
Method Article
Structural Studies of Macromolecules in Solution using Small Angle X-Ray Scattering
In This Article
Summary
Here, we present how Small Angle X-Ray Scattering (SAXS) can be utilized to obtain information on low-resolution envelopes representing the macromolecular structures. When used in conjunction with high-resolution structural techniques such as X-Ray Crystallography and Nuclear Magnetic Resonance, SAXS can provide detailed insights into multidomain proteins and macromolecular complexes in-solution.
Abstract
Protein-protein interactions involving proteins with multiple globular domains present technical challenges for determining how such complexes form and how the domains are oriented/positioned. Here, a protocol with the potential for elucidating which specific domains mediate interactions in multicomponent system through ab initio modeling is described. A method for calculating solution structures of macromolecules and their assemblies is provided that involves integrating data from small angle X-ray scattering (SAXS), chromatography, and atomic resolution structures together in a hybrid approach. A specific example is that of the complex of full-length nidogen-1, which assembles extracellular matrix proteins and forms an extended, curved nanostructure. One of its globular domains attaches to laminin γ-1, which structures the basement membrane. This provides a basis for determining accurate structures of flexible multidomain protein complexes and is enabled by synchrotron sources coupled with automation robotics and size exclusion chromatography systems. This combination allows rapid analysis in which multiple oligomeric states are separated just prior to SAXS data collection. The analysis yields information on the radius of gyration, particle dimension, molecular shape and interdomain pairing. The protocol for generating 3D models of complexes by fitting high-resolution structures of the component proteins is also given.
Introduction
Cells contain intricate networks of proteins that act as molecular machines to carry out cellular functions such as signaling cascades and maintaining structural integrity. The ways in which these different components move and interact in three-dimensional space gives rise to the specific functions of the macromolecules. The importance of protein structure, dynamics, and interactions in determining function has provided the need for continually evolving, complex techniques to measure these properties. Of these, Nuclear Magnetic Resonance (NMR), X-Ray Crystallography (XRC) and more recently, Cryo-Electron Microscopy (CEM) provide high-resolution structural information. However, XRC and CEM yield structures of one of many biomolecular states and lack information about the dynamics of the protein structure, while 3D structure determination by NMR is typically limited to smaller globular proteins. One way to overcome these limitations is to utilize Small Angle X-Ray Scattering (SAXS) to generate molecular envelopes of large, multidomain, or complexed systems, and combine the high-resolution rigid macromolecular structures to elucidate the global architecture and dynamic features.
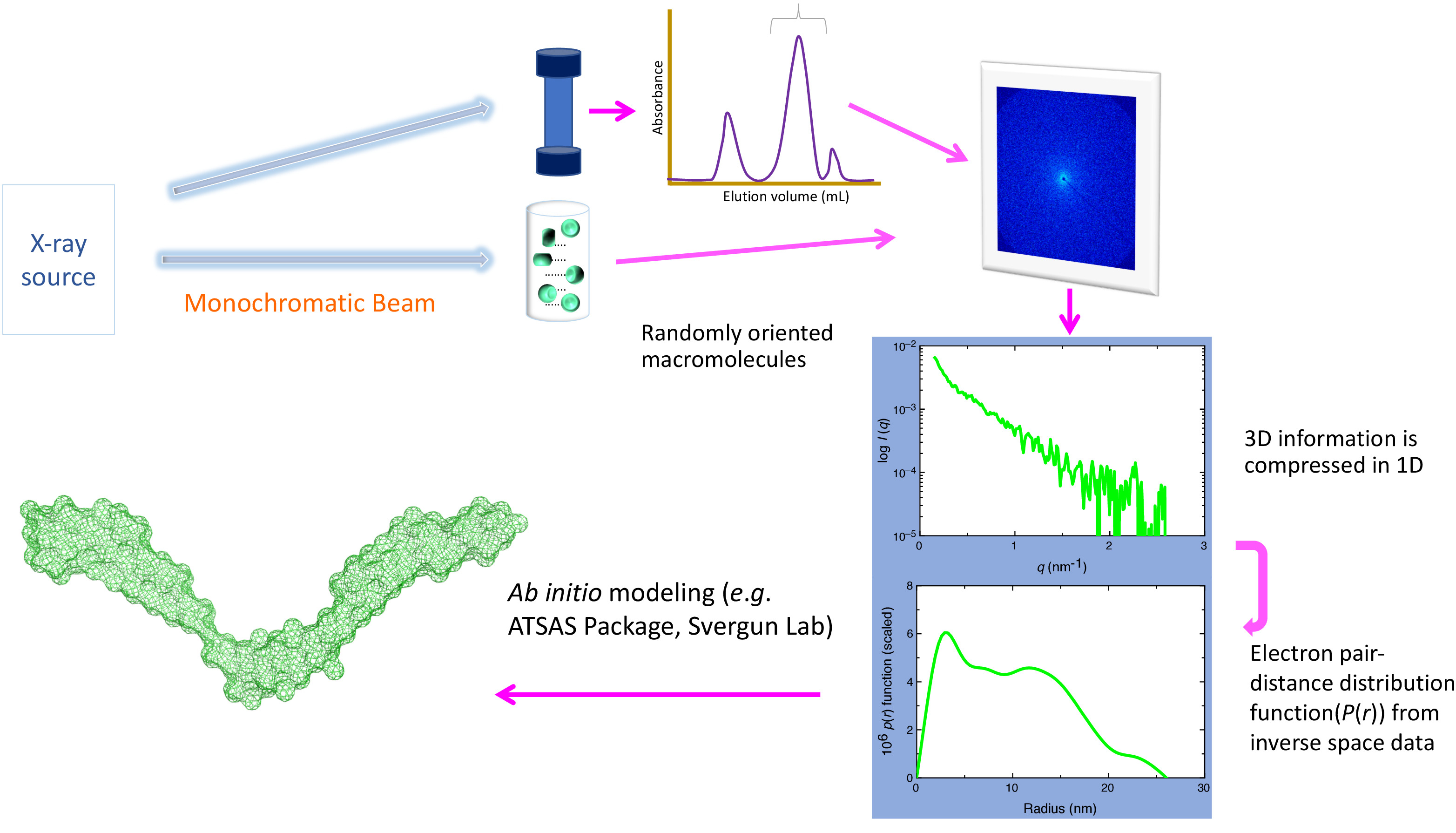
SAXS produces low-resolution envelopes of macromolecular complexes with a resolution of approximately 10-20 Å 1, giving insight not only into the structure but also the dynamic characteristics that the complex displays. Although SAXS utilizes X-rays to uncover molecular structure, it is unlike XRC in that the random isotropic orientation of the particles in solution does not lead to diffraction, but rather to scattering, which cannot yield atomic resolution. Instead, an electron “envelope” of the macromolecule is generated that represents an average of the conformations that the macromolecule displays. This information can be used in direct fitting of previously solved atomic resolution structures to infer regions of flexibility in a single protein or subunit organization, or dynamics in a larger, multi-protein complex. SAXS data is collected at synchrotrons using high-energy monochromatic X-rays or from in-house sources, which offer a weaker X-rays source requiring hours rather than seconds of sample exposure time (Figure 1). SAXS data is often collected from several samples with a single experimental setup and buffer, requiring an extended time to collect a round of useful data on a system. Samples should, therefore, be stable and non-aggregating for at least a few hours based on verifiable quality control methods such as dynamic light scattering (DLS) and/or analytical ultracentrifuge (AUC) analysis to obtain high-quality SAXS data2,3. Here we provide a practical description of SAXS, the principles behind its usage, benefits, limitations and sample preparation and focus heavily on data collection and analysis, along with touching briefly on ab initio modeling using the extracellular matrix proteins nidogen-1 and laminin γ-1 as an experimental example.
Principles, Benefits, and Limitations of SAXS:
The guiding principle(s) behind SAXS is relatively simple: a solution of the monodispersed preparation of macromolecule(s) of interest is placed within a capillary and is exposed to a high energy monochromatic X-ray beam. The photons cause electrons of the atomic shell to begin oscillating, resulting in a spherical wave being emitted of the same energy and wavelength. Since every electron will oscillate, a constant background will be achieved, and the resulting electron density of the macromolecule is contrasted to the background. The resulting scattering intensity is collected as a function of the scattering angle, 2Θ (Figure 1).
While other techniques such as XRC, NMR, and CEM provide structural information at the atomic level, there are multiple benefits to SAXS that other techniques cannot provide. SAXS can be performed in almost any buffer and does not require any special sample preparation. This is particularly important in studying the behavior and structure of macromolecules under varying conditions, such as the presence or absence of mono- or divalent cations or changes in pH4,5. SAXS has the ability to provide information about flexible regions of a macromolecule6, something the other listed techniques can struggle with. Therefore, SAXS can be used as a strong complimentary technique with the stable portions of a macromolecule being studied with XRC, NRM or CEM, and the entire macromolecule or complex analyzed in low resolution with SAXS and combined using various analysis tools such as FoXSDock7 or CRYSOL8. Since SAXS is a solution technique, it is often used to confirm if static structures such as those obtained from XRC are consistent in solution6. SAXS also has the advantage of being a technique that requires a relatively small amount of sample investment (typically 50-100 µL) and a relatively small amount of experiment time (30 min-1 h).
The largest limitation of SAXS is the vulnerability to sample aggregation and/or degradation, which can lead to incorrect structural predictions. An aggregation, even as low as 5%, can scatter light in very high amounts, leading to an overestimation of the maximal particle dimension (Dmax) and radius of gyration (Rg). On the other hand, sample degradation can lead to an underestimate of molecular properties. This vulnerability arises from SAXS being an averaging technique, which means that sample homogeneity is critical to achieving reliable and reproducible results. Any sample that is to be analyzed by SAXS should, therefore, undergo multiple methods of purification and homogeneity checks, such as denaturing and native gel electrophoresis, size exclusion chromatography, dynamic light scattering, and analytical ultracentrifugation. Often, SAXS beamlines will run samples through high-performance liquid chromatography as a final quality control step before SAXS (S-SAXS)3,9. SAXS data should be collected at multiple concentrations and the Rg of each data set should be compared, ensuring a close similarity to avoid interparticle interactions and aggregation, which results in an overestimation of particle dimensions, leading to inaccurate data analysis and modeling. Since scattering depends on both concentration and size, smaller macromolecules may require a more specific optimization of the concentration range. This is due to the Reciprocity Theorem, where large sizes scatter towards small angles and small sizes towards large angles. This manifests in data collection, where IO is proportional to R6, where R is the particle radius. A final limitation of SAXS is the potential for radiation damage to the sample during exposure, which can lead to distortion of the data. It is good practice to compare sample quality before and after SAXS sample exposure to ensure this is not occurring.
Protocol
1. SAXS Sample Preparation and Data Acquisition
- Sample Preparation:
- SAXS experiments require homogeneous, stable and non-aggregating protein samples; observe stability and oligomeric state with size exclusion chromatography (S), DLS and/or AUC prior to data collection.
- Subject samples (nidogen-1 and laminin γ-1 in this case) to DLS analysis and tricine SDS-PAGE to visualize sample purity10.
NOTE: Samples may cover a range of concentrations (1-4 mg/mL) depending on their size, their solution behavior such as self-association and aggregation along with stability; here, five concentrations of nidogen-1, (139 kDa), three of laminin γ-1 (109 kDa) and four of the S-purified equimolar complex were prepared as previously described10.
- Data Collection:
- Collect SAXS data using an in-house system or synchrotron, according to manufacturer’s or facility guidelines.
NOTE: Data used in this work was collected using an in-house system (see Table of Materials) that contains a 3-pinhole camera equipped with + 002 microfocus sealed tube (Cu Kα radiation at 1.54 Å) and Confocal Max-Flux (CMF) optics operating at 40 W. The system is also equipped with a 200 nm multi-wire 2D detector for data collection. However, with the availability of modern synchrotrons in France, Germany, UK, USA, and other countries, which provide access to an S-SAXS set-up that facilitates separation of a monodispersed preparation from possible aggregation/degradation, we now routinely collect data at synchrotron facilities. A recently published article on a DNA G-quadruplex11 is an example of an S-SAXS data collection strategy. In this case, the SAXS data were collected in the range of 0.08 ≤ q ≤ 0.26 Å for 3 hours for nidogen-1 (2.0, 2.5, 3.0, 3.5 and 4.0 mg/mL); the laminin γ-1 (1.5, 2.0 and 2.5 mg/mL) and their complex (0.8, 1.0, 1.25 and 1.5 mg/mL). - Reduce data for buffer and samples using processing software specific to the system. Subtract the buffer contribution from protein data using a program like PRIMUS/qt12 (Figure 2A).
- Collect SAXS data using an in-house system or synchrotron, according to manufacturer’s or facility guidelines.
2. Data Analysis
NOTE: Currently, there are a few software packages that are useful for SAXS data analysis: ScÅtter43 (download available at www.bioisis.net), bioXtas RAW44, and the ATSAS suite13. This section provides an overview of general steps to be taken when analyzing raw SAXS data using the ATSAS program suite and specific steps are taken from the ATSAS 2.8.1 download. Other programs can be used and are briefly discussed later.
- Buffer Subtraction
NOTE: These steps are relevant for static SAXS samples only.- Select the “OPEN” menu option in PRIMUS/qt and select the data files of interest. Be aware that data files must be in ASCII format, in which the first column is the s-vector axis and the second column is the intensity. Repeat this step for data collected for the buffer itself by inserting this data into the same menu.
- Select “SUBTRACT” in the Data Processing window, which will generate a subtracted scattering curve representing only scattering from the macromolecule of interest. Repeat this step for each concentration.
- Guinier Analysis
- To perform Guinier analysis, load a buffer subtracted scatter curve into PRIMUS/qt as previously described in step 2.1.1.
- Click “Radius of Gyration” which will proceed in opening the Primus Guinier Wizard; a plot of ln(I) vs q2 will be displayed.
- To obtain a preliminary Rg use the “AUTORG” function, which is an external module built into PRIMUS/qt. Find the "Autorg" button and click it.
- Input multiple files at once by highlighting them all and inserting them into the menu on the right hand side, very similar to 2.1.1.
- Use the Guinier plot created earlier to assess the data quality; the green line under the Guinier plot shows the residuals plot representing a linearity of the fit. Be aware that non-linearity in the Guinier analysis could be a sign of sample aggregation and further analysis should not be performed in this case.
NOTE: A linear Guinier fit gives an Rg with a small error (<5%) and suggests a high-quality sample.
- Kratky Analysis
- Load the data to be visualized in a similar manner as described above.
- Click on the “SELECT BOX” next to the data file name. This will plot the data in a separate window.
- Click the “PLOT” button followed by “Kartky Plot” selection in the dropdown menu.
- This will plot the data as “q2 x L(q) vs q”. Be aware that globular proteins display a Gaussian peak, while unfolded proteins will display a plateau instead of a peak and resemble a hyperbolic plot17.
- Data Merging
- Load buffer subtracted data for each concentration in PRIMUS/qt once again, as in step 2.1.1.
- To merge the data, simply click on the “MERGE” button in the processing window.
- Inspect each curve and the I Scale number, which correlates to the dilutions made from the original sample.
NOTE: Samples at higher concentration display less noise in the tail region of the curves.
- P(r) Distribution
- To generate the P(r) plot, load the merged data curves into PRIMUS/qt as previously described.
- Click on the “Distance Distribution” button to open a new window presenting the merged data of intensity of scattered light vs. q and pair-distance distribution function plot on the right-hand side.
NOTE: The information on the right-hand side presents the overall quality of the pair-distance distribution function calculations. - Adjust the data range of the merged data to avoid any significant noise at the tail end of the raw data.
- Omit data points close to the beam stop in the low-q region.
- To determine the Dmax, start with a range of ~5 times the Rg obtained from the Guinier analysis. Gradually decrease this value until the P(r) plot does not abruptly drop to zero on the Y-axis and does not have a long-tail before approaching zero.
- Check that the Experimental Rg/I0 (derived from Guinier approximation) and P(r) Rg/I0 numbers are similar.
NOTE: In some cases, further manipulation on the range of data, data points, and ALPHA (a regularization parameter which tells the program the ratio of how much attention is paid to the smoothness of the distribution compared to fitting the experimental data) is also required21 to obtain a good quality P(r) plot.
3. Ab initio bead modeling and averaging
- Once the data collected at multiple concentrations are merged, or the data collected using S-SAXS is minimized, and the Kratky plot, P(r) plot and Guinier analysis have been verified, calculate low-resolution structures of macromolecules and their complexes. This pipeline can be used to study solution structures and interactions of nucleic acids, proteins, and nucleic acids-protein or protein-protein complexes10,11,21,22,23,24,25,26,27,28,29,30,31,32,33,34. One of the most popular programs is DAMMIN, developed by Svergun35 and a part of the ATSAS package13, which employs simulated annealing protocols with preliminary input information on Rg and Dmax. The ab initio modeling approaches and principles are described in detail elsewhere18,36.
Results
The data analysis approach described above was utilized to calculate the Rg and Dmax for nidogen-1, laminin γ-1, and their complex using the P(r) function. We obtained Rg values of 7.20 (±0.10) nm, 8.10 (±0.20) nm, and 10.9 (±0.4) nm for nidogen-1, laminin γ-1 and their complex respectively (Figure 2A-B). In addition, Dmax values of 24 nm, 26 nm, and 35 nm for nidogen-1, laminin γ-1, and their complex respectively (Figure 2)10 were obtained. The DAMMIF program was used to obtain low-resolution structures of nidogen-1 and laminin γ-1, which suggested that both proteins adopt an extended shape in solution. The X and NSD values for nidogen-1 (~1 and 0.8) and laminin γ-1 (~0.9 and 0.8 respectively) were also in the acceptable range. The alignment of high-resolution structures, two domains of nidogen-1 and two of laminin γ-1, on their low-resolution structures obtained using SAXS allowed identification of their N- and C-terminal regions10.
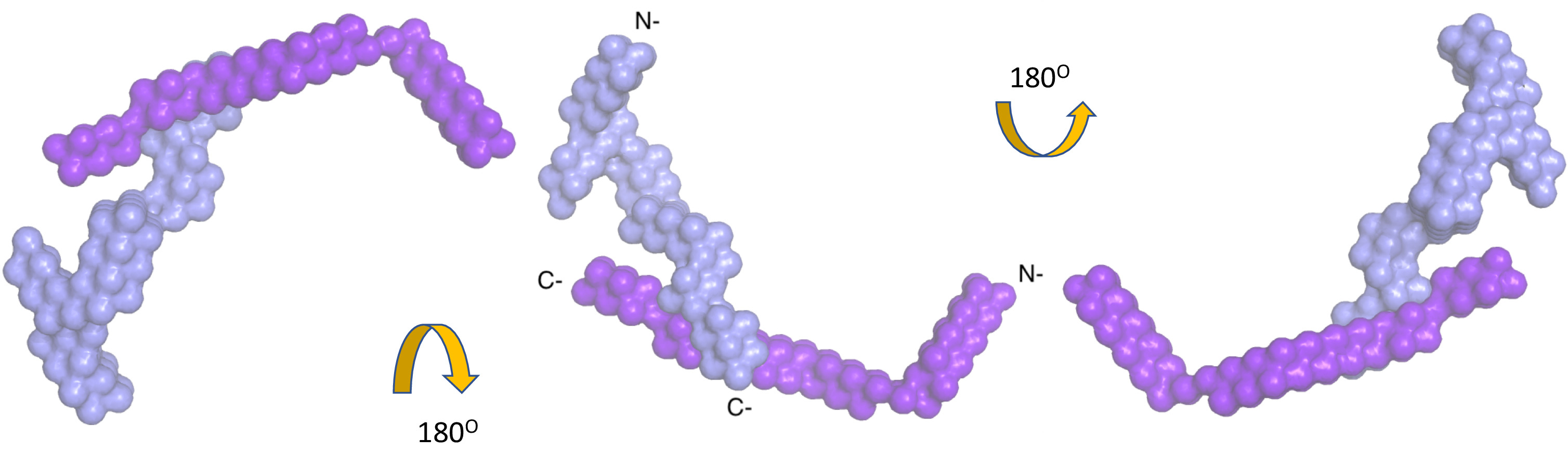
Nidogen-1 was identified as an interacting partner of laminin γ-139,40 and the interaction site was mapped using X-ray crystallography to the C-terminal domains41. However, high-resolution structures only involved interacting domains and not the full-length nidogen-1 or the entire laminin γ-1 arm. Therefore, we purified a complex containing nidogen-1 (full length) and the laminin γ-1 arm to identify the interacting regions as well as to study the relative orientation of the N-terminal domains of both proteins. The SAXS data for the complex yielded an Rg of 10.9 (±0.4) nm and a Dmax of 35 nm. We utilized MONSA to obtain the low-resolution structure of the entire complex, which suggested that indeed, only the C-terminal region of both proteins participate in mediating interactions, whereas the rest of the domains are far apart from each other (Figure 3, Video 1).

Figure 1. Schematics of SAXS set-up. A monodispersed preparation of biomolecules or their complexes is prepared, followed by exposure with high energy X-rays. Depending on the source (e.g., in-house vs. synchrotron), the energy of X-rays and the sample to source distance can vary. The X-rays’ scattering pattern (that depends on the size and shape of biomolecules) is recorded and radially averaged to obtain a 1-dimensional plot (1D) that contains information on the intensity of scattered light with respect to the scattering angle. As buffer molecules also scatter light, the contributions from these molecules are subtracted to obtain a scattering pattern of the biomolecules of interest. At the synchrotron, prior to the SAXS data collection, an additional purification step using in-line size exclusion/high-performance chromatography is also typically performed (top view). This step is critical to remove any aggregated and/or degraded product as well as to remove any unbound biomolecules from the complex. The 1D scattering plot is converted to the electron pair-distance distribution plot (P(r) plot), which provides the radius of gyration and maximum particle dimension of biomolecules. This plot is used as the input file for the ab initio modeling packages (i.e., DAMMIN/DAMMIF) to obtain low-resolution structures of biomolecules, or other packages (i.e., SASREF/CORAL) if the high-resolution structure of parts of the biomolecules or individual biomolecules of the complex is known. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. (A) A plot of an intensity of scattered light vs. scattering angle (q=4πsinθ/λ, nm-1) suggesting the quality of biomolecules (low region) and shape (high region) of biomolecules. (B) The electron pair-distance distribution P(r) determined from the scattering data suggest an elongated shape of biomolecules under investigation (laminin γ-1, nidogen-1, and their complex). (C) Kratky plot suggesting that nidogen-1 and laminin γ-1 proteins are not unfolded. (D) Guinier plot for nidogen-1, laminin γ-1 and their complex, indicating the linear region for determination of the radius of gyration using data at low-scattering angle. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Low-resolution structure of the complex of nidogen-1, and laminin γ-1 obtained by analysis of merged data sets using the program MONSA. The color scheme is the same as Figure 2. Please click here to view a larger version of this figure.
{kind=link}

Video 1. The low-resolution structure of the nidogen-1 and laminin γ-1 complex. This movie was prepared using PYMOL to visualize various structural features of the complex. The crystal structure of the laminin-nidogen complex (PDB ID: 1NPE) is shown as ribbon cartoons, highlighting the interacting sites for this complex. The color scheme is the same as Figure 2. Please click here to view this video. (Right-click to download.)
Discussion
The critical steps of SAXS data analysis outlined in the protocol section of this paper include buffer subtraction, Guinier analysis, Kratky analysis, data merging and P(r) distribution. The ab initio bead modeling is too extensive to be covered here in detail and is therefore only covered briefly.
At synchrotrons (e.g. DESY in Germany, DIAMOND in the UK and ESRF in France), it is possible to collect SAXS data for a very tiny fraction (~few µL) of each sample as the fractions are being eluted from the s column that is connected in-line (see Figure 1). The elastically scattered SAXS data is radially averaged using the packages provided by the instrument manufacturer or by the synchrotron before buffer subtraction can take place. The resulting 1D data represents the amount of scattered light (In I(q)) on the Y-axis and scattering angle (q=4πsinθ/λ, where λ is the wavelength of incident X-rays) and is outlined in Figure 1. The program PRIMUS/qt12 is used to directly subtract any background due to buffer and is described in section 1.1. Other programs such as; ScÅtter43 (download available at www.bioisis.net) with a tutorial available at https://www.youtube.com/channel/UCvFatdC5HcZOLv6OSjblfeA, and bioXtas RAW44 (available at https://bioxtas-raw.readthedocs.io/en/Latest/index.html) can be utilized as an alternative to the ATSAS package.
The Guinier analysis provides information on sample aggregation and homogeneity as well as providing the Radius of Gyration (Rg) for the macromolecule of interest based on the SAXS data from the low s region14. A plot is constructed with PRIMUS/qt for SAXS data obtained from each concentration, followed by curve fitting with the maximum range of up to 1.30 for q x Rg. A monodispersed sample preparation should provide a linear Guinier plot in this region (Figure 2D), whereas aggregation results in a nonlinear Guinier plot15,16. If the Guinier analysis is linear, the degree of “unfoldedness” of a macromolecule of interest can be observed with the Kratky plot, which is useful when deciding whether to perform rigid body modeling or construct ensembles of low-resolution models. A globular protein will appear in a Kratky plot to have a bell-shaped curve, whereas extended molecules or unfolded peptides will appear to plateau or even increase in the larger q range and lack the bell-shape (Figure 2C).
Obtaining the Rg from Guinier analysis only considers data points from the low q region of the 1D scatter plot (Figure 2D), however, it is possible to use almost the entire dataset to perform an indirect Fourier transformation to convert the reciprocal-space information of ln(I(q)) vs. (q) into a real space distance distribution function (P(r)) which provides information on Dmax and Rg (Figure 2B) The shape of the P(r) plot represents the gross solution conformation of the macromolecule of interest18,19. the conversion of reciprocal-space data to real-space data is a critical step but a detailed description is not within the scope of this paper. Therefore, refer to an article by Svergun20 to understand each parameter.
Once the buffer subtracted data at individual concentrations are processed through Guinier analysis with a consistent value for Rg, followed by investigating their folding pattern using Kratky analysis, these data can be merged. The merged data for nidogen-1, laminin γ-1, and their complex were processed as described above and the resulting P(r) plots are presented in Figure 2B. Ideally, one should also calculate the pair-distance distribution function P(r) for each concentration to determine if SAXS data collected for each concentration provides similar Rg and Dmax values. If the Rg and Dmax remain similar over a wide range of concentrations, then the user should proceed. It should be noted that depending on the signal, data can be truncated prior to data merging. This is often the case if the concentrations and/or molecular weight of the macromolecules under investigation is low.
Low-resolution shape analysis using DAMMIN can be performed in various modes (e.g. Fast, Slow, Expert modes, etc.). The Fast mode is an ideal first step to evaluate if the P(r) plot provides good quality models. Typically, at least 10 models should be obtained for each P(r) plot to check if reproducible results, in terms of the low-resolution structure, are obtained, with a low goodness of fit parameter called χ (a value of 0.5-1.0 is considered good based on our extensive work), a value that describes an agreement between experimentally collected SAXS data and model-derived data. For publication purpose, we typically use Slow or Expert mode and calculate at least 15 models. In addition to DAMMIN, a faster version of it, DAMMIF37, as well as GASBOR38 are also alternatives. Furthermore, to study protein-protein or protein-nucleic acid complexes, it is possible to use the MONSA program35, which facilitates simultaneous fitting of the individual SAXS data for both macromolecules as well as their complex. For more details on high-resolution model calculations as well for RNA-protein interaction studies, refer to a recent article by Patel et al3.
SAXS is theoretically simple but undoubtedly a highly complementary method to other structural biology tools and results in low-resolution structural data that can be used on its own or in conjunction with high-resolution techniques to elucidate information about macromolecular structure and dynamics. As long as a monodispersed preparation of macromolecules and their complexes can be obtained, SAXS can be utilized to study in-solution structure and interactions of any type of biological macromolecule. In the case of the complex discussed here, it is remarkable that less than 10% of the overall accessible surface area of nitrogen-1 and laminin γ-1 is buried in this complex, whereas the rest of the domains of both proteins are freely accessible to interact with other proteins at the extracellular matrix to maintain its structural rigidity (Figure 3). Obtaining such information for a complex with ~240kDa would be very challenging using other structural biology techniques such as X-Ray Crystallography, NMR, and Cryo-EM Microscopy.
Uncovering protein structure via X-Ray Crystallography or NMR is an inherently time-consuming process. This bottleneck in structure determination is one area where SAXS shows its strength as a structural technique; data acquisition for a single SAXS experiment can take less than an hour and with the help of streamlined analysis software, analysis can be done quickly and efficiently. SAXS has the potential to greatly increase throughput of structural studies as a stand-alone technique because it offers a low-resolution model of the macromolecular structure before high-resolution data is available. A barrier to other structural techniques is the requirement for a highly pure, concentrated sample for data acquisition, which necessitates a high level of protein expression and stability over a long period of time. While SAXS samples also need to be pure and concentrated, the sample volumes are roughly 100 µL making SAXS a relatively inexpensive method of analysis compared to other structural techniques. Moreover, SAXS coupled with size exclusion chromatography is becoming increasingly common which provides an additional quality control step. Recently there has been strong advances in the combination of NMR and SAXS data using the Ensemble Optimization Method (EOM)45,46 to elucidate flexible systems. In a recent paper by Mertens and Svergun47, the authors describe multiple recent examples of EOM SAXS in combination with NMR, along with many other examples of SAXS data being used in conjunction with NMR. Advances are continually being made in the field of SAXS, and new techniques are being developed for SAXS to be used in conjunction with, not just complimentary to, other structural techniques. Consequently, we believe that the demand for SAXS will only increase over time, especially in conjunction with NMR to characterize dynamic systems where functions are defined by flexibility.
Disclosures
Authors have no disclosures to declare.
Acknowledgements
This project has been supported by NSERC RGPIN-2018-04994, Campus Alberta Innovation Program (RCP-12-002C) and Alberta Prion Research Institute / Alberta Innovates Bio Solutions (201600018) grants awarded to M.O. TRP is a Canada Research Chair in RNA & Protein Biophysics (201704) and acknowledges NSERC Discovery grant (RGPIN-2017-04003). TM is funded by the NSERC Discovery grant to TRP.
Materials
| Name | Company | Catalog Number | Comments |
| HEK 293 EBNA Cell Line | In-Lab availability | - | Cell line used to overexpress protein(s) |
| Ni Sepharose High Performance histidine-tagged protein purification resin | GE Healthcare | 17524801 | Affinity protein purification resin |
| Superdex 200 Increase 10/300 | GE Healthcare | 28990944 | SEC Column |
| ÄKTA Pure FPLC | GE Healthcare | - | FPLC System |
| Nanodrop | Nanodrop | - | Spectrophotometer |
| Basic Reagents (NaCl, Tris-HCl etc.) | |||
| S-MAX3000 | Rigaku | - | SAXS Pinhole Camera System |
| Zetasizer Nano-S | Malvern Instruments Ltd | - | Dynamic Light Scattering instrument |
| 0.1µm Filter | Millipore | JVWP04700 | Used to Concentrate Sample Prior to DLS |
| Thrombin cleavage kit | abcam | ab207000 | Thrombin cleavage to remove His tag |
| Strep-Tactin Sepharose Column | IBA | 2-1201-010 | Strep-Tag Affinity Purification |
| D-desthiobiotin | Sigma-Aldrich | 533-48-2 | Elution of Strep Tag Protein |
| Software | |||
| SAXGUI | Rigaku | - | Data Collection for SAXS and data reduction |
| ATSAS Suit | Franke et al., 2017 | - | SAXS Data Analysis Software program suite |
| PRIMUS | Konarev et al., 2003 | - | Buffer Subtraction |
| GNOM | Svergun, 1992 | - | Rg, Dmax and p(r) Calculation |
| DAMMIF | Franke and Svergun, 2009 | - | Ab initio model calculation |
| DAMAVER | Volkov and Svergun, 2003 | - | Averaged Solution Conformation calculations |
| MONSA | Svergun, 1999 | - | Simultaneous model fitting for the complex |
| GASBOR | Svergun et al., 2001 | - | Alternative Ab initio model calculations |
| DTS Software V6.20 | Malvern Instruments Ltd | - | DLS supplied instrument software |
| PyMOL | Schrodinger, LLC. | - | The PyMOL Molecular Graphics System V2.0 |
| The Protein Data Bank | Berman et al., 2000 | - | PDB ID: 1NPE |
References
- Svergun, D. I., Koch, M. H. J. Advances in structure analysis using small-angle scattering in solution. Current Opinion in Structural Biology. 12 (5), 654-660 (2002).
- Patel, T. R., Chojnowski, G., Koul, A., McKenna, S. A., Bujnicki, J. M. Structural studies of RNA-protein complexes: A hybrid approach involving hydrodynamics, scattering, and computational methods. Methods. 118, 146-162 (2017).
- Stetefeld, J., McKenna, S. A., Patel, T. R. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophysical Reviews. 8 (4), 409-427 (2016).
- Uversky, V. N., Gillespie, J. R., Fink, A. L. Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins. 41 (3), 415-427 (2000).
- Uversky, V. N., Gillespie, J. R., Millett, I. S., Khodyakova, A. V., Vasiliev, A. M., Chernovskaya, T. V., Abramov, V. M. Natively Unfolded Human Prothymosin α Adopts Partially Folded Collapsed Conformation at Acidic pH. Biochemistry. 38 (45), 15009-15016 (1999).
- Kikhney, A. G., Svergun, D. I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Letters. 589 (19 Pt A), 2570-2577 (2015).
- Schneidman-Duhovny, D., Hammel, M., Tainer, J. A., Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Research. 44, 424-429 (2016).
- Svergun, D., Barberato, C., Koch, M. H. J. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography. 28 (6), 768-773 (1995).
- Pérez, J., Vachette, P. A Successful Combination: Coupling SE-HPLC with SAXS. Advances in Experimental Medicine and Biology. 1009, 183-199 (2017).
- Patel, T. R., Bernards, C., Meier, M., McEleney, K., Winzor, D. J., Koch, M., Stetefeld, J. Structural elucidation of full-length nidogen and the laminin-nidogen complex in solution. Matrix Biology. 33, 60-67 (2014).
- Meier, M., Moya-Torres, A., Krahn, N. J., McDougall, M. D., Orriss, G. L., McRae, E. K. S. Structure and hydrodynamics of a DNA G-quadruplex with a cytosine bulge. Nucleic Acids Research. , (2018).
- Franke, D., Petoukhov, M. V., Konarev, P. V., Panjkovich, A., Tuukkanen, A., Mertens, H. D. T. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 50, 1212-1225 (2017).
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., Svergun, D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 36 (5), 1277-1282 (2003).
- Tuukkanen, A. T., Svergun, D. I. Weak protein-ligand interactions studied by small-angle X-ray scattering. The FEBS Journal. 281 (8), 1974-1987 (2014).
- Rambo, R. P., Tainer, J. A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 496 (7446), 477-481 (2013).
- Koch, M. H., Vachette, P., Svergun, D. I. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Quarterly Reviews of Biophysics. 36 (2), 147-227 (2003).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics. 40 (3), 191-285 (2007).
- Petoukhov, M. V., Svergun, D. I. Applications of small-angle X-ray scattering to biomacromolecular solutions. The International Journal of Biochemistry & Cell Biology. 45 (2), 429-437 (2013).
- Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography. 25 (4), 495-503 (1992).
- Patel, T. R., Morris, G. A., Zwolanek, D., Keene, D. R., Li, J., Harding, S. E. Nano-structure of the laminin γ-1 short arm reveals an extended and curved multidomain assembly. Matrix Biology: Journal of the International Society for Matrix Biology. 29 (7), 565-572 (2010).
- Patel, T. R., Reuten, R., Xiong, S., Meier, M., Winzor, D. J., Koch, M., Stetefeld, J. Determination of a molecular shape for netrin-4 from hydrodynamic and small angle X-ray scattering measurements. Matrix Biology: Journal of the International Society for Matrix Biology. 31 (2), 135-140 (2012).
- Dzananovic, E., Patel, T. R., Deo, S., McEleney, K., Stetefeld, J., McKenna, S. A. Recognition of viral RNA stem-loops by the tandem double-stranded RNA binding domains of PKR. RNA. 19 (3), 333-344 (2013).
- Meier, M., Patel, T. R., Booy, E. P., Marushchak, O., Okun, N., Deo, S. Binding of G-quadruplexes to the N-terminal recognition domain of the RNA helicase associated with AU-rich element (RHAU). The Journal of Biological Chemistry. 288 (49), 35014-35027 (2013).
- Dzananovic, E., Patel, T. R., Chojnowski, G., Boniecki, M. J., Deo, S., McEleney, K. Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. Journal of Structural Biology. 185 (1), 48-57 (2014).
- Bacik, J. -. P., Tavassoli, M., Patel, T. R., McKenna, S. A., Vocadlo, D. J., Khajehpour, M., Mark, B. L. Conformational itinerary of Pseudomonas aeruginosa 1,6-anhydro-N-acetylmuramic acid kinase during its catalytic cycle. The Journal of Biological Chemistry. 289 (7), 4504-4514 (2014).
- Deo, S., Patel, T. R., Dzananovic, E., Booy, E. P., Zeid, K., McEleney, K. Activation of 2’ 5’-oligoadenylate synthetase by stem loops at the 5’-end of the West Nile virus genome. PloS One. 9 (3), e92545 (2014).
- Vadlamani, G., Thomas, M. D., Patel, T. R., Donald, L. J., Reeve, T. M., Stetefeld, J., Mark, B. L. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the D-Ala-D-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. The Journal of Biological Chemistry. 290 (5), 2630-2643 (2015).
- Deo, S., Patel, T. R., Chojnowski, G., Koul, A., Dzananovic, E., McEleney, K., McKenna, S. A. Characterization of the termini of the West Nile virus genome and their interactions with the small isoform of the 2’ 5’-oligoadenylate synthetase family. Journal of Structural Biology. 190 (2), 236-249 (2015).
- Ariyo, E. O., Booy, E. P., Patel, T. R., Dzananovic, E., McRae, E. K., Meier, M., McKenna, S. A. Biophysical Characterization of G-Quadruplex Recognition in the PITX1 mRNA by the Specificity Domain of the Helicase RHAU. PloS One. 10 (12), (2015).
- Hyde, E. I., Callow, P., Rajasekar, K. V., Timmins, P., Patel, T. R., Siligardi, G., Scott, D. J. Intrinsic disorder in the partitioning protein KorB persists after co-operative complex formation with operator DNA and KorA. The Biochemical Journal. 474 (18), 3121-3135 (2017).
- Dzananovic, E., Astha, n. u. l. l., Chojnowski, G., Deo, S., Booy, E. P., Padilla-Meier, P., McKenna, S. A. Impact of the structural integrity of the three-way junction of adenovirus VAI RNA on PKR inhibition. PloS One. 12 (10), (2017).
- Krahn, N., Meier, M., To, V., Booy, E. P., McEleney, K., O’Neil, J. D., Stetefeld, J. Nanoscale Assembly of High-Mobility Group AT-Hook 2 Protein with DNA Replication Fork. Biophysical Journal. 113 (12), 2609-2620 (2017).
- Patel, T. R., Besong, T. M. D., Meier, M., McEleney, K., Harding, S. E., Winzor, D. J., Stetefeld, J. Interaction studies of a protein and carbohydrate system using an integrated approach: a case study of the miniagrin-heparin system. European Biophysics Journal: EBJ. , (2018).
- Svergun, D. I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophysical Journal. 76 (6), 2879-2886 (1999).
- Blanchet, C. E., Svergun, D. I. Small-Angle X-Ray Scattering on Biological Macromolecules and Nanocomposites in Solution. Annual Review of Physical Chemistry. 64 (1), 37-54 (2013).
- Volkov, V. V., Svergun, D. I. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 36, 860-864 (2003).
- Kozin, M. B., Svergun, D. I. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 34 (1), 33-41 (2001).
- Franke, D., Svergun, D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 42 (Pt 2), 342-346 (2009).
- Svergun, D. I., Petoukhov, M. V., Koch, M. H. J. Determination of Domain Structure of Proteins from X-Ray Solution Scattering. Biophysical Journal. 80 (6), 2946-2953 (2001).
- Baumgartner, R., Czisch, M., Mayer, U., Pöschl, E., Huber, R., Timpl, R., Holak, T. A. Structure of the nidogen binding LE module of the laminin gamma1 chain in solution. Journal of Molecular Biology. 257 (3), 658-668 (1996).
- Stetefeld, J., Mayer, U., Timpl, R., Huber, R. Crystal structure of three consecutive laminin-type epidermal growth factor-like (LE) modules of laminin gamma1 chain harboring the nidogen binding site. Journal of Molecular Biology. 257 (3), 644-657 (1996).
- Takagi, J., Yang, Y., Liu, J. -. H., Wang, J. -. H., Springer, T. A. Complex between nidogen and laminin fragments reveals a paradigmatic beta-propeller interface. Nature. 424 (6951), 969-974 (2003).
- Förster, S., Apostol, L., Bras, W. Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Cryst. 43, 639-646 (2010).
- Hopkins, J. B., Gillilan, R. E., Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Cryst. 50, 1545-1553 (2017).
- Bernadó, P., Mylonas, E., Petoukhov, M. V., Blackledge, M., Svergun, D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. (17), 5656-5664 (2007).
- Tria, G., Mertens, H. D., Kachala, M., Svergun, D. I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2 (Pt 2), 207-217 (2015).
- Mertens, H. D. T., Svergun, D. I. Combining NMR and small angle X-ray scattering for the study of biomolecular structure and dynamics. Arch Biochem Biophys. 628, 33-41 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved