
Method Article
Выделение и идентификация мезенхимальных стволовых клеток костного мозга свиньи и их производных внеклеточных везикул
В этой статье
Резюме
В данной статье разработан метод выделения и идентификации мезенхимальных стволовых клеток костного мозга свиней (pBM-MSCs) и полученных из них внеклеточных везикул (EVs), обеспечивающий методологическую основу для доклинической оценки эффективности трансплантации BM-MSCs и их производных EV.
Аннотация
С развитием терапии стволовыми клетками в трансляционных исследованиях и регенеративной медицине мезенхимальные стволовые клетки костного мозга (БМ-МСК), как разновидность плюрипотентных стволовых клеток, пользуются популярностью благодаря их мгновенной доступности и доказанной безопасности. Сообщалось, что трансплантация БМ-МСК имеет большое значение для восстановления поврежденных тканей при различных заболеваниях, что может быть связано с модуляцией иммунных и воспалительных реакций через паракринные механизмы. Внеклеточные везикулы (ВВ), имеющие двухслойную структуру липидной мембраны, считаются основными медиаторами паракринных эффектов стволовых клеток. Признанные за их решающую роль в клеточной коммуникации и эпигенетической регуляции, ВВ уже были применены in vivo для иммунотерапии. Однако, как и в случае с материнскими клетками, большинство исследований эффективности трансплантации ВВ все еще остаются на уровне мелких животных, чего недостаточно для предоставления существенных доказательств для клинической трансляции. В данной работе мы используем центрифугирование с градиентом плотности для выделения клеток костного мозга (БМК) из костного мозга свиньи на начальном этапе, а затем получения БМ-МСК свиней (pBM-МСК) с помощью клеточной культуры, идентифицированной по результатам наблюдения под микроскопом, индуцированного анализа дифференцировки и проточной цитометрии. Кроме того, мы изолируем EV, полученные из pBM-MSCs, в надосадочной жидкости клеток с помощью ультрацентрифугирования, что успешно доказано методами просвечивающей электронной микроскопии (TEM), анализа отслеживания наночастиц (NTA) и вестерн-блоттинга. В целом, pBM-MSCs и их производные EV могут быть выделены и эффективно идентифицированы с помощью следующих протоколов, которые могут быть широко использованы в доклинических исследованиях эффективности трансплантации BM-MSC и их производных EV.
Введение
За последние 10 лет терапия стволовыми клетками обещала большую пользу пациентам, страдающим от различных заболеваний и травм, таких как травмы, респираторные и сердечно-сосудистые заболевания. По мере прогресса в этой области, мезенхимальные стволовые клетки костного мозга (БМ-МСК) постепенно завоевывают популярность у людей за их доступность и небольшое количество этических споров1, которые считаются золотым стандартом для клинических исследований, несмотря на другие типы клеток2. Методы лечения, основанные на МСК-БМ, также привлекательны для все большего числа исследователей благодаря их уникальной способности модулировать иммунные и воспалительные реакции и восстанавливать поврежденные ткани с помощью дифференцировки или паракринныхмеханизмов.
Внеклеточные везикулы (ВВ), как утверждает Международное общество внеклеточных везикул (ISEV)4, относятся к общим частицам с липидной бислойной структурой, которые естественным образом высвобождаются из клеток. С недавними открытиями различных компонентов, таких как белки, липиды и генетические материалы (например, микроРНК, мРНК, молекулы ДНК, а также длинные некодирующие РНК) в ВВ из различных типов клеток5, была признана их решающая роль в клеточной коммуникации и эпигенетической регуляции6. В качестве нового заменителя материнских клеток ВВ применяются в иммунотерапии и регенеративной медицине с исследованиями in vivo, которые служат основой для продолжающихся доклинических исследований и последующих клинических испытаний7.
Однако в настоящее время большая часть исследований по эффективности трансплантации БМ-МСК и их производных ВВ все еще остается на уровне мелких животных, которых недостаточно для получения необходимых доказательств для клинической трансляции. В связи с этим крайне актуальным является проведение доклинических исследований по трансплантации БМ-МСК и их производных ВВ на уровне крупных животных, таких как свиньи.
Сообщалось, что МСК присутствуют в чрезвычайно низком количестве в костном мозге, составляя всего от 0,01% до 0,001% от общегочисла клеток8. Однако для доклинического введения БМ-МСК требуется большое количество клеток (≥10,7 на животное)9; количество необходимых ВВ еще больше, средняя доза которых составляет 0,25 мг белка на килограмм массы тела у10 свиней. Для достижения этих больших цифр существует острая необходимость в безопасном и эффективном методе выделения и культивирования МСК из костного мозга свиньи для достижения их массивного расширения in vitro и последующего получения их ВВ с высокой концентрацией белка.
На сегодняшний день существуют различные методы выделения BM-MSC и производных от них EV. Современные методы выделения БМ-МСК включают прямую посадку клеток костного мозга (БМК)11, центрифугирование по градиенту плотности, сортировку молекулярных меток на поверхности клетки и скрининг проточной цитометрии. Сообщалось, что сортировка молекулярных меток на поверхности клетки и скрининг проточной цитометрии приводят к снижению скорости клеточной адгезии, увеличению смертности за 24 часа и ингибированиюпролиферации12, в то время как прямое культивирование BMC может привести к высокому количеству смешанных гемопоэтических клеток. Таким образом, центрифугирование с градиентом плотности в настоящее время широко используется для получения BM-MSC. Современные методы выделения EV из надосадочной жидкости клеток включают ультрацентрифугирование, ультрафильтрацию, осаждение полимеров и исключение размера13. По сравнению с другими методами, ультрацентрифугирование имеет преимущество в низкой стоимости, простоте использования и совместимости с подготовкой больших объемов без сложной предварительной обработки, что является «золотым стандартом» для разделения EV14. Тем не менее, существует большая неоднородность в реагентах и методах в различных лабораториях втечение процесса, что может ввести читателей в заблуждение. В данной статье подробно описывается ряд последовательных шагов по выделению pBM-MSC и полученных из них EV, а последующие результаты идентификации доказывают, что метод осуществим для получения pBM-MSC и их EV для дальнейшего анализа в доклинических исследованиях. Мы надеемся, что эта систематическая работа может послужить методологической основой для исследователей, занимающихся доклинической оценкой трансплантации pBM-MSCs и их производных EV, чтобы клинические испытания могли быть проведены как можно скорее.
протокол
Согласно Руководству по уходу и использованию лабораторных животных, опубликованному Национальным институтом здравоохранения США, все экспериментальные процедуры были одобрены Комитетом по институциональному уходу за животными и их использованию (IACUC), больницей Фувай, Китайской академией медицинских наук.
1. Предоперационная подготовка животных
- Приобретите взрослых самцов китайских минипигов (30 ± 5 кг) в возрасте около 12 месяцев из Института зоологии Китайской академии наук и разместите их в помещениях Экспериментального центра для животных больницы Фувай, по крайней мере, за 2 недели. Проводите предоперационные осмотры, такие как регулярные исследования крови, чтобы убедиться, что животные здоровы.
- Очистите и побрейте кожу в области бедер минипига за день до операции. Чтобы избежать аспирации, голодайте на мини-пиги в течение 12 ч перед экстракцией костного мозга.
2. Подготовка к выделению и культивированию клеток
- Используйте метод Перколла (1,130 г/мл) для выделения мезенхимальных стволовых клеток из костного мозга. Смешайте исходный раствор с 10-кратным концентрированным PBS в соотношении 9:1 для получения изотонической среды. Затем смешайте изотоническую среду с PBS в соотношении 3:2 для получения 60% раствора Percoll (1,077 г/мл), который можно использовать в качестве окончательного разделительного раствора для выделения pBM-MSCs.
- Приготовьте модифицированную среду Dulbecco's Medium (IMDM) от Iscove с 10% эмбриональной бычьей сывороткой и 1,0% пенициллин-стрептомицином для получения полноценной среды. Предварительно нагрейте всю среду и PBS на водяной бане при температуре 37 °C для последующего культивирования клеток.
3. Обезболивание животных
- Введите общую анестезию кетамином (10 мг/кг) и ксилазином (2 мг/кг) внутримышечно. Проводите эндотрахеальную интубацию быстро, когда у минипига медленное дыхание и меньшая активность конечностей, и сохраняйте спонтанное дыхание, чтобы предотвратить неудачную интубацию.
- Поддерживающую анестезию проводят путем ингаляции 2% изофлурана с кислородом (1,5 л/мин) в качестве газа-носителя. Во время анестезии контролируйте частоту сердечных сокращений, дыхание и насыщение крови крови кислородом в режиме реального времени.
4. Извлечение костного мозга из минипига
- Поместите минипига в боковое положение. Чтобы обеспечить достаточное количество костного мозга для последующей культивирования клеток, расположите точку пункции костного мозга в проксимальном отделе бедренной кости минипига (рис. 1). Перед проведением операции продезинфицируйте и подкините кожу области прокола.
- Подтяните кожу вокруг точки прокола недоминирующей рукой и проткните иглу биопсии костного мозга вертикально в этой точке доминирующей рукой. При ощущении контакта иглы с кортикальной костью осторожно поворачивайте ручку влево и вправо, чтобы просверлить иглу. При попадании иглы в полость костного мозга часто возникает ощущение потери сопротивляемости.
- После того, как прокол будет на месте, извлеките иглу стержня. Затем приложите одноразовый стерильный шприц объемом 50 мл к концу наружной иглы, и предварительно промойте внутреннюю стенку шприца гепарином.
- Медленно извлеките 20 мл костного мозга и осторожно переложите их в стерильную центрифужную пробирку объемом 50 мл.
- Когда процедура будет завершена, вытащите иглу и снимите простыню. Продезинфицируйте место прокола и прижмите его на 20 минут для гемостаза.
- Экстубируйте минипига после восстановления спонтанного дыхания. Когда он полностью придет в себя со свободным движением конечностей, верните минипига в клетку для продолжения кормления.
5. Выделение мезенхимальных стволовых клеток из костного мозга
- Добавьте равный объем разогретых ПБС в костный мозг и тщательно перемешайте их. Затем с помощью стерильной пипетки осторожно перенесите 20 мл разбавленного костного мозга выше уровня градиента плотности 60% раствора в соотношении 1:1 по объему в стерильную центрифужную пробирку объемом 50 мл.
- Центрифугируйте пробирку при 600 x g (ускорение (ACC) = 5, замедление (DEC) = 5) в течение 20 мин при комнатной температуре (RT).
ПРИМЕЧАНИЕ: После центрифугирования в пробирке образуются четыре фазы, включая фазу сыворотки, фазу мононуклеарной клетки, фазу среды с градиентом плотности и фазу осаждения сверху вниз. Фаза мононуклеарной клетки представляет собой тонкий флокулянтный слой между сывороткой и средой градиента плотности. Мезенхимальные стволовые клетки, полученные из костного мозга, находятся в этой фазе. - Наберите фазу мононуклеарной клетки в стерильную центрифужную пробирку объемом 15 мл и дважды промойте PBS при плотности 800 x g в течение 5 минут.
- Повторно суспендируйте промытые клетки в 2 мл готовой среды и посадите ресуспендированные клетки в колбу для культуры2 см размером 175 см с плотностью 3-5 x 105/мл.
ПРИМЕЧАНИЕ: Все вышеуказанные процедуры выделения мезенхимальных стволовых клеток можно увидеть на рисунке 2.
6. Культивирование мезенхимальных стволовых клеток in vitro
- Инкубировать колбу с культурой при температуре 37 °C в насыщенной увлажненной атмосфере с содержанием 5%СО2. Осторожно встряхивайте колбу с культурой каждые 24 часа, чтобы предотвратить адгезивный рост осажденных гемопоэтических стволовых клеток, и наблюдайте за ростом, морфологией и контаминацией клеток под микроскопом.
- Заменяйте питательную среду в первый раз через 3 дня, а затем меняйте среду каждые 2-3 дня. Когда клеточные колонии достигнут 80%-90% слияния, субкультивируйте клетки в соотношении 1:2.
7. Адипогенная, остеогенная и хондрогенная дифференцировка пБМ-МСК
- Адипогенный дифференцировочный анализ
- Приготовьте среду для адипогенной дифференцировки А (среда А) и В (среда В) для БМ-МСК в соответствии с инструкцией к набору. Конкретную информацию о наборе можно найти в Таблице материалов.
- Добавьте 1 мл 0,1% желатина в шестилуночную пластину и осторожно встряхните, чтобы она могла равномерно покрыть дно каждой лунки. Затем поместите планшет с шестью лунками в чистый стол или инкубатор сCO2 не менее чем на 30 минут.
- Через 30 минут отсадите желатин и добавьте по 2 мл общей готовой среды в каждую лунку. Затем посадите pBM-MSCs в шестилуночный планшет с плотностью ячеек 2 x 104 ячейки/см2. После этого инкубируйте тарелку при температуре 37 °C в насыщенной влажности 5%CO2.
- Когда клетки достигнут 100% слияния, осторожно удалите всю среду и добавьте по 2 мл среды А в каждую лунку планшета. Через 3 дня достаньте Medium A из тарелки и добавьте по 2 мл Medium B в каждую лунку.
- После выдержки в течение 1 дня удалите среду B и замените ее средой A для индукции. В соответствии с принципом «Среда А в течение 3 дней, среда В в течение 1 дня» используйте среду А и В последовательно для индукции.
- Наблюдайте за состоянием клеток каждый день в течение периода. Если клетки сжимаются или умирают в процессе индукции среды А, замените ее средой В до тех пор, пока состояние клеток не восстановится.
- Повторите процесс индукции и поддержания и подготовьтесь к окрашиванию, когда под микроскопом будет обнаружено достаточное количество липидных капель подходящего размера.
- Удалите среду для адипогенной дифференцировки в шестилуночном планшете и аккуратно промойте 1x PBS. Добавьте в каждую лунку по 2 мл 4% раствора параформальдегида и зафиксируйте на 30 мин при RT.
- Снимите фиксатор параформальдегида и промойте его 1x PBS два или три раза, чтобы убедиться, что фиксатор полностью удален. Добавьте 2 мл красителя Oil Red O в каждую лунку и окрашивайте в течение 30 минут при температуре RT.
- Удалите краситель Oil Red O и промойте 1x PBS два или три раза. Добавьте по 2 мл 1x PBS в каждую лунку, а затем наблюдайте за эффектом адипогенной дифференцировки под микроскопом.
- Остеогенный дифференцировочный анализ
- Подготовьте готовую среду для остеогенной дифференцировки в соответствии с инструкцией набора.
- Выполните шаги 7.1.2-7.1.3 для посадки и культивирования pBM-MSC.
- Когда клетки достигнут 70% слияния, осторожно удалите общую полную среду и добавьте 2 мл среды для остеогенной дифференцировки в каждую лунку планшета.
- Меняйте на свежую остеогенную среду для дифференцировки каждые 3 дня. Продолжайте индукцию в течение 2-4 недель и приготовьтесь к окрашиванию красителем Ализарин Ред при появлении явных кальциевых узелков во время остеогенеза.
- Удалите среду для остеогенной дифференцировки в шестилуночном планшете и аккуратно промойте 1x PBS. Добавьте в каждую лунку по 2 мл 4% раствора параформальдегида и зафиксируйте на 30 мин при RT.
- Снимите параформальдегидный фиксатор и промойте его 1x PBS два или три раза, чтобы убедиться, что фиксатор тщательно очищен. Добавьте 2 мл красителя Ализарин Красный в каждую лунку и окрашивайте на 10 минут при температуре RT.
- Удалите краситель Alizarin Red и умойтесь 1x PBS два или три раза. Добавьте в каждую лунку по 2 мл 1x PBS, а затем наблюдайте за эффектом остеогенной дифференцировки под микроскопом.
- Анализ хондрогенной дифференцировки
- Подготовьте премикс для хондрогенной дифференциации в соответствии с инструкцией набора.
- Перенесите 3-4 x 105 pBM-MSC в стерильную центрифужную пробирку объемом 15 мл. Центрифугируйте при 250 x g в течение 4 мин при 20 °C.
- Удалите надосадочную жидкость и добавьте 0,5 мл предварительной смеси для ресуспендирования гранулы, полученной центрифугированием на предыдущем этапе, а затем центрифугируйте при 150 x g в течение 5 минут при 20 °C. Повторите этот шаг, чтобы снова промыть клетки.
- Подготовьте всю среду для хондрогенной дифференцировки.
- Ресуспендируют клетку, полученную на предыдущем этапе, с помощью 0,5 мл полной среды и центрифугируют при 150 x g в течение 5 мин при 20 °C.
- Открутите крышку трубки центрифуги, чтобы облегчить газообмен. Поместите его в вертикальное положение в инкубатор при температуре 37 °C, 5%CO2 и насыщенной влажности.
- Когда кажется, что клетки агрегируются (обычно через 24-48 часов, в зависимости от фактической ситуации), поверните дно центрифугной трубки, чтобы хрящевые шарики отделились от дна и подвешивали в среде.
- Меняйте на свежую полноценную среду для хондрогенной дифференцировки каждые 2-3 дня. Продолжайте индукцию до тех пор, пока в трубке не сформируются хрящевые шарики диаметром 1,5-2 мм, а затем подготовьте срезы для окрашивания.
- Подготовьте парафиновые срезы хрящевых шариков в соответствии с обычными этапами патологических экспериментов. Добавьте синий краситель Alicia на очищенные от воска участки и окрашивайте при температуре 37 °C в течение 1 часа.
- Промойте предметное стекло проточной водой в течение 5 минут, а затем наблюдайте за окрашивающим эффектом Алисии голубой под микроскопом после высыхания.
8. Идентификация фенотипа клеток методом проточной цитометрии
- Когда клеточные колонии достигнут 80%-90% слияния в проходах 3-5, удалите питательную среду и дважды промойте клетки предварительно подогретыми PBS. Затем расщепляют клетки с 3-4 мл 0,25% трипсина/ЭДТА и инкубируют их при 37 °С в насыщенной увлажненной атмосфере с 5%СО2 в течение 2-3 мин до тех пор, пока они не будут отделены от дна колбы под микроскопом.
- Соберите клетки с готовой средой объемом 10 мл и переложите клеточную суспензию в стерильную центрифужную пробирку объемом 15 мл. Центрифугируйте клеточную суспензию при 800 x g в течение 5 мин при RT. Выбросьте надосадочную жидкость и промойте клетки 4 °C PBS.
- Ресуспендируйте клетки до 10 мл при 4 °C PBS. Клеточную суспензию делят на девять групп объемом 1 мл в каждой микропробирке объемом 1,5 мл, называемых отрицательным контролем, контролем изотипа FITC, контролем изотипа PE, контролем изотипа APC, группой CD105, CD29, CD90, CD14 и CD45 соответственно. Убедитесь, что количество клеток в каждой микропробирке находится в диапазоне от 1 x 105 до 1 x 106.
- Центрифугируйте суспензию при 800 x g в течение 5 мин при 4 °C и снова суспендируйте клетки в каждой микропробирке 100 мкл 4 °C PBS. За исключением отрицательной контрольной группы, добавьте в каждую микропробирку по 5 мкл соответствующего изотипа контроля (FITC, PE и APC Mouse IgG1 kappa Isotype Control) и антител (CD105, CD29, CD90, CD14 и CD45 Monoclonal Antibody) для проточной цитометрии в соответствии с порядком на шаге 8.3. Аккуратно перемешайте и выдерживайте в течение 1,5 часов при температуре 4 °C в темноте.
- Добавьте 1 мл PBS 4 °C в каждую микропробирку и центрифугируйте при 300 x g в течение 10 минут при 4 °C. Выбросьте надосадочную жидкость и добавьте 200 мкл 4 °C PBS для ресуспендирования клеток.
- Протестируйте не менее 10 000 клеток на проточной цитометрии после фильтрации клеточной суспензии16 и проанализируйте данные с помощью программного обеспечения проточной цитометрии.
9. Выделение внеклеточных везикул (ВВ), полученных из мезенхимальных стволовых клеток костного мозга свиньи
- Когда слияние pBM-MSCs достигнет 80%-90%, выбросьте надосадочную жидкость и промойте клетки PBS 2x. Затем добавьте 25 мл IMDM без сыворотки в каждую колбу для культур и продолжайте инкубацию при 37 °C во влажной атмосфере с содержаниемCO2 5% в течение 48 часов.
- Соберите надосадочную жидкость клеток (кондиционированная среда, КМ) в центрифужную пробирку объемом 50 мл и центрифугируйте при 300 x g в течение 10 минут при 4 °C для удаления клеточного мусора.
- Снова соберите надосадочную жидкость в другую центрифужную пробирку объемом 50 мл. Проводите изоляцию электромобилей как можно скорее после сбора надосадочной жидкости. При длительном хранении надосадочную жидкость храните в холодильнике при температуре -80 °C, чтобы предотвратить потерю электромобилей.
- Центрифугируйте надосадочную жидкость на стадии 9.3 при 2000 x g в течение 20 мин при 4°С.
- Перенесите надосадочную жидкость в стерильную пробирку, используемую для высокоскоростной центрифуги и центрифуги при давлении 16 500 x g в течение 30 минут при 4 °C.
- Снова перенесите надосадочную жидкость в ультрацентрифужную пробирку и центрифугируйте при давлении 120 000 x g в течение не менее 70 минут при 4 °C с помощью ротора с фиксированным углом наклона.
- Полностью выбросьте надосадочную жидкость. Добавьте 1 мл PBS 4 °C в каждую ультрацентрифужную пробирку и повторно суспендируйте осадок с помощью микропипетки. Смешайте раствор из той же группы в ультрацентрифужную пробирку, а затем добавьте 4 °C PBS, чтобы объем был больше 3/4 пробирки.
- Центрифугируйте при давлении 120 000 x g в течение 60 минут при 4 °C и максимально удалите надосадочную жидкость.
- Снова суспендируйте осадок со стерильным PBS и храните электромобили в холодильнике при температуре -80 °C.
ПРИМЕЧАНИЕ: Количество PBS для ресуспендирования может быть определено как 100 мкл для осадков из каждых двух колб с культурой размером 175см2 . Все вышеперечисленные шаги по изоляции электромобилей систематически показаны на рисунке 3 .
10. Идентификация ВВ с помощью просвечивающей электронной микроскопии (ПЭМ), анализа отслеживания наночастиц (NTA) и вестерн-блоттинга
- Поместите образец EVs на лед после его размораживания на водяной бане при температуре 25 °C, а затем разбавьте до половины предыдущей концентрации PBS для обнаружения NTA17.
- Нанесите пипеткой 10 мкл образца на медную сетку, дайте ему отстояться в течение 1 минуты и впитайте плавающую жидкость с помощью фильтровальной бумаги. Затем добавьте 10 μл фосфовольфрамовой кислоты в медную сетку, дайте ей постоять 1 минуту и впитайте лишнюю жидкость.
- После высыхания в течение нескольких минут на РТ проводят электронную микроскопию при условии ускорения напряжения 100 кВ18.
- Ресуспендируйте образец EVs с этапа 9.9 в буфере для лизиса RIPA (25 мМ Трис· HCl (pH 7,6), 150 мМ хлорида натрия [NaCl], 1% нонилфеноксиполиэтоксителанола (NP-40), 1% дезоксихолата натрия, 0,1% додецилсульфата натрия (SDS), 1 мМ фенилметансульфонилфторида (PMSF), 1x ингибитор протеазы) и определяют экспрессию специфических маркеров для EV, таких как Alix, TSG101, CD81 и CD63, с помощью вестерн-блоттинга19.
Результаты
Создание мезенхимальных стволовых клеток костного мозга свиньи
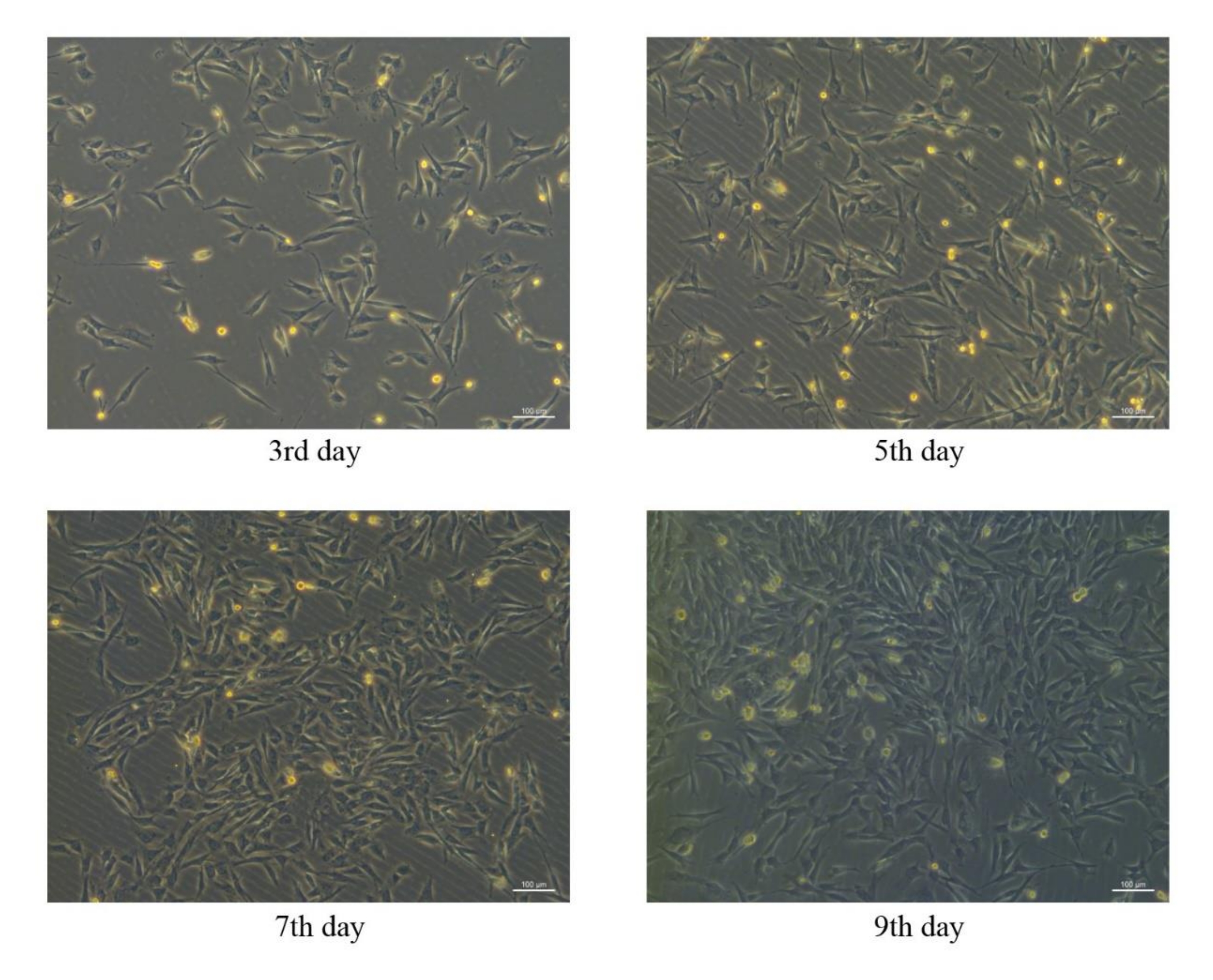
Мезенхимальные стволовые клетки, полученные из костного мозга свиней, были успешно выделены и культивированы in vitro, а морфология pBM-MSCs в разные дни показана на рисунке 4. В первичной культуре pBM-MSCs микроскопическое наблюдение показало, что адгезия клеток происходит через день после посадки, а адгезивные клетки обычно имеют круглую форму. Первичные pBM-МСК обычно оставались в фазе покоя в течение 3 дней после посадки, а пролиферация клеток начиналась на4-й день. Морфология клеток изменилась с круглого на веретенообразный, многосторонний или звездчатый тип после пролиферации, а ядра расположены центрально, с двойными ядрышками в некоторых клетках. Клеточные колонии образовывались через 7-9 дней после начала пролиферации клеток, а 80-90% слияние клеток достигалось через 12-14 дней. Микроскопическое наблюдение показало, что адгезивные клетки росли как разрозненные колонии и располагались в виде закрученного узора.
Пролиферация клеток значительно ускорялась после пассирования, и за неделю можно было достичь 80-90% слияния. Морфология клеток была гомогенной веретенообразной от второго прохода, напоминающей фибробласты, с соотношением длины к ширине примерно 2-3:1. Если бы клетки были дифференцированы, они могли бы казаться многоугольными или звездообразными. После пассирования клетки уже не росли разрозненными колониями, а равномерно и радиально в параллельном расположении.
Идентификация потенциала дифференцировки клеток путем окрашивания
В анализе адипогенной дифференциации окрашивание Oil Red O показало, что вокруг ядра появились круглые оранжево-красные капли липидов разного размера (рис. 5А); В анализе остеогенной дифференцировки окрашивание Ализарином красным показало красные узелки на поверхности клеток (рис. 5B), что было вызвано цветовой реакцией с солями кальция, осажденными остеобластами, дифференцированными из pBM-MSCs. В анализе хондрогенной дифференцировки окрашивание Алисии синим показало, что весь участок ткани был синим (рис. 5C), что было вызвано окрашиванием эндокислотного мукополисахарида в хрящевых шариках.
Идентификация фенотипа клеток методом проточной цитометрии
Были проведены анализы маркеров клеточной поверхности для создания фенотипа pBM-МСК. По результатам проточной цитометрии (рис. 6) три положительных маркера, таких как CD105, CD29 и CD90, были достоверно экспрессированы на поверхности pBM-МСК, составляя 96,5%, 99,8% и 92% соответственно (рис. 6A-C). Тем не менее, экспрессия CD14 и CD45 была отрицательной (рис. 6D, E). Между тем, результаты контроля соответствующих изотипов были отрицательными, что уже было наложено на рисунок, исключая возможность неспецифического связывания антител.
Идентификация ВВ, полученных из pBM-MSC, с помощью NTA, TEM и вестерн-блоттинга
Результат NTA показал, что средний размер частиц составил 126,9 нм, что было в пределах диапазона EV; кроме того, исходная концентрация образца EVs составляла 1,5 x 1010 частиц/мл, а точное значение, присвоенное размеру, можно найти на рисунке 7A. Диаграмма траектории частиц показана на рисунке 7B, иллюстрируя, что частицы находились в нерегулярном броуновском движении. Кроме того, дисковидные пузырьки, как классическая структура EV, можно было ясно увидеть под электронным микроскопом при увеличении в 50 000 раз (рис. 7C). Кроме того, экспрессия специфических маркеров для ВВ, таких как Alix, TSG101, CD81 и CD63, была обнаружена в образце с помощью вестерн-блоттинга (рис. 7D).

Рисунок 1: Точка прокола костного мозга минипига. Красной областью отмечена точка прокола извлечения костного мозга, расположенная в проксимальном отделе бедренной кости минипига. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 2: Выделение мезенхимальных стволовых клеток из костного мозга свиней. Процесс выделения мезенхимальных стволовых клеток из костного мозга свиньи показан на технологической схеме, а после центрифугирования с градиентом плотности четко проиллюстрированы четыре жидкие фазы. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 3: Изоляция EV, полученных из pBM-MSC. На принципиальной схеме показаны конкретные шаги по изоляции электромобилей от кондиционированной среды с помощью ультрацентрифугирования. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 4: Морфологические характеристики pBM-MSCs в разные дни. Сходные морфологические характеристики pBM-MSC можно наблюдать на3-й,5-й,7-й и9-й день после посадки под 100-кратное микроскопическое поле, а клеточные колонии образовались на9-е сутки. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 5. Определение дифференцировочного потенциала pBM-MSCs методом окрашивания. (А) адипогенный, (В) остеогенный и (В) хондрогенный дифференцировочный анализ рБМ-МСК соответственно. По результатам окрашивания можно определить дифференцировочный потенциал pBM-MSCs. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 6: Результаты идентификации pBM-MSCs методом проточной цитометрии. CD105, CD29 и CD90 в значительной степени экспрессируются на поверхности pBM-МСК, составляя 96,5%, 99,8% и 92,0% соответственно, в то время как экспрессия CD14 и CD45 отрицательна. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}

Рисунок 7: Результаты идентификации ВВ, полученных из пБМ-МСК, с помощью морфологии и молекулярной биологии. (А) результат НТА ВВ, полученных из пБМ-МСК, с графиком распределения частиц по размерам и (В) диаграммой траектории частиц, соответственно; (C) Изображение ПЭМ сделано с увеличением 50 000x, а белой стрелкой показана классическая структура дисковидных везикул. (D) Экспрессия специфических маркеров для ВВ методом вестерн-блоттинга. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этой цифры.
{kind=link}
Обсуждение
Традиционная точка пункции костного мозга минипигов располагалась на гребне подвздошной кости20. Несмотря на то, что его легко обнаружить, объем экстракции костного мозга ограничен21 мл (всего около 5 мл в целом), поэтому трудно удовлетворить потребность в большом количестве расширений in vitro для трансплантации in vivo. В этом методе мы переместили точку пункции костного мозга в проксимальный отдел бедренной кости, и из этого участка можно извлечь не менее 20 мл костного мозга, что гарантирует достаточное количество pBM-MSCs для последующей культивирования клеток.
Двумя основными разделительными решениями, используемыми для выделения BM-MSC методом центрифугирования с градиентом плотности, являются Percoll и Ficoll. Percoll состоит из силиконизированного поливинилпирролидона (PVP), который является новым нетоксичным и не раздражающим центробежным разделителем градиента плотности. Низкая диффузионная константа Перколла приводит к относительно стабильному градиенту плотности; Таким образом, удовлетворительное разделение ячеек обычно может быть достигнуто в течение десятков минут при низких центробежных силах (200-1000 x g). Метод выделения pBM-MSCs с использованием метода Фиколла был описан ранее21. По сравнению с Фиколлом, Перколл постепенно стал использоваться благодаря своим преимуществам легкой изопроницаемости, низкой вязкости, нетоксичности и непричинности клеточной агрегации, что может дополнить существующие методы выделения pBM-MSCs.
При выделении и культивировании pBM-MSC нельзя игнорировать некоторые критически важные этапы. Во-первых, успешная стратификация различных жидких фаз после центрифугирования с градиентом плотности является ключом к выделению очищенных pBM-MSC. БМ-МСК, как тип мононуклеарных клеток костного мозга (БМ-МНК), имеют удельный вес, аналогичный удельному весу лимфоцитов и моноцитов, около 1,075 г/мл. Исходная плотность Перколла составляет 1,130 г/мл, и для успешного получения клеточного слоя, содержащего BM-МСК, после центрифугирования с градиентом плотности необходимо заранее сконфигурировать 60% раствор для разделения изотонического градиента плотности (1,077 г/мл) в соответствии с соотношением плотность-концентрация Перколла22. Кроме того, соответствующие условия центрифугирования также способствуют успешной стратификации. Учитывая низкую константу диффузии Перколла, мы центрифугировали извлеченный костный мозг при 600 x g в течение 20 мин при относительно низких уровнях ускорения/замедления (ACC = 5, DEC = 5), что позволило достичь хорошего эффекта стратификации. Во-вторых, соответствующая плотность посадки также имеет важное значение для клеточной культуры. Для того чтобы получить достаточное количество МСК (обычно более 107 на 9 животных) для последующей трансплантации, мы используем колбы для культуры 175 см2 для культивирования клеток. В предыдущем исследовании20 полученные BM-MNC обычно высаживали в колбы для культивирования при плотности 5 x 105/см2. Сообщалось, что после центрифугирования с градиентом плотности на каждые 5 мл костного мозга свиньи23 можно получить 2-3 x 107 BM-MNC. Таким образом, в этом протоколе мы рекомендуем высаживать общее количество BM-MNC, выделенных из каждого 20 мл свиного костного мозга, в колбу для культуры объемом 175см2 для получения подходящей плотности. В-третьих, следует избегать примесей во время выделения и культивирования pBM-MSCs. При вытягивании фазы мононуклеарной ячейки пипетку не следует вставлять в фазу Перколла, чтобы она не смешивалась с разделяющейся жидкостью. Кроме того, через 24 ч после посадки клеток колбу с культурой следует осторожно встряхнуть, чтобы уменьшить адгезию эритроцитов.
В процессе ультрацентрифугирования высокие уровни загрязнения белковых агрегатов и липопротеинов с помощью этого метода неизбежно ставят под угрозу количественную оценку и функциональный анализ EVs14. Чтобы максимально снизить загрязнение в процессе, при переносе надосадочной жидкости перед этапом ультрацентрифугирования необходимо каждый раз сохранять глубину 5 мм жидкости на дне. Между тем, после первого ультрацентрифугирования суспендирование гранулы в стерильном PBS и последующее повторное выполнение ультрацентрифугирования может эффективно снизить загрязнение липопротеинов.
Несмотря на то, что центрифугирование с градиентом плотности и ультрацентрифугирование широко используются для выделения BM-MSC и их производных EV соответственно, эти два метода также имеют свои ограничения. С одной стороны, метод Перколла является длительным и громоздким, и получение образца концентрата БМ с помощью устройства для концентрации клеток у постели больного сообщалось в качестве альтернативного метода выделения МСК24. С другой стороны, метод ультрацентрифугирования требует не только высококвалифицированных техников, но и дорогостоящего оборудования; Таким образом, совместное применение двух или более методов может представлять собой разумную стратегию для более эффективной изоляции EV25. Кроме того, идентификация pBM-MSC и производных от них EV также нуждается в улучшении. Например, в соответствии с международными критериями определения МСК26, экспрессия некоторых положительных или отрицательных маркеров, таких как CD73, CD34 и HLA-DR, до сих пор отсутствует в результатах идентификации фенотипов БМ-МСК методом проточной цитометрии в данном исследовании. Кроме того, несмотря на то, что были приняты меры для предотвращения загрязнения в процессе изоляции электромобилей, из-за ограничений нашей лаборатории мы не можем оценить чистоту образца электромобилей, чтобы улучшить последующую работу.
Данное исследование объединяет методы последовательного выделения pBM-MSC и их производных ВВ, что доказано последующими результатами идентификации на систематической основе. В частности, мы выделили ключевые операции в этой серии шагов, пояснив некоторые конкретные экспериментальные условия, которые могут в определенной степени решить проблему неоднородности, существующую в разных лабораториях во время этого процесса. Эта методическая работа может быть широко использована в доклинических исследованиях эффективности трансплантации МКМ-МСК и их производных ВВ, что может обеспечить экспериментальную базу с достаточным уровнем для клинических исследований.
Раскрытие информации
У всех авторов нет конфликта интересов, о котором можно было бы заявить.
Благодарности
Мы благодарим Ян Цзяньчжуна и Ван Сюэминя за их вклад в операцию по экстракции костного мозга. Эта работа была поддержана грантами Инновационного фонда медицинских наук CAMS (CIFMS) [номер гранта 2016-I2M-1-009], Национального фонда естественных наук Китая (No: 82070307; No: 81874461).
Материалы
| Name | Company | Catalog Number | Comments |
| 175 cm2 cell culture flask | Thermo Fisher | 159910 | used for cell culture |
| 0.25% Trypsin/EDTA | Thermo Fisher | 25200056 | used to digest cells |
| Adipogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90031 | used for adipogenic differentiation assay |
| Alix Monoclonal Antibody | Thermo Fisher | MA1-83977 | used to identify extracellular vesicles(Evs) by western blotting |
| APC Mouse IgG1 kappa Isotype Control | Thermo Fisher | 17-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| CD105 (Endoglin) Monoclonal Antibody | Thermo Fisher | 17-1057-42 | used to identify pBM-MSCs by flow cytometry |
| CD14 Monoclonal Antibody | Thermo Fisher | MA1-82074 | used to identify pBM-MSCs by flow cytometry |
| CD29/IGTB1 Monoclonal Antibody | Thermo Fisher | MA1-19458 | used to identify pBM-MSCs by flow cytometry |
| CD45 Monoclonal Antibody | Thermo Fisher | MA5-28383 | used to identify pBM-MSCs by flow cytometry |
| CD63 Monoclonal Antibody | Thermo Fisher | 10628D | used to identify EVs by western blotting |
| CD81 Monoclonal Antibody | Thermo Fisher | MA5-13548 | used to identify EVs by western blotting |
| CD90 Monoclonal Antibody | Thermo Fisher | A15794 | used to identify pBM-MSCs by flow cytometry |
| Chondrogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90041 | used for chondrogenic differentiation assay |
| Fetal Bovine Serum | Gibco | 10099141C | used to prepare complete medium |
| FITC Mouse IgG1 kappa Isotype Control | Thermo Fisher | 11-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| Flow cytometry | BD | Accuri C6 | used for identification of cell phenotype |
| FlowJo software | BD | V10 | used to analyze data from flow cytometry |
| High-speed centrifuge tube (50 mL) | Beckman | 357003 | used for high-speed centrifugation |
| Iscove's Modified Dulbecco's Medium | Gibco | C12440500BT | used for cell culture |
| Low-temperature high-speed floor centrifuge | Avanti | J-26XPI | used for high-speed centrifugation to obtain clean conditioned medium |
| Nonyl phenoxypolyethoxylethanol (NP-40) | Sigma-Aldrich | NP40S | used for the composition of RIPA lysis buffer |
| Osteogenic Differentiation Kit for Bone Marrow Mesenchymal Stem Cell | OriCell | GUXMX-90021 | used for osteogenic differentiation assay |
| PE Mouse IgG1 kappa Isotype Control | Thermo Fisher | 12-4714-42 | used to eliminate the effects of non-specific staining in flow cytometry |
| Percoll | Cytiva | 17089102 | used to isolate porcine bone marrow mesenchymal stem cells(pBM-MSCs) |
| Phenylmethanesulfonyl fluoride (PMSF) | Thermo Scientific | 36978 | used for the composition of RIPA lysis buffer |
| Phosphate Buffered Saline(10x) | Beyotime | ST476 | used to prepare isotonic Percoll solution |
| Phosphate Buffered Saline(1x) | Cytiva | AF29561133 | used to dilute Percoll and wash cells |
| Protease inhibitor (1x) | Thermo Scientific | A32955 | used for the composition of RIPA lysis buffer |
| sodium chloride | Sigma-Aldrich | S9888 | used for the composition of RIPA lysis buffer |
| sodium deoxycholate | Sigma-Aldrich | D6750 | used for the composition of RIPA lysis buffer |
| sodium dodecyl sulfate (SDS) | Sigma-Aldrich | L3771 | used for the composition of RIPA lysis buffer |
| Transmission electron microscopy | Hitachi | HT7700 | used for electron microscopy imaging |
| Tris·HCl | Sigma-Aldrich | 93363 | used for the composition of RIPA lysis buffer |
| TSG101 Monoclonal Antibody | Thermo Fisher | MA1-23296 | used to identify EVs by western blotting |
| Ultracentrifuge (Type 50.2 Ti Rotor) | Beckman | optima L-100XP | used for ultracentrifugation to isolate exosomes |
| Ultracentrifuge tube (26.3 mL) | Beckman | 355654 | used for ultracentrifugation |
| ZetaVIEW | Particle Metrix | S/N 17-310 | used for Nanoparticle Tracking Analysis |
Ссылки
- Heslop, J. A., et al. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cells Translational Medicine. 4 (4), 389-400 (2015).
- Berebichez-Fridman, R., Montero-Olvera, P. R. Sources and clinical applications of mesenchymal stem cells: State-of-the-art review. Sultan Qaboos University Medical Journal. 18 (3), 264-277 (2018).
- Antebi, B., Mohammadipoor, A., Batchinsky, A. I., Cancio, L. C. The promise of mesenchymal stem cell therapy for acute respiratory distress syndrome. The Journal of Trauma and Acute Care Surgery. 84 (1), 183-191 (2018).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750 (2018).
- Valadi, H., et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature Cell Biology. 9 (6), 654-659 (2007).
- Farooqi, A. A., et al. Exosome biogenesis, bioactivities and functions as new delivery systems of natural compounds. Biotechnology Advances. 36 (1), 328-334 (2018).
- Lener, T., et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. Journal of Extracellular Vesicles. 4 (1), 30087 (2015).
- Castro-Malaspina, H., et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood. 56 (2), 289-301 (1980).
- vander Spoel, T. I. G., et al. Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovascular Research. 91 (4), 649-658 (2011).
- Gupta, D., Zickler, A. M., El Andaloussi, S. Dosing extracellular vesicles. Advanced Drug Delivery Reviews. 178, 113961 (2021).
- Mareschi, K., et al. Multipotent mesenchymal stromal stem cell expansion by plating whole bone marrow at a low cellular density: a more advantageous method for clinical use. Stem Cells International. , 920581 (2012).
- Van Vlasselaer, P., Falla, N., Snoeck, H., Mathieu, E. Characterization and purification of osteogenic cells from murine bone marrow by two-color cell sorting using anti-Sca-1 monoclonal antibody and wheat germ agglutinin. Blood. 84 (3), 753-763 (1994).
- Yang, D., et al. and perspective on exosome isolation - efforts for efficient exosome-based theranostics. Theranostics. 10 (8), 3684-3707 (2020).
- Li, P., Kaslan, M., Lee, S. H., Yao, J., Gao, Z. Progress in exosome isolation techniques. Theranostics. 7 (3), 789-804 (2017).
- Willms, E., Cabañas, C., Mäger, I., Wood, M. J. A., Vader, P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Frontiers In Immunology. 9, 738 (2018).
- Huang, P., et al. Combinatorial treatment of acute myocardial infarction using stem cells and their derived exosomes resulted in improved heart performance. Stem Cell Research & Therapy. 10 (1), 300 (2019).
- Akbar, N., Pinnick, K. E., Paget, D., Choudhury, R. P. Isolation and characterization of human adipocyte-derived extracellular vesicles using filtration and ultracentrifugation. Journal of Visualized Experiments: JoVE. (170), e61979 (2021).
- Jung, M. K., Mun, J. Y. Sample preparation and imaging of exosomes by transmission electron microscopy. Journal of Visualized Experiments: JoVE. (131), e56482 (2018).
- Menck, K., Bleckmann, A., Schulz, M., Ries, L., Binder, C. Isolation and characterization of microvesicles from peripheral blood. Journal of Visualized Experiments: JoVE. (119), e55057 (2017).
- Yang, Y. J., et al. Atorvastatin treatment improves survival and effects of implanted mesenchymal stem cells in post-infarct swine hearts. European Heart Journal. 29 (12), 1578-1590 (2008).
- Lee, W. -. J., et al. Isolation and cellular phenotyping of mesenchymal stem cells derived from synovial fluid and bone marrow of minipigs. Journal of Visualized Experiments: JoVE. (113), e54077 (2016).
- Lindqvist, R., Wilson, I. D. Food Microorganisms: Buoyant Density Centrifugation. Encyclopedia of Separation Science. , 2843-2849 (2000).
- Yao, Y. L., Zhang, H., Gong, D. J., Song, Z. G., Xu, Z. Y. Transfecting hyperpolarization-activated cyclic nucleotide-gated channel 2 gene into porcine bone marrow mesenchymal stem cells. Journal of Clinical Rehabilitative Tissue Engineering Research. 13 (49), 9673-9676 (2009).
- Hegde, V., et al. A prospective comparison of 3 approved systems for autologous bone marrow concentration demonstrated nonequivalency in progenitor cell number and concentration. Journal of Orthopaedic Trauma. 28 (10), 591-598 (2014).
- Gallet, R., et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. European Heart Journal. 38 (3), 201-211 (2017).
- Dominici, M., et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 8 (4), 315-317 (2006).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены