecules for generation of graded patterns. Nature Protocols. 1 (3), 1322-1328 (2006).
- Ricoult, S. G., Kennedy, T. E., Juncker, D. Substrate-bound protein gradients to study haptotaxis. Frontiers in Bioengineering and Biotechnology. 3, (2015).
- Bietsch, A., Michel, B. Conformal contact and pattern stability of stamps used for soft lithography. Journal of Applied Physics. 88 (7), 4310-4318 (2000).
- Hui, C. Y., Jagota, A., Lin, Y. Y., Kramer, E. J. Constraints on microcontact printing imposed by stamp deformation. Langmuir. 18 (4), 1394-1407 (2002).
- Sharp, K. G., Blackman, G. S., Glassmaker, N. J., Jagota, A., Hui, C. Y. Effect of stamp deformation on the quality of microcontact printing: theory and experiment. Langmuir. 20 (15), 6430-6438 (2004).
- Delamarche, E., Schmid, H., Michel, B., Biebuyck, H. Stability of molded polydimethylsiloxane microstructures. Advanced Materials. 9 (9), 741-746 (1997).
- Perl, A., Reinhoudt, D. N., Huskens, J. Microcontact Printing: Limitations and Achievements. Advanced Materials. 21 (22), 2257-2268 (2009).
- Chakra, E. B., Hannes, B., Dilosquer, G., Mansfield, C. D., Cabrera, M. A new instrument for automated microcontact printing with stamp load adjustment. Review of Scientific Instruments. 79 (6), (2008).
- Pasturel, A., Strale, P., Studer, V. Tailoring 3D cell culture templates with common hydrogels. bioRxiv. , (2019).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados