
Method Article
Fabricating Degradable Thermoresponsive Hydrogels on Multiple Length Scales via Reactive Extrusion, Microfluidics, Self-assembly, and Electrospinning
In This Article
Summary
Protocols are described for the fabrication of degradable thermoresponsive hydrogels based on hydrazone cross-linking of polymeric oligomers on the bulk scale, microscale, and nanoscale, the latter for preparation of both gel nanoparticles and nanofibers.
Abstract
While various smart materials have been explored for a variety of biomedical applications (e.g., drug delivery, tissue engineering, bioimaging, etc.), their ultimate clinical use has been hampered by the lack of biologically-relevant degradation observed for most smart materials. This is particularly true for temperature-responsive hydrogels, which are almost uniformly based on polymers that are functionally non-degradable (e.g., poly(N-isopropylacrylamide) (PNIPAM) or poly(oligoethylene glycol methacrylate) (POEGMA)). As such, to effectively translate the potential of thermoresponsive hydrogels to the challenges of remote-controlled or metabolism-regulated drug delivery, cell scaffolds with tunable cell-material interactions, theranostic materials with the potential for both imaging and drug delivery, and other such applications, a method is required to render the hydrogels (if not fully degradable) at least capable of renal clearance following the required lifetime of the material. To that end, this protocol describes the preparation of hydrolytically-degradable hydrazone-crosslinked hydrogels on multiple length scales based on the reaction between hydrazide and aldehyde-functionalized PNIPAM or POEGMA oligomers with molecular weights below the renal filtration limit. Specifically, methods to fabricate degradable thermoresponsive bulk hydrogels (using a double barrel syringe technique), hydrogel particles (on both the microscale through the use of a microfluidics platform facilitating simultaneous mixing and emulsification of the precursor polymers and the nanoscale through the use of a thermally-driven self-assembly and cross-linking method), and hydrogel nanofibers (using a reactive electrospinning strategy) are described. In each case, hydrogels with temperature-responsive properties similar to those achieved via conventional free radical cross-linking processes can be achieved, but the hydrazone cross-linked network can be degraded over time to re-form the oligomeric precursor polymers and enable clearance. As such, we anticipate these methods (which may be generically applied to any synthetic water-soluble polymer, not just smart materials) will enable easier translation of synthetic smart materials to clinical applications.
Introduction
Smart materials have attracted significant attention due to their potential for reversible "on-demand" responses to external and/or environmental signals. Temperature-responsive materials have attracted particular interest due to their lower critical solution temperature (LCST) behavior, resulting in temperature-driven precipitation at temperatures T>LCST1,2. In the context of thermoresponsive hydrogels, this lower critical solution temperature behavior is manifested by reversible swelling/de-swelling events that result in temperature-tunable bulk sizes (larger at T<LCST)3, pore sizes (larger at T<LCST)4, and interfacial properties (more hydrophilic at T<LCST)5. Such transitions have been widely applied in drug delivery (for external or environmentally-triggerable drug release4,6,7), tissue engineering and cell culture (for thermoreversible cell adhesion/delamination8,9,10), separations (for switchable membrane porosities and permeabilities or thermally-recyclable diagnostic supports11,12,13), microfluidic processes (for on-off valves regulating flow14,15), and rheological modifiers (for temperature-tunable viscosities16). The most commonly investigated thermoresponsive hydrogels are based on poly(N-isopropylacrylamide) (PNIPAM)17, although significant (and increasing) work has also been conducted on poly(oligoethylene glycol methacrylate) (POEGMA)2,18 and poly(vinylcaprolactam) (PVCL)19,20. POEGMA has attracted particular recent interest given its anticipated improved biocompatibility21,22and its facile-to-tune LCST behavior, in which linearly-predictable mixtures of monomers with different numbers of ethylene oxide repeat units in their side chains can alter the LCST from ~20 °C to >90 °C2,23. However, each of these polymers is prepared by free radical polymerization and thus contains a carbon-carbon backbone, significantly limiting the potential utility and translatability of such polymers in the context of biomedical applications in which degradation (or at least the capacity for clearance through renal filtration) is typically a requirement.
In response to this limitation, we have recently reported extensively on the application of hydrazone chemistry (i.e., the reaction between hydrazide and aldehyde-functionalized pre-polymers) to prepare degradable analogues of thermoresponsive hydrogels24,25,26,27,28,29. The rapid and reversible reaction between hydrazide and aldehyde groups upon mixing of the functionalized precursor polymers30 enables both in situ gelation (enabling facile injection of these materials without the need for surgical implantation or any type of external polymerization stimulus such as UV irradiation or chemical initiation) as well as hydrolytic degradation of the network at a rate controlled by the chemistry and density of the crosslinking sites. Furthermore, by maintaining the molecular weight of the pre-polymers used to prepare the hydrogels below the renal filtration limit, hydrogels made using this approach degrade back into the oligomeric precursor polymers that can be cleared from the body25,27,28. Coupled with the low cytotoxicity and low inflammatory tissue response induced by these materials25,26,27, this approach offers a potentially translatable method for the use of thermoresponsive smart hydrogels in medicine, particularly if well-controlled degradable analogues of such hydrogels on all length scales (bulk, micro, and nano) can be fabricated.
In this protocol, we describe methods for making synthetic thermoresponsive pre-polymers functionalized with controlled numbers of hydrazide and aldehyde groups as well as methods to apply these polymers to create hydrogels with well-defined dimensions on various length scales. In particular, this manuscript describes four distinct approaches we have developed to control the mixing of the reactive hydrazide and aldehyde-functionalized pre-polymers and thus create thermoresponsive hydrogel networks with well-defined geometries and morphologies:
To create degradable bulk hydrogels with defined sizes, a templating strategy is described by which the reactive pre-polymers are loaded into separate barrels of a double-barrel syringe equipped at its outlet with a static mixer and subsequently co-extruded into a silicone mold with the desired hydrogel shape and dimensions21,27 (Figure 1).

Figure 1: Schematic of bulk hydrogel formation. Hydrazide and aldehyde-functionalized polymer solutions (in water or aqueous buffer) are loaded into separate barrels of a double barrel syringe and then co-extruded through a static mixer into a cylindrical silicone mold. Rapid in situ gelation upon mixing forms a hydrazone crosslinked hydrogel, which is free standing (once the mold is removed) within seconds to minutes depending on concentration and functional group density of the precursor polymers. Please click here to view a larger version of this figure.
{kind=link}
To create degradable gel particles on the micron-scale, a reactive microfluidics method is described in which precursor polymer solutions are simultaneously mixed and emulsified using a soft lithography-templated microfluidic chip design, enabling the formation of mixed reactive polymer droplets that subsequently gel in situ to form gel microparticles with sizes templated by the emulsion (Figure 2)31,32.

Figure 2: Schematic of gel microparticle formation via reactive microfluidics. (A,B) Hydrazide and aldehyde-functionalized polymer solutions (in water or aqueous buffer) are fed by syringe pump into separate reservoirs that are connected downstream across a zig-zag series of channels designed to create a pressure gradient preventing backflow. The polymers are then mixed just before being sheared by paraffin oil flowing from both sides (also driven by a syringe pump) and forced through a nozzle, resulting in flow-focusing production of aqueous (polymer solution) droplets in a continuous paraffin oil phase (see (B) for an illustration of the nozzle area and the droplet formation process). An additional two paraffin oil inlets are positioned after the nozzle to further separate the droplets in the collection channel to allow for complete gelation prior to particle removal from laminar flow, after which the resulting microparticulate gels are collected in a magnetically stirred beaker; (C) Picture of droplet generation process at the nozzle (note that hydrazide polymer is labeled as blue to illustrate mixing)
To create degradable gel particles on the nanoscale, a thermally-driven reactive self-assembly method is described in which a solution of one of the reactive precursor polymers (the "seed" polymer) is heated above its LCST to form a stable nanoaggregate that is subsequently crosslinked by the addition of the complementary reactive precursor polymer (the "crosslinking" polymer); the resulting hydrazone crosslinked nanogel has a size templated directly by the nanoaggregate (Figure 3)28.

Figure 3: Schematic of nanogel formation via thermally-driven reactive self-assembly. An aqueous solution containing the (thermoresponsive) hydrazide-functionalized polymer is heated above its lower critical solution temperature to create a stable uncrosslinked nanoaggregate. Following, an aldehyde-functionalized polymer is added to crosslink the nanoaggregate via hydrazone bond formation and thus stabilize the nanogel particle upon cooling below the LCST. Please click here to view a larger version of this figure.
{kind=link}
To create degradable nanofibers, a reactive electrospinning technique is described in which a double barrel syringe equipped with a static mixer at its outlet (as used for making bulk hydrogels) is attached to a standard electrospinning platform (Figure 4)33.

Figure 4: Schematic of hydrogel nanofiber formation via reactive electrospinning. A double barrel syringe with a static mixer (loaded as described for bulk hydrogels but also including a fraction of high molecular weight poly(ethylene oxide) as an electrospinning aid) is mounted on a syringe pump, with the needle at the end of the syringe connected to a high voltage power supply. Hydrazone crosslinking occurs during the fiber spinning process so that when the stream hits the collector (either aluminum foil or a rotating aluminum disk) the nanofibrous morphology is maintained. Please click here to view a larger version of this figure.
{kind=link}
The application of such methods for creating degradable smart hydrogel networks is demonstrated in this protocol using either PNIPAM or POEGMA as the polymer of interest; however, the basic approaches described can be translated to any water-soluble polymer, albeit with suitable adjustments for viscosity and (in the case of the self-assembly nanogel fabrication method) the stability of the pre-polymer in forming the seed nanoaggregate.
Protocol
1. Synthesis of Hydrazide-functionalized Polymers
Note: The following specific recipe is provided for the PNIPAM-mimetic thermoresponsive POEGMA precursor polymer (PO10) with 30 mol% hydrazide functionalization. PNIPAM and POEGMA precursor polymers with different phase transition temperatures can be prepared using this same general method but modifying the type and ratio of the core monomers used (see section 1.2 for modifications for various POEGMA polymers)21,25,27.
- Weigh out 37 mg of 2,2'-azobis(2-methylpropionate) (AIBMe, initiator), 3.1 g of diethylene glycol methacrylate (M(EO)2MA), 0.9 g of oligoethyleneglycol methacrylate (OEGMA475, 475 g/mol n=7-8 ethylene oxide repeat units), 523 µL of acrylic acid (AA, comonomer), and 7.5 µL of thiolglycolic acid (TGA, chain transfer agent) into a 20 mL glass scintillation vial.
- For PO0 (room temperature transition temperature POEGMA), use 4.0 g of M(EO)2MA (no OEGMA475). For PO100 (no transition temperature POEGMA), use 4.0 g of OEGMA475 (no M(EO)2MA).
Note: Intermediate phase transition temperatures can be achieved based on the use of intermediate mixtures of M(EO)2MA and OEGMA475, according to Lutz et al.23 - Dissolve all reagents in dioxane (5 mL/g total monomer) in a round bottom flask with one or more necks.
- Purge the reaction with nitrogen (UHP grade) flow for 30 min.
- Once purged, place the flask in a pre-heated oil bath maintained at 75 °C for 4 h under nitrogen and 400 rpm magnetic stirring.
- After 4 h, remove the solvent using a rotary evaporator set to 50 °C and 200 rpm.
- Dissolve the resulting polymer product in 150 mL of deionized water.
- Add adipic acid dihydrizide (ADH) at a five-fold molar excess to the number of AA residues incorporated into the polymer (in this example, AA comprises 29 mol% of the monomer units in the polymers produced, as per conductometric titration).
- Adjust the pH of the solution to pH 4.75 using 0.1 M HCl.
- Once the pH has stabilized, add N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide (EDC) at a 5-fold molar excess to the number of AA residues present).
- Maintain the reaction pH 4.75 with dropwise addition of 0.1 M HCl over 4 h.
- Leave the reaction to stir overnight.
- Pour the product solution into three ~30 cm long dialysis tubes (3500 Da molecular weight cut-off, 1 inch thickness), using a funnel to minimize spillage. Use a pinch clamp to close the bottom of the tube prior to filling by folding a small (~2 cm) segment of the tube to improve the clamp integrity; repeat at the top (pressing to remove air bubbles) when filling is complete. Place the tubes inside a 100-fold excess volume of deionized water and leave for at least 6 h, fully replacing the water over six cycles of dialysis to achieve desired purity.
- Lyophilize the dialyzed sample to obtain a final dried polymer product.
2. Synthesis of Aldehyde-functionalized Polymers
- Synthesis of Aldehyde-Precursor Monomer N-(2,2-Dimethoxyethyl) Methacrylate (DMEMA)
- Place 200 mL of a 20% w/v NaOH solution into a 500 mL 3 neck round-bottom flask.
- Cool the solution in an ice bath and maintain a temperature of 0 °C with ice during the reaction.
- Add 50 mL of aminoacetyl aldehyde dimethyl acetal to the cooled NaOH solution.
- Add in 0.1 g of TEMPO ((2,2,6,6-Tetramethylpiperidin-1-yl)oxyl) and stir at 400 rpm using a magnetic stir bar until the TEMPO fully dissolves.
- Add 48 mL of methacryloyl chloride dropwise using a burette over 2 h.
- After 2 h, cover the reaction vessel with aluminum foil and leave to stir overnight.
- Extract the product by adding the reaction product to 75 mL of petroleum ether in a 1 L separation funnel, shaking, degassing, and discarding the top layer.
- Repeat step 2.1.7 three times by adding the bottom layer product from each extraction step as the raw product to the next extraction cycle.
- Remove the final bottom layer product and transfer to a 100 mL beaker.
- Add ~5 g magnesium sulfate (Mg2SO4) to beaker with monomer until a "snow globe" effect is observed.
- Filter through a 100 mL Buchner funnel to remove the Mg2SO4.
- Rinse the beaker twice with ~75 mL of tert-butyl methyl ether, pouring the rinse solution through the funnel each time.
- Transfer the product to a 500 mL round-bottom flask and evaporate the solvent using a rotary evaporator at room temperature 200 RPM to collect the final product.
- Synthesis of Aldehyde-Functionalized Polymers
Note: The following specific recipe is provided for the PNIPAM-mimetic POEGMA precursor polymer (PO10) with 30 mol% aldehyde functionalization. PNIPAM and POEGMA precursor polymers with different phase transition temperatures can be prepared using the same general method but modifying the type and ratio of the core monomers used (see section 1.2 for modifications for various POEGMA polymers)21,25,27.- Weigh out 37 mg of 2,2'-azobis(2-methylpropionate) (AIBMe), 3.10 g of diethylene glycol methacrylate M(EO)2MA, 0.1 g of oligo ethyleneglycol methacrylate (OEGMA475, 475 g/mol, n=7-8 ethylene oxide repeat units), 1.30 g of N-(2,2-dimethoxyethyl) acrylamide (DMEMA) and 7.5 µL of thiolglycolic acid (TGA) into a 20 mL glass scintillation vial.
- For PO0 (room temperature transition temperature POEGMA), use 4.0 g of M(EO)2MA (no OEGMA475). For PO100 (no transition temperature POEGMA), use 4.0 g of OEGMA475 (no M(EO)2MA).
Note: Intermediate phase transition temperatures can be achieved based on the use of intermediate mixtures of M(EO)2MA and OEGMA475, according to Lutz et al.23 - Dissolve all reagents in dioxane (5 mL/g total monomer) in a round bottom flask with one or more necks.
- Purge the reaction with nitrogen (UHP grade) flow for 30 min.
- Once purged, place flask in a pre-heated oil bath maintained at 75 °C for 4 h under nitrogen and 400 rpm magnetic stirring.
- After 4 h, remove the solvent using a rotary evaporator set to 50 °C and 200 rpm.
- Dissolve the resulting polymer product in 100 mL of deionized H2O.
- Add 50 mL of 1 M HCl into the dissolved solution and stir under magnetic stirring (400 RPM) for 24 h to fully hydrolyze the acetal functionalities in DMEMA.
- After reaction completion, transfer the polymer solution into dialysis tubing, as per step 1.13.
- Lyophilize the dialyzed sample to obtain a final dried polymer product.
3. Fabrication of Hydrazone Crosslinked Bulk Hydrogels
- Dissolve hydrazide and aldehyde functionalized polymers separately in 10 mM phosphate buffered saline (PBS), or any desired aqueous buffer, to create solutions of desired concentrations.
Note: Mass concentrations between 5-40 wt% are typically used, with gelation at lower concentrations possible if higher functional group fractions are present on the polymers. - Using a single barrel syringe to transfer the solutions, load each precursor solution (~1 mL each) into separate barrels of a double barrel syringe (2.5 mL volume, 1:1 ratio syringe) attached to a static mixer (1.5" length) and (optionally) a syringe (typically 18 G, 1.5” length for in vitro studies) and (optionally) a syringe (typically 18 G, 1.5” length for in vitro studies).
- Prepare molds of desired thickness, shape, and diameter by punching holes into a silicone rubber sheet.
Note: In a typical experiment, a standard punch set is used to punch a 7 mm diameter cylindrical hole inside a 1/16" thick silicone rubber sheet (total volume of reservoir ~300 µL). - Mount the silicone mold on a standard glass microscope slide such that the holes punched in the mold are completely backed by glass. A 0.1 M HCl wash of the glass is recommended but not required prior to mounting of the silicone mold.
- Co-extrude the double barrel syringe contents through the static mixer to completely fill (or slightly overfill, with a meniscus at the top) the silicone mold.
Note: Multiple samples can be prepared during one extrusion sample provided the gelation time is on the same order of magnitude or longer than the total time required to fill multiple molds. - Place another standard glass microscope slide on top of the mold and wait for the gelation to complete.
Note: The standard recipes described in the synthesis section gel within <1 minute; slower gelation times (and thus longer required wait times) are observed at lower functional group densities, lower polymer concentrations and/or higher fractions of OEGMA475 relative to M(EO)2MA (for POEGMA hydrogels). - Remove the top microscope slide and use a spatula to push the hydrogel away from the silicone rubber mold.
- Lift the mold from lower microscope slide to recover the hydrogels for further testing.
4. Fabrication of Hydrazone Crosslinked Gel Microparticles
- Fabrication of Microfluidic Chip
- Dehydrate a silicon wafer (D = 76.2 mm, 380 µm thickness, P-doped, <100> orientation) by heating on a hotplate at 200 °C for 5 min.
- Center the wafer on a spin coater and coat a ~100 µm thick layer of SU-8 100 photoresist by applying ~7 mL of SU-8 resist, ramping the spin speed up to 3000 rpm at a rate of 500 rpm/s, and then holding the speed at 3000 rpm for 30 seconds.
- Pre-bake the coating at 65 °C for 10 min and then soft-bake the coating at 95 °C for 30 min.
- Print a photomask on a transparency with the microfluidic pattern defined by Figure 2A, such that the transparent sections are the desired pattern of the polymerized photoresist layer.
- Insert the photoresist-coated silicon wafer and the photomask in a mask aligner and expose the wafer to 365 nm light for 95 s (6.5 W exposure power).
- Bake the patterned wafer for 10 min at 95 °C, first by placing it on a hotplate at 65 °C and subsequently heating the hotplate to 95 °C at 10 °C/min.
- Remove the wafer from the hotplate and place in a 500 mL beaker containing 100 mL SU-8 developer for at least 10 min, swirling the wafer slowly in the solution throughout to remove non-exposed photoresist. After 10 min, rinse the patterned wafer with isopropanol and dry with air. Store the patterned wafer in a cool, dry environment away from light when not in use for soft lithography replica molding.
- Place the patterned microfluidic mold in a Petri dish. Position ~10 mm lengths of L/S 13 silicone tubing on the inlets and outlets of the chip.
- Pour ~10 mL of poly(dimethyl siloxane) (PDMS; prepared by mixing Silicone Elastomer Base and Silicone Elastomer Curing Agent in a 10:1 ratio) on top of the chip, carefully avoiding incorporating any PDMS within the placed silicone tubing.
- Place the Petri dish in a vacuum chamber for ~10 min to remove air bubbles persisting in and around the patterned structure during curing.
- Cure the PDMS by placing the Petri dish containing the patterned mold and uncured PDMS on a hotplate at 85 °C for 2-3 h.
- Carefully peel off the cured PDMS from the patterned silicon wafer to expose the soft lithographic patterned PDMS replica of the microfluidic mold.
- Place the patterned PDMS and a glass slide upside-down in a high-power plasma cleaner with an air feed. Apply the plasma at 200 mTorr and 45 W for 90 s to bond the PDMS to the glass slide and create the final microfluidic chip.
- Synthesis of Gel Microparticles
- Prepare hydrazide-functionalized PNIPAM (PNIPAM-Hzd) by dissolving NIPAM (4.5g), acrylic acid (0.5 g - 15 mol% total monomer), thioglycolic acid (TGA, 80 µL), and 2,2-azobisisobutyric acid dimethyl ester (AIBME, 0.056 g) in 20 mL of anhydrous ethanol and subsequently following steps 1.4-1.14 to complete the synthesis, although changing the reaction temperature to 56 °C in step 1.5.
- Prepare aldehyde-functionalized PNIPAM (PNIPAM-Ald) by dissolving NIPAM (4 g), N-(2,2-dimethoxyethyl)methacrylate (DMEMA, 0.95 g - 13.4 mol% total monomer), thioglycolic acid (TGA, 80 µL), and 2,2-azobisisobutyric acid dimethyl ester (AIBME, 0.056 g) in 20 mL of ethanol and subsequently following steps 2.2.4-2.2.10 to complete the synthesis, although changing the reaction temperature to 56 °C in step 2.2.5.
- Dissolve PNIPAM-Hzd and PNIPAM-Ald at 6 wt% in deionized water and load into separate standard 5 mL syringes.
- Dissolve 1 wt% nonionic surfactant (e.g., Span 80) in heavy paraffin oil and load the solution into a standard 60 mL syringe.
- Connect the two precursor polymer solution syringes individually to the two separate polymer inlet channels on the microfluidic chip and the paraffin oil solution to the oil inlet channel on the microfluidic chip via 1/32” ID silicone tubing (~30 cm length per inlet, ~45 cm length per outlet).
- Using two seperate infusion syringe pumps (one for the oil upstream, one for the oil added after the nozzle), deliver the oil into the chip at a flow rate between 1.1 mL/h and 5.5 mL/h without starting the polymer flow to prime the chip and ensure the chip is defect-free and operational (typically maintained over a 30 min period).
- Using a separate infusion syringe pump, deliver each of the aqueous polymer solutions to the chip at a flow rate of 0.03 mL/h.
- Following an initial stabilization period to ensure that the flow has equilibrated and uniform particles are formed (30 min - 1 h), collect the particles in a magnetically stirred round bottom flask.
- Collect the particles until all oil is consumed (12-55 h, depending on flow). Stop the syringe pumps and, if desired, immediately pump water in place of the precursor polymer solutions through the chip to clean. However, given the rapid in situ gelation of these materials when the flow is stopped, it is recommended to use a new chip for each separate experiment.
- Turn off the magnetic stirring and allow the gel microparticles to settle. Decant off all available paraffin oil using a pipette.
- To remove the remaining paraffin oil, wash the gel microparticles with pentane (applied at a volume of 10 mL for every 0.5 mL of microparticle volume), vigorously mix the emulsion for ~ 1 minute, allow the gel microparticles to re-settle for ~1-2 hours, and decant off the residual organic phase using a pipette. Repeat at least 5 times to ensure full paraffin oil removal.
- Resuspend the gel microparticles in 10 mL deionized water inside a 20 mL glass scintillation vial and purge the vial with nitrogen overnight to remove any residual pentane.
5. Fabrication of Hydrazone Crosslinked Nanogels
- Dissolve stock solutions of PNIPAM-Hzd (1 w/v%) and PNIPAM-Ald (1 w/v%) in deionized water. Prepare PNIPAM-Hzd and PNIPAM-Ald as described in sections 4.2.1 and 4.2.2, respectively.
- Heat a 5 mL aliquot of the PNIPAM-Hzd stock solution to 70˚C using an oil bath under magnetic stirring (350 RPM) inside a 20 mL glass scintillation vial.
NOTE: The solution should become opaque (i.e. the temperature exceeds the lower critical solution temperature of PNIPAM-Hzd), but no visible precipitate should be formed. - Add a 0.25 mL aliquot of PNIPAM-Ald (5-20 wt% of the mass of PNIPAM-Hzd present in the seed solution) drop-wise into the heated PNIPAM-Hzd solution over a period of 5-10 s.
- Continue mixing the solution in the scintillation vials for an additional 15 minutes, after which remove the sample from the oil bath and allow the product to cool to room temperature overnight.
- Dialyze the resulting nanogels over 6x6 hour cycles (using a 3500 kDa MWCO dialysis membrane) against deionized water to remove any non-crosslinked polymer. If desired, lyophilize for storage.
6. Fabrication of Hydrazone Crosslinked Nanofibers
- Prepare hydrazide-functionalized POEGMA (POEGMA-Hzd) by dissolving 37 mg dimethyl 2,2'-azobis(2-methylpropionate) (AIBMe), 4.0 g oligoethyleneglycol methacrylate (OEGMA475, 475 g/mol, n=7-8 ethylene oxide repeat units), and 0.25 g acrylic acid (AA) in 20 mL dioxane and following steps 1.3-1.14 to complete the synthesis.
- Prepare aldehyde-functionalized POEGMA (POEGMA-Ald) by dissolving 50 mg dimethyl 2,2'-azobis(2-methylpropionate) (AIBMe), 4.0 g oligoethyleneglycol methacrylate (OEGMA475, 475 g/mol, n=7-8 ethylene oxide repeat units), and 0.60 g N-(2,2-dimethoxyethyl) methacrylate (DMEMA) in 20 mL dioxane and following steps 2.2.3-2.2.10 to complete the synthesis.
- Dissolve POEGMA-Hzd (15 wt%) and POEGMA-Ald (15 wt%) in separate deionized water solutions.
- Dissolve poly (ethylene oxide) (PEO, Mw=600x103 g/mol, 5 wt%) in deionized water. Mix 1 mL of the PEO solution with each reactive POEGMA solution prepared in step 6.3 to create final precursor solutions of 7.5 wt% POEGMA precursor polymer and 2.5 wt% PEO.
- Load the two solutions into separate barrels of the same double-barrel syringe described in section 3 (also including the 1.5” static mixer) and mount the double barrel syringe on an infusion syringe pump.
- Attach a static mixer and a blunt-tip 18G needle to the double-barrel syringe.
- Connect a high-voltage power supply to the blunt-tip needle, grounded at the collector.
NOTE: Collectors consist either of a 10 mm x 10 mm square of aluminum foil or a ~10 mm diameter aluminum disk spinning at a rate of 200 rpm, both mounted perpendicular to the needle at a distance of 10 cm from the end of the needle. - Start the syringe pump at a rate of 0.48 mL/h and, simultaneously, switch on a high voltage of 8.5 kV to perform the electrospinning and create nanofibers.
- Continue electrospinning as desired to make scaffolds of different thicknesses or until the inlet solutions are exhausted.
- To remove the PEO electrospinning aid, soak the collected scaffolds for 24 h in deionized water.
Results
Bulk hydrogels extruded from a double barrel syringe into a silicone mold conform to the dimensions of the mold and become free-standing upon mold removal; gelation typically occurs seconds to minutes following co-extrusion depending on polymer precursors used. Typical characterization via swelling (measured gravimetrically using a cell culture insert to easily remove the hydrogel from the swelling solution), thermoresponsivity (measured using the same technique but cycling the incubation temperature above and below the phase transition temperature), degradation (measured using the same technique but over longer time periods), and shear or compressive modulus (measured using 2 mm thick and 7 mm diameter molded samples) demonstrates the tunability of the hydrogel responses depending on the chemistry of the precursor polymer (specifically, for POEGMA, the ratio of short to long chain OEGMA monomers used to prepare the hydrogel), the mole fraction of functional groups on the precursor polymers, and the concentration of those precursor polymers (Figure 5)27.
Microfluidics leads to the formation of well-defined gel microparticles on the size scale of 25-100 µm, with the size controllable based on the flow rates of the oil and/or the combined aqueous polymer phases (Figure 6A)31. Hot stage optical microscopy confirms that the gel microparticles maintain the thermoresponsive nature of the bulk hydrogels, showing reversible temperature-dependent swelling-deswelling with only a slight hysteresis on cycle 1 (attributable to irreversible hydrogen bond formation between neighboring amide groups in the collapsed state34) consistent with that observed in bulk PNIPAM hydrogels (Figure 6B)32. Furthermore, the gel microparticles degrade back to their oligomeric precursors over time, enabling renal clearance (Figure 6C)32.
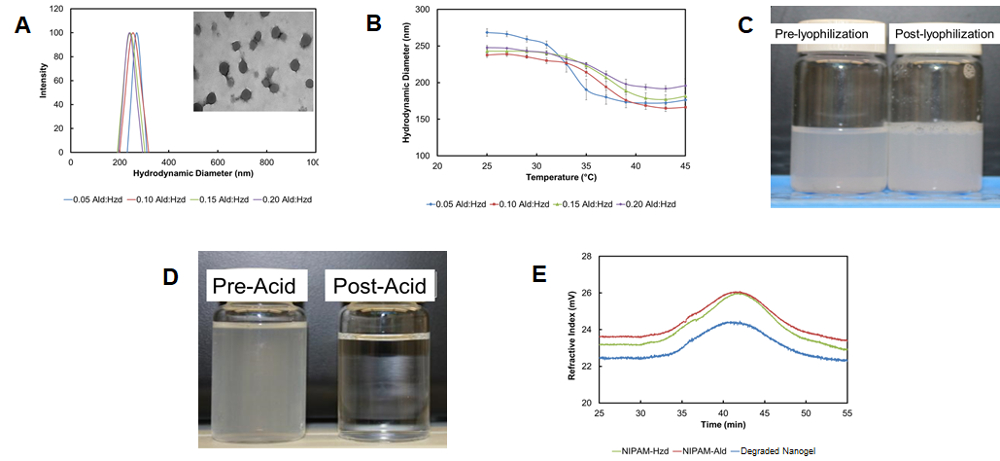
Self-assembly driven by the nanoaggregation of a hydrazide-functionalized PNIPAM polymer in a heated solution followed by crosslinking with an aldehyde-functionalized PNIPAM polymer results in highly monodisperse nanogels (polydispersity <0.1) on the size range of 180-300 nm, depending on the process conditions used (Figure 7A)28. The nanogels retain the typical thermoresponsive behavior of conventional free-radical crosslinked PNIPAM nanogels, with lower degrees of thermal deswelling observed as more cross-linking polymer was added (Figure 7B). The nanogels can be lyophilized and redispersed without a change in particle size (Figure 7C) and degrade over time via hydrolysis to re-form the oligomeric precursor polymers used to formulate the nanogels (Figure 7D).
Reactive electrospinning creates a nanofibrous hydrogel structure (Figure 8A), with nanofiber diameters on the order of ~300 nm achievable without visible electrosprayed particles present33. Soaking the POEGMA-based nanofibers in water results in rapid hydration (roughly two orders of magnitude faster than achieved with a bulk gel of the same composition, Figure 8B) but maintains the nanofibrous morphology over 8-10 weeks prior to hydrolytic degradation at physiological conditions; faster degradation is observed in acid-catalyzed environments, as expected due to the potential for acid-catalyzed hydrazone bond degradation (Figure 8C). The nanofibrous structures are also mechanically robust in both the dry and swollen states over multiple cycles, enabling easy handling and repetitive straining (Figure 8D).

Figure 5: Properties of in situ-gelling bulk degradable thermoresponsive hydrogels. (A) Representative POEGMA gel network microstructures and bulk hydrogel images with corresponding gelation times as a function of the mole % incorporation of OEGMA475 in the precursor polymers; (B-C) Storage modulus of PO100 hydrogels by varying (B) precursor polymer concentration and (C) mole % functional group incorporation per precursor polymer; (D-F) Physiochemical properties of POEGMA hydrogels as a function of OEGMA475 mole % incorporation: (D) storage modulus (E) degradation profile in 1 M HCl, and (F) volume phase transition temperature in response to temperature change over the range 20-60 °C. All error bars represent the standard deviation of n=4 replicate measurements. Adapted from reference27 with permission from Elsevier. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Properties of degradable gel microparticles from reactive microfluidics. (A) Effect of paraffin oil flow rate on (purified) gel microparticle size in water; (B) Thermoresponsivity of purified gel microparticles in water following a single thermal cycle above and below the volume phase transition temperature; (C) Visual assessment (photos) and gel permeation chromatography traces (graph) confirming degradation of gel microparticles back to their precursor polymer components (here, in 1 M HCl to facilitate accelerated degradation on the time scale of imaging); scale bar = 100 µm. Adapted from reference32. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Properties of degradable nanogels from reactive self-assembly. (A) Particle size distributions of nanogels prepared with different aldehyde:hydrazide polymer mass ratios from dynamic light scattering (inset: transmission electron micrograph confirming the spherical nature of the nanogels); (B) Thermosensitivity of self-assembled particles as a function of the mass ratio between aldehyde:hydrazide polymer used to prepare the nanogels (from dynamic light scattering), with error bars representing the standard deviation of n=4 replicates; (C) Visual confirmation of the lack of nanogel aggregation both pre and post-lyophilization; (D) Visual confirmation of the acid-catalyzed degradation of nanogels (here in 1 M HCl for consistency with other studies above); (E) Gel permeation chromatograph traces of nanogel degradation products indicating their similarity to the hydrazide and aldehyde-functionalized precursor polymers. Adapted with permission from reference28. Copyright 2015, American Chemical Society. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: Properties of degradable nanofibers from reactive electrospinning. (A) Scanning electron microscopy images of nanofibers in the dry state (left), half dipped in water (middle, thin film), and fully soaked in water overnight (right, thick scaffold); (B) Swelling of nanofibrous hydrogel (red) relative to a bulk hydrogel (blue) of the same composition, with error bars representing the standard deviation of n=4 replicates; (C) Scanning electron microscopy and (inset) visual images tracking the acid-catalyzed degradation of nanofibers in 1M HCl; (D) Tensile cycling of dry (80 cycles, 20% elongation/cycle) and swollen (325 cycles, 10% elongation/cycle in 10 mM PBS) electrospun nanofibrous hydrogels. Figure modified from reference33 and reproduced with permission from the Royal Society of Chemistry. Please click here to view a larger version of this figure.
{kind=link}
Discussion
We have successfully applied all these fabrication techniques to multiple polymer systems using only slight variations of the methods described in detail above for PNIPAM and POEGMA; however, users of these protocols must be cognizant of the potential issues that can arise when other polymers are substituted into these processes. In particular, increasing the viscosity of the precursor polymers can negatively impact both the processibility (especially in the microfluidic method) as well as the efficiency of mixing of the two precursor polymers. In addition, the gelation time of the polymers must be controlled at a rate dependent on the morphology targeted in order to avoid premature gelation that serves to inhibit flow or prevent interdiffusion of the reactive pre-polymers, essential to form the desired homogeneous gel structures. The specific limitations of each strategy, as well as approaches we have used to adapt these approaches to address such limitations at each fabrication length scale, are described below.
Bulk hydrogels via double barrel syringe co-extrusion
Gelation time is the key variable to control to ensure the efficacy of the double barrel syringe technique for forming bulk hydrogels. Polymers that gel too fast upon contact (<1-2 s) can clog the static mixer, either stopping flow entirely or resulting in non-stoichiometric amounts of the two precursor polymers being extruded from the syringe. We have found that gelation times >5 s are preferable (although not required) for the use of this technique; this is particularly important if replicate hydrogels are being cast for physical or mechanical analysis to ensure that each hydrogel cast has the same composition. Gelation time can be easily altered by changing the density of reactive functional groups on one or both precursor polymers (lower functional group density leading to slower gelation) or changing the concentration of the precursor polymers used to form the gel (lower concentrations leading to slower gelation)21. Alternately, replacing the (more reactive) aldehyde group with the (less reactive) ketone group as the electrophile in the gelling pair significantly reduces the gelation time without significantly changing the composition of the resulting hydrogel35; polymers prepared with mixtures of aldehyde and ketone monomeric precursors can be used to tune the gelation time as desired without changing the concentration of precursor polymers used (and thus the mass percentage of solids in the resulting gel formed).
We would also note that the first hydrogel cast does not always have the same properties as subsequent hydrogels cast, an observation attributed to slight differences in the rate at which the contents of the two barrels actually reach the static mixer. As a result, we typically prime the double barrel syringe by extruding a small (<0.3 mL) fraction of gel prior to initiating the casting process to minimize such variability. Finally, while not typically problematic when using oligomeric synthetic pre-polymers, the viscosity of one or more precursor polymer solutions can pose a challenge in the context of this technique, both in terms of facilitating flow using simple thumb depression as well as promoting effective mixing within the static mixer. However, somewhat surprisingly, even precursor polymer solutions with sharply different viscosities still form relatively homogeneous hydrogels using the static mixer attachments described in the parts list (e.g., PNIPAM with a high molecular weight carbohydrate26), suggesting that concerns about inefficient mixing as a result of mis-matched viscosities may not be significant at least on the bulk scale. If required, the use of a syringe pump (instead of the thumb) to drive flow and/or the use of a larger gauge needle at the outlet can help overcome issues associated with extrudability in these systems.
Microscale hydrogels via reactive microfluidics
The key step associated with the microfluidics approach for gel microparticle fabrication is the priming of the microfluidics chip with the two reactive polymers. If the polymers are delivered with different pressures or at different rates into the chip, the differential pressure can drive the backflow of one precursor polymer solution into the reservoir (or at least toward the reservoir) of the other precursor polymer. This results in gelation upstream from particle formation, effectively blocking flow and thus requiring chip disposal. The torturous path imprinted between each reservoir and the mixing point creates a significant resistance to backflow; however, even a trained operator will occasionally gel a chip before a stable flow regime is achieved. Based on our experience, between 1-2 min is typically required to stabilize the flows following the initiation of droplet formation (over which time relatively polydisperse gel microparticles are produced); if no problems are observed within the first 5-10 minutes of operation, it is likely that several hours of continuous monodisperse particle production can be achieved. The use of precursor polymers with relatively well-matched viscosities as well as non-instantaneous gelation times (at least >15 s preferable) greatly assists in avoiding such problems and promoting the formation of stable flows.
Note that various flow rates ranging from 0.01-0.1 mL/h in the aqueous phase and 1.1-5.5 mL/h in the oil phase have been tested using this chip design, leading to the fabrication of particles on the size range of ~25-100 µm according to the shear applied at the flow-focusing junction; faster flow rates equate to higher shear and thus smaller particles formed31,32. Varying the oil flow rate while keeping the total aqueous flow rate low (~0.03 mL/h, as cited in the protocol) was found to be most efficient to control gel microparticle size without compromising either monodispersity or the lifetime of the device, both of which were observed to significantly decrease at the higher end of the cited total aqueous flow rates. Larger oil flow rates (>5.5 mL/h) to create smaller particles are possible, but increased the risk of chip delamination (a common limitation encountered with plasma-bonded PDMS microfluidic chips). Bonding the chips using another method may enable faster flow rates and thus smaller gel microparticle production, a strategy we are currently exploring. Decreasing the size of the nozzle may also help to reduce the size of the microparticles that could be produced, albeit at a heightened risk of premature gelation prior to particle formation. Slower flow rates tended to lead to flow instabilities and thus higher polydispersities and an increased risk of chip gelation; this limitation could be overcome by using a multichannel microfluidic flow control system that has higher stability and higher resolution than the standard syringe pumps used in this protocol.
The choice of oil was critical to the success of this protocol, as heavier oils (favorable in terms of preventing gel microparticle agglomeration after collection) led to much less consistent particle formation at the nozzle than the light silicone oil reported in the protocol. We hypothesize this reduced reproducibility is a result of lower consistency of syringe pumping of heavier oils, leading to more variable shear at the mixing point. Avoiding gel microparticle aggregation in the collection flask was also a challenge, particularly immediately at the exit from the microfluidic device at which point in situ gelation was not complete and large numbers of available reactive functional groups were available to form bridges between colliding particles in the collection bath. This challenge is addressed by: increasing the length of the exit channel on the microfluidic chip itself, maintaining the gel microparticles in laminar flow for a longer period of time to promote more complete gelation; adding the side channels after the nozzle to feed more oil into the chip and thus better separate the gel microparticles in this post-mixing channel without affecting the shear fields at the nozzle itself or the particle production rate; and adding a magnetic mixer to the collection flask to avoid gel microparticle sedimentation and maintain a larger average separation between adjacent particles. While very slow gelling polymers would likely improve the device stability and minimize issues with priming, such systems also were observed to significantly increase the risk of gel microparticle aggregation, as a larger number of reactive functional groups remains unreacted (and thus able to form inter-particle bridges) over a longer period of time. As such, gelation times on the order of 15-60 s appear to be optimal for this technique: slow enough to enable priming but fast enough to ensure most reactive functional groups are consumed prior to the gel microparticles exiting the laminar flow channel into the collection flask.
Finally, removal of the templating oil is essential to ensure that the resulting particles maintain the smart properties anticipated based on the composition of the pre-polymers added and enable use of these particles in a biomedical context. The pentane washing procedure described was highly effective in this regard for general gel microparticle production. However, the application of this technique in a direct biomedical context (e.g., on-chip cell encapsulation) would require re-evaluation of this protocol. We have also explored the use of olive oil, suggested to be a more inert oil in the context of contacting cells36, as the dispersant. While particle formation was possible, the gel microparticle populations were significantly more polydisperse than could be achieved with mineral oil, at least with the current chip design. Thus, while the chip appears to be adaptable to both synthetic polymer and natural polymer gel microparticle formation31, a modified design may be required to exploit this technique more broadly across all possible material combinations.
Nanoscale hydrogels via reactive self-assembly
Nanogels have been formed using a very wide range of processing conditions, including different concentrations of seed polymer (0.5-2 wt%), different ratios of crosslinking:seed polymer (0.05-0.2), different temperatures (40-80 °C), different mixing speeds (200-800 rpm), and different heating times following the addition of the crosslinker polymer (2-60 min)28. In terms of concentrations, the trends observed are generally as would be predicted, as higher concentrations of seed polymer lead to larger nanogels and higher ratios of crosslinker:seed polymer lead to nanogels with higher crosslink densities and thus lower thermoresponsivities. It should be emphasized that increasing the seed polymer concentration too high ultimately leads to bulk aggregation as opposed to nanoaggregation, consistent with what is observed in the conventional free radical precipitation process for forming thermoresponsive nanogels3. Shorter heating times were also found to be favorable for forming smaller and more monodisperse particles. We hypothesize that holding the nanoaggregate at longer times at a temperature above the LCST one or both of the precursor polymers increases the probability of aggregation upon nanogel collision, with the increased hydrophobicity of the hydrazone bond relative to either the precursor aldehyde or hydrazide functional groups making this aggregation more likely as the degree of crosslinking achieved is increased. Ultimately, shorter heating times are favorable from a process perspective, as a monodisperse nanogel population can be formed in as little as 2 min after crosslinker polymer addition; 10 min was found to be the longest time that could consistently produce monodisperse nanogels while also allowing for the production of more highly crosslinked nanogels. Interestingly, the method is remarkably insensitive to mixing, with nearly identical particle sizes and particle size distributions resulting from mixing at different speeds or even scaling the process to larger volumes. While initially surprised by this result, it likely speaks to the primary role of thermodynamics in regulating nanogel production.
To achieve low polydispersities, the colloidal stability and the degree of hydration of the nanoaggregate appear to be the key variables. For example, nanoaggregates prepared using the more hydrophilic hydrazide-functionalized polymers as the seed as opposed to the less hydrophilic aldehyde-functionalized polymers lead to nanogels with significantly lower polydispersities. The difference between the experimental assembly temperature and the LCST of the seed polymer is also critical. Operating at a temperature just above the seed polymer LCST ((T - LCST) < 5 °C) offers the highest probability of monodisperse nanogel formation; operating well above the LCST creates more hydrophobic and collapsed nanoaggregates that are more likely to aggregate and less likely to crosslink, while operating below the LCST results in a relatively non-compact seed polymer that cannot be effectively or reproducibly crosslinked. For the best prediction of particle monodispersity, we recommend first performing a UV/vis scan to measure the onset LCST of the seed polymer and subsequently performing the self-assembly process at a temperature 1-2 °C above that LCST.
Note that nanogels produced using this method could be lyophilized and redispersed without any change in colloidal stability, often not possible for self-assembled structures and in our view attributable to our crosslinking stabilization method. We also anticipate that only the seed polymer needs to be thermoresponsive for this method to work; use of cross-linking polymers that are either non-responsive or responsive to other stimuli may further broaden the ultimate applicability of this technique. Finally, since the mixing of the two reactive precursor polymers is in this case passive as opposed to active, gelation time is much less important in terms of process control relative to the other fabrication strategies described. However, even in this technique, keeping the total crosslinking time <30 min is desirable to minimize the risk of particle aggregation.
Nanofibrous hydrogels via reactive electrospinning
Controlling the gelation time of the reactive pre-polymers is again essential to the success of gel nanofiber production. In particular, approximately matching the residence time of the precursor polymers in the static mixer (controlled by changing the flow rate of solution from the double-barrel syringe as well as the length and tortuosity of the static mixer) with the bulk gelation time of the precursor polymers is essential both to preserve spinnability as well as ensure effective crosslinking of the spun fibers between the needle and the collector. Faster gelation leads to ineffective Taylor cone development and thus poor spinnability, while slower gelation results in an aqueous solution instead of a gel hitting the collector, resulting in spreading and the ultimate formation of a thin film gel instead of nanofibers. Working at residence times slightly below the bulk gelation time has also been found to be effective (and indeed preferable to reduce the risk of needle clogging) since water evaporation as the solution is spun effectively concentrates the precursor polymers in the stream and thus accelerates gelation kinetics during the spinning process. In this same vein, operating at higher needle-to-collector distances (>10 cm) is generally favorable in this process, as shorter distances reduce the time available for water evaporation and thus require more stringent control over the relationship between residence time and gelation time in order to preserve a nanofibrous product.
Note that the use of PEO (or another high molecular weight and easily electrospun polymer) is essential in this protocol to promote nanofiber formation, as the short and highly branched POEGMA oligomers cannot alone reach an adequate degree of entanglement to induce electrospinning; instead, electrospray results at all process conditions tested for POEGMA-only formulations (although this may also have applications for making degradable gel particles using this same chemistry). A minimum PEO concentration of 1 wt% (1 MDa molecular weight) is required to maintain a fully nanofibrous morphology. Note that the PEO can be removed from the fibers following a simple soaking procedure (deionized water, 24 h) without disrupting the integrity of the nanofibrous network; in this way, PEO acts more as a transient electrospinning aid than an essential component of the final nanofibrous product. Note also that various types of collectors, including simple aluminum foil (to create thin layer hydrogels that can delaminate from the collector upon soaking) as well as a rotating aluminum disk (to create thicker scaffolds) can be used in conjunction with this same technique, provided the other process variables controlling the rate of gelation, the rate of electrospinning, and the rate of water evaporation during electrospinning remain unaltered.
Interestingly, depending on the method used to prepare the different morphologies, significant differences have been observed in the degradation times of hydrogels prepared from the same hydrogel precursors. For example, POEGMA nanofibrous hydrogels degrade slower than bulk POEGMA hydrogels with the same composition despite their significantly higher surface area and thus access to water to hydrolyze the hydrazone bonds. We relate these differences to the inherent contrasts between the described protocols in terms of the geometry of mixing the precursor polymers, which may lead to internal gel homogeneities and/or morphologies that are significantly different and/or the in situ concentration of polymer precursors on the same time scale as gelation, particularly relevant in electrospinning due to the simultaneous water evaporation and crosslinking observed in this process. While this may somewhat complicate the choice of the precursor polymers if one polymer is targeted for use in each protocol, it may also offer a technical opportunity in terms of making hydrogels with one chemical composition but very different physical properties.
Overall, the methods described provide a strategy for fabricating degradable (or at least renally clearable) analogues of thermoresponsive polymers on multiple length scales (bulk, micro, and nano) and with multiple types of internal structures (particles or fibers). Such protocols address the key barriers to the successful translation of conventionally-prepared synthetic thermoresponsive materials to the biomedical field: injectability and degradability. We are continuing to explore the application of such materials in both drug delivery and tissue engineering applications ranging from the physical targeting of cancers, the transport of drugs across the blood-brain barrier, the therapeutic delivery of proteins at the back of the eye, the directional growth of tissues, and the thermoreversible adhesion and differentiation of cells, among other applications.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Funding from the Natural Sciences and Engineering Research Council of Canada (NSERC), the NSERC CREATE-IDEM (Integrated Design of Extracellular Matrices) program, 20/20: NSERC Ophthalmic Biomaterials Research Network, and the Ontario Ministry of Research and Innovation Early Researcher Awards program is acknowledged.
Materials
| Name | Company | Catalog Number | Comments |
| Chemicals | |||
| 2,2 - azobisisobutryic acid dimethyl ester | Wako Chemicals | 101138 | |
| Di(ethylene glycol) methyl ether methacrylate (M(EO)2MA) | Sigma Aldrich | 447927 | 188.2 g/mol, n=2 ethylene oxide repeat units |
| Oligo (ethylene glycol) methyl ether methacrylate (OEGMA475) | Sigma Aldrich | 447943 | 475 g/mol, n=8-9 ethylene oxide repeat units |
| Acrylic acid (AA), 99% | Sigma Aldrich | 147230 | |
| Thioglycolic acid (TGA), 98% | Sigma Aldrich | T3758 | |
| Dioxane, 99% | Caledon Labs | 360481 | |
| Nitrogen, UHP grade | Air Liquide | Alphagaz1 765A-44 | |
| Adipic acid dihydrazide (ADH), 98% | Alfa Aesar | A15119 | |
| N'-ethyl-N-(3- dimethylaminopropyl)-carbodiimide (EDC, x%) | Carbosynth | FD05800 | |
| Hydrochloric acid (HCl), 37% | Sigma Aldrich | 320331 | |
| Sodium hydroxide (NaOH), 97% | Sigma Aldrich | 221465 | |
| Aminoacetyl aldehyde dimethyl acetal, 99% | Sigma Aldrich | 121967 | |
| 4-Hydroxy-TEMPO, 97% | Sigma Aldrich | 176141 | |
| Methacryloyl chloride,97x% | Sigma Aldrich | 523216 | |
| Petroleum ether, 95% | Sigma Aldrich | 32047 | |
| Magnesium sulfate, 99.5% | Sigma Aldrich | M7506 | |
| tert-Butyl methyl ether, >99.0% | Sigma Aldrich | 443808 | |
| Phosphate buffered saline | BioShop | PBS405.1 | 1x, pH 7.3-7.5 |
| N-isopropylacrylamide, 99% | J&K Scientific | 258717 | Recrystallized from 60% hexanes/40% toluene |
| Ethanol, anhydrous | Commerical Alchols | P016EAAN | |
| Span 80 | Sigma Aldrich | S6760 | |
| Heavy paraffin oil | Caledon Labs | 1326197 | |
| Pentane, reagent grade | Caledon Labs | 1/10/7800 | |
| Poly (ethylene oxide) average Mv 600,000 | Sigma Aldrich | 182028 | |
| Supplies essential for synthesis and hydrogel fabrication | |||
| Rotary evaporator | Heidolph | G3 | |
| Dialysis tubing (3500 Da molecular weight cut-off) | Spectrum Labs | 28170-166 | Vol/length= 6.4mL/cm |
| Double barrel syringe | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio |
| Static mixer | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio, 1.5" length |
| Silicone rubber sheet, 1/16" thickness | McMaster-Carr | 9010K12, 30A Durometer (Super Soft) | |
| Syringe pump | KD Scientific | KDS Legato 200 | Infuse Only Dual Syringe Pump |
| High voltage power supply | Spellman | 230-20R | 0 to 20 kV |
| Microfluidic Chip Fabrication | |||
| Silicon wafer | University Wafer | 2080 | D = 76.2 mm; 380 µm thickness; P-doped; <100> orientation |
| SU-8 100 | MicroChem | Y131273 | |
| SU-8 Developer | MicroChem | Y020100 | |
| Custom 2.5" spincoater | Built in-house | N/A | |
| Mask Aligner | KARL SUSS | MJB3 UV400 (with a 276 W lamp) | |
| Masterflex L/S 13 Silicone Tubing | Cole Parmer | OF-96400-13 | Peroxide-cured |
| Dow Corning Sygard 184 Silicone Elastomer Base | Ellsworth Adhesives | 4019862 | |
| Dow Corning Sygard 184 Silicone Elastomer Curing Agent | Ellsworth Adhesives | 4019862 | |
| High Power Plasma Cleaner | Harrick | PDC-002-HP | |
| Characterization Instruments | |||
| Mach 1 micromechanical tester | Biomomentum | LB007-EN | |
| Cellstar tissue culture 12 well plate | Greiner Bio-one | 665 180 | |
| Cell culture insert for 12 well plate | Corning | 08-771-12 | 8 µm pore size |
| Optical microscope | Olympus BX51 optical microscope | BX51 | |
| Temperature-controlled microscope stage | Linkam Scientific | THMS600 | |
| Gel permeation chromatograph (GPC) | Waters | 590 HPLC Pump | Waters Styragel columns (HR2, HR3, HR4; 30 cm x 7.8 mm (ID); 5 mm particles), Waters 410 refractive index detector |
| Dynamic light scattering (DLS) | Brookhaven | 90Plus Particle Size Analyzer | |
| Transmission electron microscopy (TEM) | TEMSCAN | JEOL 1200EX | Accelerating voltage 100 kV |
| Scanning electron microscopy (SEM) | Tescan | Vega II LSU | Accelerating voltage 10 kV |
| Microsquisher | CellScale Biomaterials Testing | MS-50M-01 |
References
- Heskins, M., Guillet, J. E. Solution Properties of Poly(N-isopropylacrylamide). J. Macromol. Sci. A. 2 (8), 1441-1455 (1968).
- Lutz, J. -F., Akdemir, Ö, Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over. J. Am. Chem. Soc. 128 (40), 13046-13047 (2006).
- Pelton, R. H., Chibante, P. Preparation of Aqueous Lattices with N-Isopropylacrylamide. Colloids Surf. 20 (3), 247-256 (1986).
- Palasis, M., Gehrke, S. H. Permeability of Responsive Poly(N-Isopropylacrylamide) Gel to Solutes. J. Controlled Release. 18 (1), 1-11 (1992).
- Kawaguchi, H., Fujimoto, K., Mizuhara, Y. Hydrogel Microspheres .3. Temperature-Dependent Adsorption of Proteins on Poly-N-Isopropylacrylamide Hydrogel Microspheres. Colloid Polym. Sci. 270 (1), 53-57 (1992).
- Okuyama, Y., Yoshida, R., Sakai, K., Okano, T., Sakurai, Y. Swelling Controlled Zero-Order and Sigmoidal Drug-Release from Thermoresponsive Poly(N-Isopropylacrylamide-Co-Butyl Methacrylate) Hydrogel. J. Biomater. Sci. Polym. Ed. 4 (5), 545-556 (1993).
- Snowden, M. J. The Use of Poly(N-Isopropylacrylamide) Lattices as Novel Release Systems. J. Chem. Soc. - Chem. Comm. (11), 803-804 (1992).
- Haraguchi, K., Takehisa, T., Ebato, M. Control of cell cultivation and cell sheet detachment on the surface of polymer/clay nanocomposite hydrogels. Biomacromolecules. 7 (11), 3267-3275 (2006).
- Lee, B., et al. Initiated chemical vapor deposition of thermoresponsive poly(N-vinylcaprolactam) thin films for cell sheet engineering. Acta Biomater. 9 (8), 7691-7698 (2013).
- Cole, M. A., Voelcker, N. H., Thissen, H., Griesser, H. J. Stimuli-responsive interfaces and systems for the control of protein-surface and cell-surface interactions. Biomaterials. 30 (9), 1827-1850 (2009).
- Feil, H., Bae, Y. H., Feijen, J., Kim, S. W. Molecular Separation by Thermosensitive Hydrogel Membranes. J. Membrane Sci. 64 (3), 283-294 (1991).
- Kim, J., Park, K. Smart hydrogels for bioseparation. Bioseparation. 7 (4-5), 177-184 (1998).
- Yamashita, K., Nishimura, T., Nango, M. Preparation of IPN-type stimuli responsive heavy-metal-ion adsorbent gel. Polym. Adv. Tech. 14 (3-5), 189-194 (2003).
- Ziolkowski, B., Czugala, M., Diamond, D. Integrating stimulus responsive materials and microfluidics: The key to next-generation chemical sensors. J. Intelligent Mater. Syst. Struct. 24 (18), 2221-2238 (2013).
- Zhang, Y., Kato, S., Anazawa, T. A flap-type hydrogel actuator with fast responses to temperature. Smart Mater. Struct. 16 (6), 2175-2182 (2007).
- Suzuki, D., Taniguchi, H., Yoshida, R. Autonomously Oscillating Viscosity in Microgel Dispersions. J. Am. Chem. Soc. 131 (34), 12058-12059 (2009).
- Schild, H. G. Poly(N-isopropylacrylamide): Experiment, Theory and Application. Prog. Polym. Sci. 17, 163-249 (1992).
- Oh, J. K., Min, K., Matyjaszewski, K. Preparation of poly (oligo (ethylene glycol) monomethyl ether methacrylate) by homogeneous aqueous AGET ATRP. Macromolecules. 39 (9), 3161-3167 (2006).
- Vihola, H., Laukkanen, A., Tenhu, H., Hirvonen, J. Drug Release Characteristics of Physically Cross-Linked Thermosensitive Poly(N-vinylcaprolactam) Hydrogel Particles. J. Pharm. Sci. 97 (11), 4783-4793 (2008).
- Zhang, L. F., Liang, Y., Meng, L. Z. Thermo-sensitive amphiphilic poly(N-vinylcaprolactam) copolymers: synthesis and solution properties. Polym. Adv. Tech. 21 (10), 720-725 (2010).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable and tunable poly(ethylene glycol) analogue hydrogels based on poly(oligoethylene glycol methacrylate). Chem. Comm. 50 (25), 3306-3309 (2014).
- Lutz, J. -F. Polymerization of oligo (ethylene glycol)(meth) acrylates: toward new generations of smart biocompatible materials. J. Polym. Sci. A. 46 (11), 3459-3470 (2008).
- Lutz, J. -F., Hoth, A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules. 39 (2), 893-896 (2006).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Patenaude, M., Hoare, T. Injectable, Degradable Thermoresponsive Poly(N-isopropylacrylamide) Hydrogels. ACS Macro Lett. 1 (3), 409-413 (2012).
- Patenaude, M., Hoare, T. Injectable, Mixed Natural-Synthetic Polymer Hydrogels with Modular Properties. Biomacromolecules. 13 (2), 369-378 (2012).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable poly(oligoethylene glycol methacrylate)-based hydrogels with tunable phase transition behaviours: Physicochemical and biological responses. Acta Biomater. 10 (10), 4143-4155 (2014).
- Sivakumaran, D., Mueller, E., Hoare, T. Temperature-Induced Assembly of Monodisperse, Covalently Cross-Linked, and Degradable Poly(N-isopropylacrylamide) Microgels Based on Oligomeric Precursors. Langmuir. 31, 5767-5778 (2015).
- Bakaic, E., Smeets, N. M. B., Dorrington, H., Hoare, T. "Off-the-shelf" thermoresponsive hydrogel design: tuning hydrogel properties by mixing precursor polymers with different lower-critical solution temperatures. RSC Adv. 5 (42), 33364-33376 (2015).
- Bulpitt, P., Aeschlimann, D. New strategy for chemical modification of hyaluronic acid: Preparation of functionalized derivatives and their use in the formation of novel biocompatible hydrogels. J. Biomed. Mater. Res. 47 (2), 152-169 (1999).
- Kesselman, L. R. B., Shinwary, S., Selvaganapathy, P. R., Hoare, T. Synthesis of Monodisperse, Covalently Cross-Linked, Degradable "Smart" Microgels Using Microfluidics. Small. 8 (7), 1092-1098 (2012).
- Sivakumaran, D., Mueller, E., Hoare, T. Microfluidic production of degradable thermoresponsive microgels based on poly(N-isopropylacrylamide). Soft Matter. , (2016).
- Xu, F., Sheardown, H., Hoare, T. Reactive Electrospinning of Degradable Poly(oligoethylene glycol methacrylate)-Based Nanofibrous Hydrogel Networks. Chem. Comm. 52 (7), 1451-1454 (2016).
- Troll, K., et al. The collapse transition of poly(styrene-b-(N-isopropyl acrylamide)) diblock copolymers in aqueous solution and in thin films. Colloid Polym. Sci. 286 (8), 1079-1092 (2008).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Kelly, T. A., Felder, M. S., Ollar, R. A. Inducing Apoptosis in a Mammalian Cell by Contacting with Paraffin or Agar. US Patent. , 6,274,377 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved