
Method Article
High Sensitivity Measurement of Transcription Factor-DNA Binding Affinities by Competitive Titration Using Fluorescence Microscopy
In This Article
Summary
Here we present a novel method for determining binding affinities at equilibrium and in solution with high sensitivity on a large scale. This improves the quantitative analysis of transcription factor-DNA binding. The method is based on automated fluorescence anisotropy measurements in a controlled delivery system.
Abstract
Accurate quantification of transcription factor (TF)-DNA interactions is essential for understanding the regulation of gene expression. Since existing approaches suffer from significant limitations, we have developed a new method for determining TF-DNA binding affinities with high sensitivity on a large scale. The assay relies on the established fluorescence anisotropy (FA) principle but introduces important technical improvements. First, we measure a full FA competitive titration curve in a single well by incorporating TF and a fluorescently labeled reference DNA in a porous agarose gel matrix. Unlabeled DNA oligomer is loaded on the top as a competitor and, through diffusion, forms a spatio-temporal gradient. The resulting FA gradient is then read out using a customized epifluorescence microscope setup. This improved setup greatly increases the sensitivity of FA signal detection, allowing both weak and strong binding to be reliably quantified, even for molecules of similar molecular weights. In this fashion, we can measure one titration curve per well of a multi-well plate, and through a fitting procedure, we can extract both the absolute dissociation constant (KD) and active protein concentration. By testing all single-point mutation variants of a given consensus binding sequence, we can survey the entire binding specificity landscape of a TF, typically on a single plate. The resulting position weight matrices (PWMs) outperform those derived from other methods in predicting in vivo TF occupancy. Here, we present a detailed guide for implementing HiP-FA on a conventional automated fluorescent microscope and the data analysis pipeline.
Introduction
Given the central role of transcription factors (TFs) in gene regulation, determining their binding preferences in a quantitative manner is of paramount importance. Seminal studies by von Hippel introduced the notion that regulatory TFs rapidly recognize DNA, such that their binding is well described by the thermodynamic equilibrium, while the downstream events of recruiting RNA polymerase to the promoter are controlled by slower kinetics1. Recent in vivo binding studies suggest that this picture is likely more complex2,3; nevertheless, these general assumptions serve as good approximations and have supported many computational approaches to find cis-regulatory elements and predict expression from sequences4,5,6. While equilibrium binding has thus been successfully employed as a concept, current methods for determining TF-DNA interactions focus on binding specificity and typically do not directly measure binding affinities at equilibrium. The systematic measurement of TF-DNA binding represents a considerable technical challenge, and the existing methods have several different limitations.
Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq)7, the most prevalent in vivo technique, does not permit the measurement of binding affinities or the precise localization of binding sites within genomic fragments. Several in vitro methods, including DNase footprinting8, electrophoretic mobility shift (EMSA)9, surface plasmon resonance (SPR)10, and microscale thermophoresis11 are able to measure binding affinities, but they are relatively low throughput. Conversely, high throughput techniques including protein binding microarrays12, HT-SELEX13,14, and bacterial one-hybrid (B1H)15 are not able to measure binding affinities and typically yield overly specific binding sequences, which is mainly due to the stringent selection or washing steps necessary. More recent developments include the deep sequencing based HiTS-FLIP16, SELEX-seq17, and the microfluidics-based MITOMI18 or SMILE-Seq19, which allow for extraction of absolute binding affinities; however, they rely on measuring fluorescence intensities of labeled TF and DNA. Fluorescence signals, therefore, become limiting at low protein concentrations and in determining low KD values (< ~10 nM). Moreover, the TF-DNA binding in these methods takes place on thin surfaces, raising issues with unspecific binding and/or auto-fluorescence background, which makes it difficult to accurately quantify weak binding.
To address these limitations, we have developed a new method to determine TF-DNA affinity landscapes at equilibrium and in solution, which we called high performance fluorescence anisotropy (HiP-FA)20. The technique is based on the established fluorescence anisotropy (FA) assay21 but modified to measure binding constants with high sensitivity and at large-scale using a customized automated microscope and analysis setup.
The FA assay monitors the interaction of fluorescently labeled species (like a DNA oligomer) to a binding partner, in this case a TF, by measuring the molecular rotation of the labeled molecule. Upon binding to the TF, its rotational speed decreases due to the higher hydrodynamic radius and molecular weight of the bound complex, which results in increased FA. The accurate measurement of very strong binding (KD < ~1 nM) requires the use of low concentrations of labeled, reference DNA (c < ~1 nM). This is difficult to achieve with a commercial instrument such as a standard microplate reader. In addition, a large size difference (10-100 fold) between the bound and unbound complexes is usually necessary, prohibiting measurement of interactions between TF binding domains and short DNA oligomers, which are typically of roughly similar molecular weights. Finally, a full titration curve normally requires the preparation and measurement of multiple wells containing a concentration series for the titrating species.
To address these issues, we use a widefield microscopy setup, modified to achieve high detection sensitivity and allow FA measurements at different z-positions of a single well. This enables us to monitor binding interactions between species of similar molecular weight and with high affinities. Higher throughput is achieved by measuring FA in multi-well plate formats and carrying out an entire titration series in a single well using a controlled delivery system (Figure 1a). Furthermore, by employing a competitive binding assay, we extract not only the binding constants but also the concentration of active protein. This is an important feature of the assay, since only a portion of the expressed TF molecules are active due to protein misfolding or degradation. The experimental setup is based on a commercial epifluorescence microscope equipped with XY- and Z- piezo stages. We upgraded the system with external laser excitation, then detected the two emitted linear polarization components on the chip of an EM-CCD camera with high quantum efficiency for light detection (Figure 1b and 1c). The system uses a high numerical aperture (NA) objective coupled to an ultra-sensitive sensor and thus affords highly sensitive FA measurements. By recording fluorescence z-stacks, binding interactions can be measured along the optical z-axis when using a heterogeneous matrix for the reactants. All these modifications can be readily implemented on an existing system and are cost-effective.
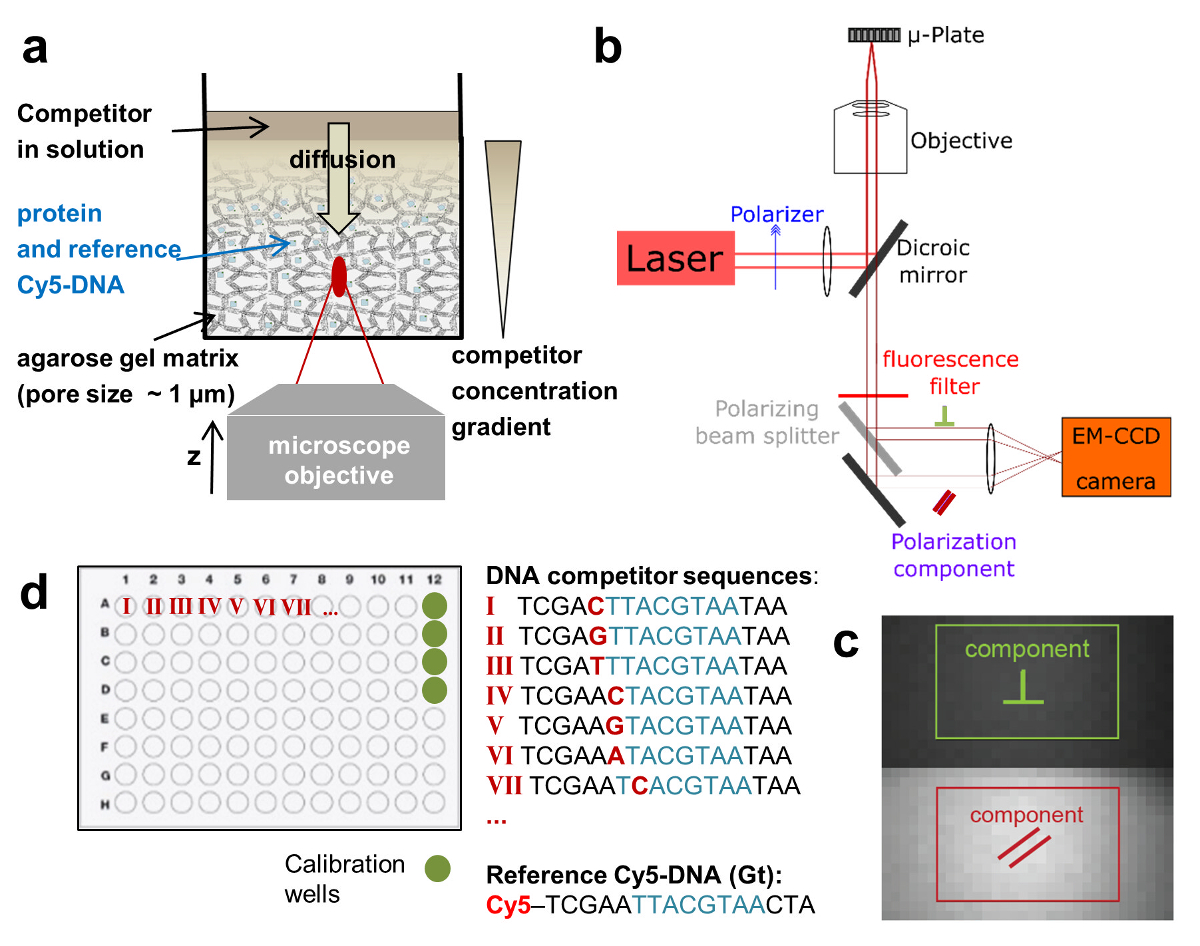
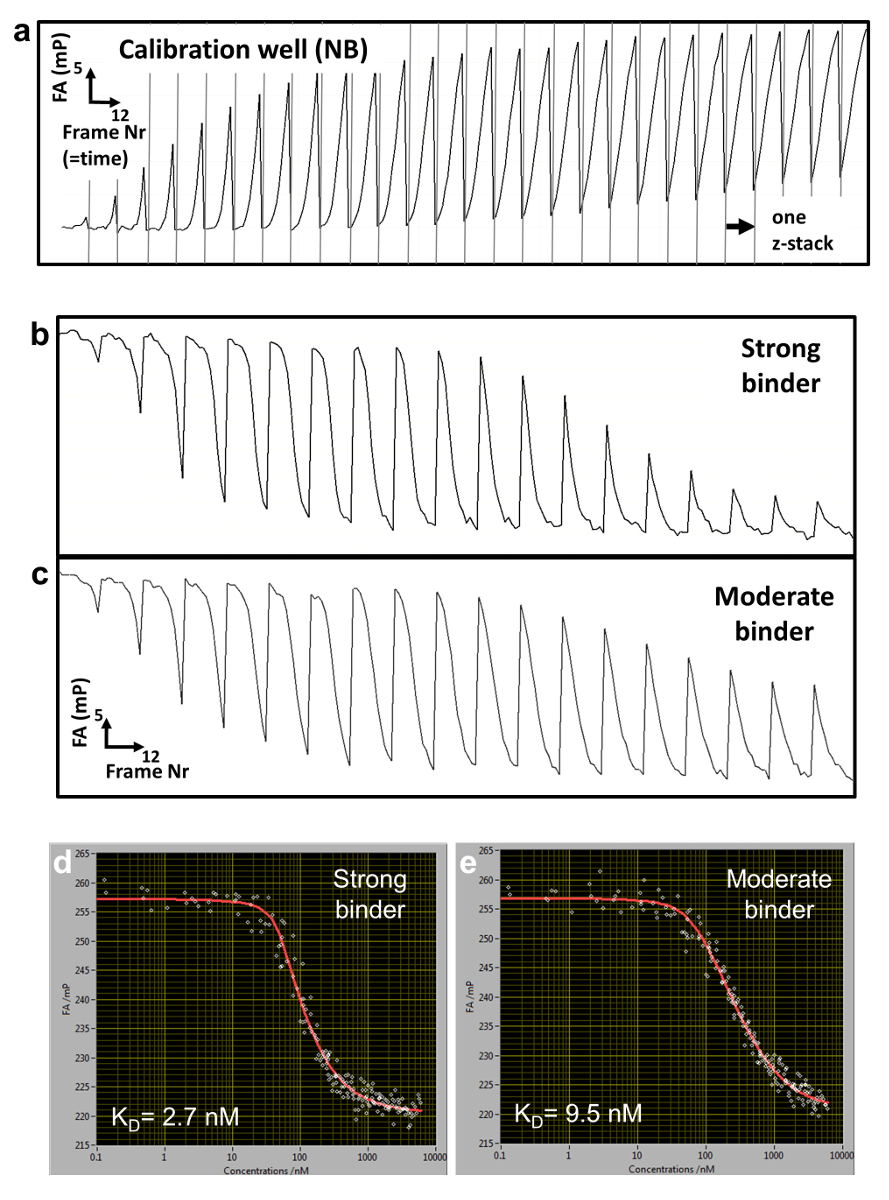
We employ a competitive binding assay in which the binding affinity of an unlabeled DNA oligomer is measured in comparison to the fluorescently labeled DNA, which serves as a reference. TF and reference DNA are incorporated at fixed concentrations in a porous agarose gel matrix (pore size ~1 µm) that constitutes a non-interacting environment for the binding. The reference DNA is labeled with Cy5. This dye proved to be well-suited for FA measurements due to its relatively long fluorescence lifetime (~1ns) and fluorescence emission in the far-red of the visible spectrum (low auto-fluorescence background). The TF concentration is in molar excess over Cy5-reference DNA, ensuring that all reference DNA is bound to protein. A solution of unlabeled competitor DNA is then deposited on the gel surface and diffuses inside the porous matrix, establishing a concentration gradient c(z, t) that changes over the z-position of the focal plane and time t (Figure 1a, Figure 2a-2c). The TF bound to the Cy5-reference DNA is thus locally exposed to different concentrations of the competitor DNA that competes for binding, leading to a dynamically changing FA of the Cy5-reference DNA FAREF(z, t) (Figure 2b and 2c).
To determine the competitor concentration c(z,t), we measure in separate wells ( calibration wells) the dynamically changing FA signal of Nile Blue (NB) FANB(z,t) (Figure 2a and 3). This dye intercalates into DNA and thereby acts as a DNA sensor for the competitor DNA. With this controlled delivery system, tens to hundreds of different DNA-protein binding affinities can be measured within one multi-well plate (96- or 384-well plate format). Measurement is then performed sequentially until complete displacement of the labeled reference DNA from the TF. We determined the binding specificity for a given factor by measuring the affinities of all 3 N single-base mutations of the consensus sequence of length N. HiP-FA requires low amounts of protein (~pmols per titration curve) and shows low variability in the determination of KDs [coefficient of variation (CV) < 20%], while allowing measurements at a relatively large scale. The method can be conducted manually or fully automated using a robotic system, resulting in even lower CVs (Figure 4, upper panel). Dissociation constants are measured with high accuracy down to 0.5 nM. For extremely high affinities (KD < 500pM), we use a standard competitive titration (Figure 5) due to the inaccuracies in measuring competitor DNA concentrations at low levels (< 100 nM).
HiP-FA can be implemented on nearly any standard, inverted, epifluorescence fluorescent microscope, provided the availability of an automated XY-stage and a piezo z-axis stage. Optical components were built around an automated widefield setup equipped with a long-distance objective. In practice, the assay can be adapted to objectives with other characteristics (in particular working, distance and numerical aperture). However, this requires optimization of the parameters (distances between the z-slices, porosity and height of the agarose gel, etc.). The use of other kinds of lasers or camera is also possible. A detailed description of the entire experimental procedure and data analysis is given below in the protocol section.
Protocol
1. Polarization Microscopy
- For widefield laser illumination, focus a 638 nm line of a continuous diode laser (40 mW) on the aperture of multimode optical fiber for beam cleanup. Mount a linear polarizer at the output of the fiber to set the polarization of laser light.
- Block the excitation component of the emitted light with a dichroic mirror (640 nm cut-off) and a bandpass filter (bandpass 700/75).
- Let the fluorescence signal pass through a polarizing beam splitter, which splits the emitted light into its perpendicular and parallel polarized components. Then, focus the non-reflected beam (parallel component) and the reflected beam (perpendicular component) with an achromatic lens of 200 mm focal length on the chip of a back-illuminated EM-CCD camera (Figure 1b and 1c). Use a mirror to adjust the direction of the perpendicular beam toward the lens.
2. Design and Testing of Fluorescent-Labelled Reference DNA Oligomer
- Determine the core sequence of the reference DNA: the method is based on a competitive assay that measures the dissociation constant (KD2) between a transcription factor and unlabeled competitor DNA oligomer that competes for binding with a fluorescently labeled DNA whose affinity to the TF acts as a references (KD1). The consensus sequence obtained from other sources like DNase footprinting or bacterial 1-hybrid can serve as a starting point5,15.
NOTE: As a rule of thumb, a suitable reference DNA has a 3 to 7-fold decrease in binding affinity to the TF compared to the consensus sequence. - Measure by HiP-FA the KD1s of 2-3 tentative single mutations of the consensus sequence derived in the previous step. Try to mutate positions in consensus sequence that are not too specific to avoid complete loss of binding.
NOTE: It is important that the reference sequence is bound by the transcription factor of interest (we used in this protocol Giant Gt), but not too strongly, so that weaker competitors can outcompete it at high concentrations. - Extend the core motif (8-12 base pairs generally) to a length of 16 base pairs or more by adding symmetrically flanking sequence at both sides (add side chains for proper binding). If necessary (for longer biding domains, for example), use longer sequences (up to ~50 base pairs in length were tested with the HiP-FA assay).
CAUTION: Be careful not to add bases that are expected to create ectopic binding sites. Use computational tools that predict binding sites from available PWMs to facilitate this process (e.g., PySite22). - As labeled reference DNA, order oligomers that are fluorescently labeled on either forward or reverse strand at the 3' or 5' end. Use, for example Cy5, Bodipy-650 or any other suitable dye at a concentration of 10 µM (100 µM 10x stock) in water, and dilute stepwise as described in step 3.1.
- Prepare 500 mL of 1x binding buffer by adding 33 mM potassium phosphate buffer (pH = 7.0), 90 mM NaCl, and 0.01% non-ionic detergent in distilled water. Also prepare 3x binding buffer, which contains the same components, except at threefold concentrations. If using 3x binding buffer as stock solution for the 1x binding buffer, prepare volumes > 500 mL; otherwise, prepare 250 mL.
NOTE: This composition was optimized for transcription factor stability and to prevent glutathione S-transferase (GST) dimerization. - Measure with the microscopy setup described in step 1 the FA of 200 µL of binding buffer containing 0.8 nM labeled reference DNA in the presence of different amount of TF in a glass bottom microscopy 96-well plate (5-6 wells with different TF concentrations) to determine the TF concentration to use. Perform a titration series with increasing amounts of TF and choose for the assay the concentration for which the curve reaches a plateau, indicating complete binding of the DNA reference oligomer.
NOTE: The optimal TF concentration depends on the values of the TF-DNA dissociation constants. Generally, lower KDs require lower concentrations.
3. Oligomer Annealing
- To anneal the DNA oligomers of the labeled reference DNA (sequence determined in the previous step), mix 7 µL of a 10 mM dye-labeled forward single-stranded DNA solution and 7 µL of a 10 mM concentration of its unlabeled reverse complement in 186 µL of water.
- For the competitor DNA sequences, mix 20 µL of 100 mM solutions (in water, provided by the manufacturer) of forward single-stranded DNA with 20 µL of 100 mM of the corresponding reverse single-stranded DNA for each individual competitor sequence to be measured.
- Perform the annealing separately in a standard PCR cycler by heating up the solutions to 70 °C for 3 min and decreasing the temperature to RT at a rate of 0.1 K/s. If the PCR machine used does not support temperature gradients at that rate, simply do stepwise incubations with decreasing temperatures (tested were 99 cycles of 3 s with -0.4 K per cycle).
4. Gel Preparation
NOTE: The following section explains the preparation of two different kind of gels: 1) the titration wells contain gels with protein and are used to determine the KDs for the respective competitor DNA sequences, and 2) the calibration wells make use of NB to determine the DNA concentration at every given time point and acquisition height. The focus is on the preparation of the experiment in a 96-well plate, but the corresponding volumes for a 384-well plate format are also indicated.
- Dissolve 0.5% w/v low melting point agarose in the binding buffer by boiling it in a laboratory microwave oven. After complete dissolution, adjust the volume again with ddH2O to compensate for possible evaporation.
NOTE: For convenience, prepare a stock of 10-20 10 mL aliquots of the gels and melt them at 75°C when they are needed. Gel stocks can be stored at RT.
CAUTION: Be careful to avoid superheating of the gel solution in the microwave oven. Short heating time intervals with shaking in between are preferable. - To prepare titration and calibration wells, first melt two 10 mL gel stock aliquots at 75 °C under shaking.
- Use 240 µL (including 20% overhead) for each competitor (n = number of competitor sequences).
- Use the same volume of gel for the NB calibration well to ensure an equal temperature and viscosity of both gels.
- Then set the temperature to 35 °C and wait for the temperature to equilibrate.
- For the titration wells, add 1.4 nM (final concentration) hybridized reference DNA (obtained in step 3), TF protein (final concentration CTF = 20-60 nM, as determined in step 2.6), DTT (0.2 mM), and binding buffer in a total volume of n x 200 µL or n x 13 µL in a 96- or 384-well plate format, respectively (plus overhead). Mix thoroughly by inverting/shaking (do not vortex).
- Slowly add 200 µL per well in 96-well plate format (13 µL/well for 386 wells) of the gel solution prepared in the previous step into the titration wells the well plate.
- For the calibration wells, first add 5 nM NB to the melted gel outside the wells (total volume depending on the well plate format used and on the number of calibration wells; usually 5-6 per well plate is enough).
- Pipette 200 µL (13 µL for 384-well plate format) of NB containing gel slowly within the titration wells of the well plate and make sure to avoid air bubbles.
NOTE: The usage of electronic pipets or robotics significantly increases reproducibility. - Let the gel solidify for 10 min at RT, and another 10 min at 4 °C (remove condensation from the glass afterwards if necessary). Make sure to conduct all these steps on a perfectly horizontal surface to avoid inhomogeneous gel surfaces.
NOTE: The protein containing gels are usually stable for at least several hours at 4 °C.
5. Adding the Competitor DNA Solution
NOTE: The following solutions should be prepared before starting the titration and are added on top of the calibration and titration wells simultaneously.
- Add the annealed labeled reference DNA and protein in 3x binding buffer at 3 times higher concentrations than the gel stock aliquots.
- Mix 20 µL of the obtained solution with 40 µL of each annealed competitor DNA solution obtained in step 3.
- For each calibration well, mix 20 µL of 3x binding buffer containing 15 mM NB solution with 40 µL of annealed competitor DNA (any sequence of the same length is suitable).
NOTE: For the 384-well plates, use 21 µL instead of 60 µL in total.
- Optionally, check the homogeneity of the gel height levels in the different wells of the plate spectroscopically by measuring the absorbance at 380 nm, using a multi-well plate reader (the absorbance values are proportional to the gel heights).
- Add 50 µL (7 µL for 384-well plate format) of the mixed competitor DNA solutions (annealed in step 3) on top of the gels. Try to add all the competitor solutions as simultaneously as possible by using electronic multichannel pipets or a 96-channel pipetting head, if available. After addition of the competitor solutions, place the plate on the microscope stage and start the measurements immediately (step 7).
6. Image Acquisition
- Sequentially acquire times series of z-stacks (e.g., use 12 planes and 100-300 ms of illumination time). Avoid taking images too close to the well surface (< ~1.4 µm with the plates used herein) to exclude any polarization bias.
- Perform 10-25 cycles of measurements until complete unbinding of the labeled reference DNA from the TF. The endpoint is typically reached after 1-2 h, depending on the binding kinetics and diffusivity of the competitor DNA.
7. Extraction of FA(z,t) from Raw Data
- Once a well plate has been imaged, compute from the raw fluorescence images the average pixel values of regions of interest for the parallel (I=) and perpendicularly (I+) polarized intensity components (Figure 1c). This can be done automatically using the HiP-FA software23.
NOTE: The HiP-FA software, an instruction manual, and a test dataset can be downloaded23. Alternatively, use any other custom-written software to extract I= and I+ and perform the downstream analysis of the titration curves, as described in detail below. - Calculate FA for each well. For each well, the script computes FA(z,t) at each z-position and time point t according to:
Equation 1:
Where G is the instrument G-factor that corrects for any bias toward the perpendicular channel. - Determine the G-factor of the microscopy setup by measuring the FA of any solutions containing a fluorescent dye of known anisotropy. Extract the two polarization components of the signal and then use Equation 1 to obtain G, knowing the FA of the solution (G = 1.15 in this setup).
8. Calibration Curve for Determination of Competitor DNA Concentration from FANB
- Anneal 120 µL of each forward and reverse reference oligomer (100 mM stock concentration; any random sequence with the same length as the competitor sequence can be used) and mix with 120 µL of 3x binding buffer containing NB (15 nM).
- Prepare a dilution series with 1:2 dilutions in 1x binding buffer with 6 dilutions in total. Mix 50 µL of these dilutions with 200 µL of 0.5% low melting point agarose (T > 35 °C) gel in 1x binding buffer containing 5 nM of NB in triplicates.
- Add 200 µL of each of the 6 solutions prepared in the previous step in a 96-well plate and store the plate for 1 h at 4 °C to ensure complete gelation, then 1 h at RT. Measure the FANB of the solutions by using the HiP-FA setup.
- Extract FANB (z,t) according to the previous step and fit the data using a Hill equation:
Equation 2:
Where CDNA is the concentration of the DNA oligomer; k the concentration of the DNA oligomers at which half of the binding sites are occupied; FAmax is a normalization constant; and n is the Hill coefficient. k, FAmax, and n are set as free parameters during the fitting procedure. - Enter the three parameters obtained from the fitting procedure in the HiP-FA software (in the left lower panel).
- Repeat the determination of the calibration curve every few months or after making changes in the microscopy setup.
9. Determination of Competitor DNA Concentrations
- Use the HiP-FA software to extract c(z,t) from the FANB(z,t) measurements (Figure 3). First obtain the calibration curve as described in the previous section and enter the fitting parameters to the software (see manual for details).
- Use the program to automatically extrapolate c(z,t) for c < 100 nM (see manual for details) using Equation 3 (Figure 3b), which describes the one-dimensional diffusion of the competitor DNA within the agarose gel matrix, assuming free diffusion.
Equation 3:
Where C0 is the initial concentration of the competitor DNA; erf is the error function; z is the position; D is the diffusion coefficient of the competitor DNA; and t0 is the starting time of the measurements. The free parameters used are C0 and z/ .
.
10. Conventional Competitive Titration with HIP-FA for Very Strong DNA Binding
- Serially dilute the different competitor DNA oligomers in the rows of a 96-well plate (or 384-well plate) at the concentrations of: 0, 1.25, 3.5, 9, 19, 45, 90, 190, 425, 900, 1900, and 4000 nM. Add the Cy5-labeled reference DNA (1 nM) and TF (20-50 nM) at a constant concentration, with a total volume of 200 µL per well in binding buffer (Figure 5a). Wait 40 min until thermodynamic equilibrium is achieved and acquire (with the HiP-FA setup) z-stacks for each well (acquiring several images per well reduces variability by averaging the calculated FA values).
- Construct the equilibrium binding titration curves and fit them with Equation 4 (Figure 5b). The KDs determined by conventional competitive titration are identical to those obtained by HIP-FA using an agarose gel matrix20.
11. Fitting Procedure of the FA Titration Curves
- Display in the HiP-FA software the reconstructed titration curves for the individual competitor sequences FA(z,t) = f[c(z,t)] and visually check the data quality (see manual for details). If needed, refine the parameters used for the determination of the competitor DNA concentrations in step 10.
- Fit each individual titration curve automatically using Equation 4, which gives an analytical solution for competitive titration assays24.
Equation 4:
With:



Where RT is the protein concentration; LT is the unlabeled and LST is the labeled DNA concentration; KD2 is the dissociation constant to be determined; RT is the concentration of active protein; and A and B are normalization parameters.
First determine KD1, which serves as a reference for the determination of the different KD2 values. KD1 is can be readily determined with the assay by choosing the sequence of dye-reference DNA as the sequence of the unlabeled competitor DNA (see manual). - Enter the obtained value of KD1 in the software and compute the KD2 values for all the competitor DNA on the plate.
NOTE: The free parameters of the fitting procedure are KD2, RT, A, and B. - Export the dissociation constant KD2 and concentration of active protein RT for all the individual titration wells of the plate by clicking the "Export" button in the software.
12. PWM Construction, Specificity of Protein-DNA Interaction, and Pseudo Counts
Create the sequence logos for the different PWMs using the online tool WebLogo 3.0 (http://weblogo.threeplusone.com/create.cgi) as described previously20.
Results
We applied HiP-FA to TFs of the segmentation gene network25,26, which generates the anterior-posterior body pattern of Drosophila embryos, largely through transcriptional regulation. From this network, we chose the bZIP domain TF Giant (Gt) for a detailed analysis (Figure 4). Since full-length transcription factors are difficult to express and yield mostly the same binding preferences as their DNA binding domains (DBD)13, we used the DBD fused to GST and expressed the construct in E.coli GST fusion proteins to deliver the same results as DBDs alone20.
The Gt consensus sequence (ATTACGTAAC) represents the strongest binding sequence that we determined as described above. We then investigated the influence on the binding energy of all possible single-point mutations within the 10-mer Gt consensus sequence (a total of 30), flanked by additional bases at the 5' and 3' ends. We measured two replicates whose gel samples were produced using automation, and one additional replicate produced manually for comparison. The KDs ranged from 0.6 to > 2000 nM for singly mutated sequences, and we also confirmed complete lack of binding to a "non-binding" sequence (data not shown).
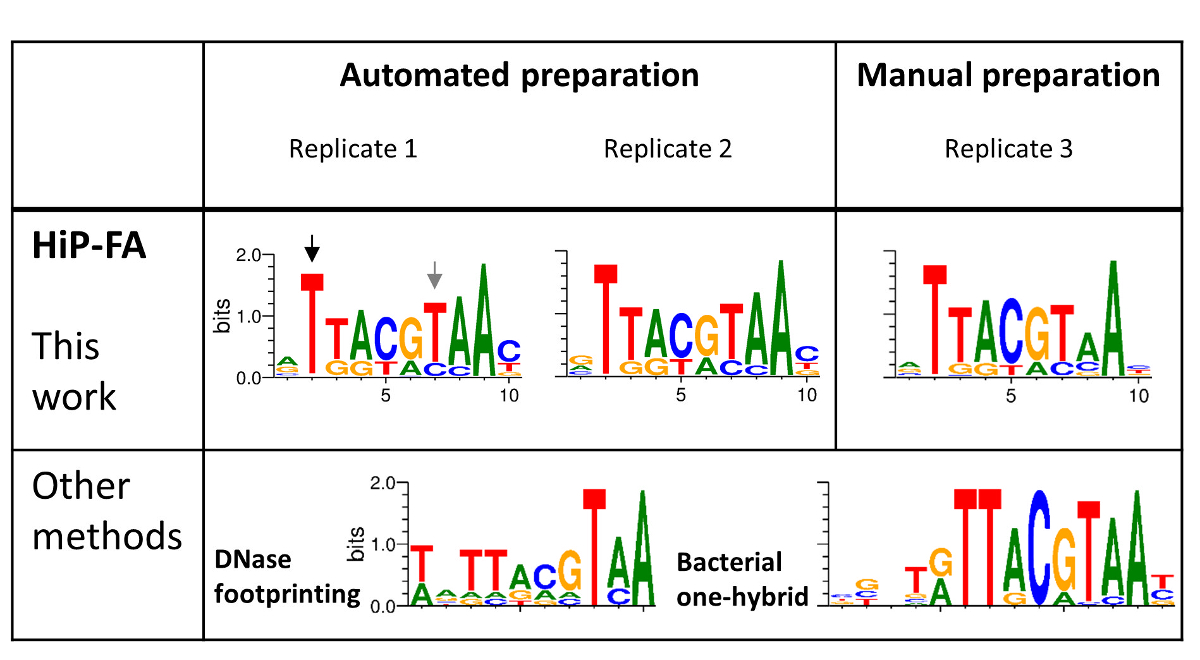
TF-DNA binding specificities are typically modelled using a position weight matrix (PWM), in which a score is assigned to each possible nucleotide at each position in the binding motif. The PWM assumes that each position contributes to binding strength independently and in most cases constitutes a sufficient model for TF binding preferences27. We generated revised PWMs based on our affinity measurements following established procedures28,29 and compared them to two PWMs previously reported in the literature. The first is based on nucleotide frequency counts from binding sites identified by DNA footprinting4,5, and the second PWM was obtained by bacterial one-hybrid (B1H) selection.15
The high similarity of the PWMs for the three replicates (Figure 4, upper panel), including for the sample prepared manually (replicate 3), demonstrates the high reproducibility of the HiP-FA method. While the PWMs obtained by HiP-FA are overall similar to the PWMs obtained with the other methods (Figure 4, lower panel), there are significant deviations: at position 2 (black arrow), the start of the core bZIP motif, mutations T>(G,C,A) lead to complete loss of binding in the motif obtained by HiP-FA, which is consistent with the B1H motif but not with that obtained with DNase footprinting, in which the binding remains relatively strong for bases (G,A). Conversely, at position 7 (gray arrow), the mutation T>C leads to much stronger binding than what was expected based on the previously measured PWMs.
Other deviations are subtler but no less important. Overall, the HiP-FA PWM is less specific than the other two, reflecting the fact that many mutations from the consensus still result in moderately strong binding. This can be quantified using the information content (IC). The IC is 11.5 bits for the HiP-FA matrix (average of the three replicates), compared to IC = 13.4 bits and 16.8 bits for the DNase footprinting and B1H matrices, respectively. Generally (though not universally), the PWMs obtained by HiP-FA are less specific than those obtained by other methods, based on 26 TFs investigated20.

Figure 1:Schematic depictions of the HIP-FA assay and experimental setup. (a) Gel delivery system for titrating competitor DNA in single wells. (b) HIP-FA microscopy setup.Customized automated widefield microscope with polarized fluorescence light detection on an EM-CCD camera. (c) Raw fluorescence image with the two regions of interest used to determine the parallel (red) and the perpendicularly (green) polarized components. (d) Typical layout of a 96-well plate. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Raw FA data and reconstructed titration curves. (a) Typical FA(z,t) trajectory for a calibration well containing NB. (b,c) FA(z,t) time trajectories for two titration wells measuring binding to a strong (b) and more moderate (c) DNA binding competitor. (d, e) Corresponding reconstructed FA titration measurements and fitted curves for the strong (d) and moderate (e) binding. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Determining the concentration of the competitor DNA c(z,t) using Nile Blue (NB). (a) FA-concentration calibration curve for 16 base pairs DNA oligomers. In a conventional titration series, NB is embedded in agarose gel together with different concentrations of competitor DNA. The affinity of NB to DNA is sequence-independent20; therefore, the same calibration curves can be used for determining c(z,t) for different DNA sequences of the same length. (b) Competitor DNA time diffusion profile at an arbitrary z height determined using five calibration wells. For each measurement cycle, the average FAs of four NB-containing wells are displayed as white dots. The curves are fitted using Equation 3 (white line, individual wells in color) and the c(z,t) at low concentrations (C < ~100 nM) is determined by the extrapolated fitting curve. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Binding specificities of the bZIP domain family TF Giant (Gt). HIP-FA PWMs of three replicates: two prepared using automation and one manually prepared (upper panel) are compared with PWMs generated by DNase footprinting and by the bacterial one-hybrid (B1H) selection method (lower panel). Overall, the HIP-FA binding motifs agree with previous data but also show significant differences, as highlighted with black and gray arrows. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Conventional competitive titration with HIP-FA. (a) Plate design shows 8 different competitor DNA serially diluted in binding buffer in a single row of a 96-well plate. An FA heat map is shown for different arbitrary binding strengths. (b) Competitive titration of three competitor DNAs binding to the Bcd TF with different affinities. Please click here to view a larger version of this figure.
{kind=link}
Discussion
HiP-FA is a comprehensive new method for determining the binding preference landscapes of TF-DNA interactions. It measures binding affinities of mutational DNA motif variants directly, avoiding any underlying assumption that binding preferences are reflected in the frequency of nucleotide occurrence in a set of above-threshold binders. Measurement takes place in the solution without immobilization and mechanical or chemical interference with the binding reaction, approximating equilibrium conditions as closely as possible. The controlled delivery system permits the measurement of a full titration curve within a single well and increases both throughput and reliability while saving protein. Using an objective with a high numerical aperture and EM-CCD camera with a high light collection efficiency allows for highly sensitive fluorescent light detection. Hence, with this setup, small FA changes as low as 10-15 mP can be accurately detected; in practice, this means that any binding reaction for which the mass increase after binding is minimal (as low as a mass ratio of 2) is readily detected. This is usually not the case with commercial systems like microplate readers. Due to its high sensitivity, HiP-FA extends the range of dissociation constants that can be measured reliably into the picomolar range. Binding energies are determined accurately over multiple orders of magnitude.
To evaluate the quality of the revised PWMs, we performed two types of analysis20. We tested, for five factors of the segmentation gene network, how well different PWMs can predict experimental ChIP-seq profiles in the genomic regions of 21 segmentation genes. As a second test, we used a sequence-to-expression model4 that predicts expression patterns of segmentation enhancers on the basis of the binding preference and protein concentration of participating TFs. In both exercises, we found that the less specific HiP-FA PWMs perform significantly better than the more specific footprinting and B1H PWMs20.
Unlike de novo methods, HiP-FA requires some prior knowledge of a given TF's binding preference. However, consensus sequences are known for many TFs, and many existing methods can supply them13,14,15. If needed, the true optimal binding sequence can be found iteratively.
We used DNA reference oligomers fluorescently labelled with Cy5 and Bodipy-650. These dyes have proven to perform well for FA measurements since the anisotropy of bound and unbound labelled-reference DNA were the largest among the different tested dyes. This ensures a maximum dynamical range for the FA values. Generally, any fluorescent dye with a fluorescence lifetime ≥ 1 ns is likely to be suitable but needs to be tested first. If possible, it is advised to use dyes fluorescing in the near-IR range to minimize protein autofluorescence.
The most critical step of the experimental procedure is the pipetting of the gel into the well plates. Good reproducibility requires the gel volumes to be as uniform as possible. Changes in gel height are translated into changes of diffusivity for the competitive DNA oligomer, and thus in apparent changes of affinity when evaluating the data. This is the main source of variance in a technical replicate. The use of an electronic pipet or automation techniques improves reproducibility. Air bubbles within the gel can be avoided by slow and careful pipetting. It is also important to add the all competitor solutions on top of the titration wells with as little delay as possible. For best reproducibility, the whole process can be automated using a pipetting robot with heat incubators. A critical part for transferring the protocol to automation is the necessary optimization of incubator temperature and the incubation times. Make sure to find an optimal balance between the viscosity of the gel (i.e., not too cold) and the stability of proteins (i.e., not too hot). This depends both on the dispensing speed of the gels into the wells and stability of the protein used.
HiP-FA makes use of a controlled delivery system for the competitor DNA oligomers. To construct the titrations curves, it is necessary to determine the competitor DNA concentration c(z,t) for each given z-position within the gel matrix and time point t. This is another critical step, since the determination of the KDs depends directly on c(z,t). Calibration wells containing the NB dye as a sensor for DNA concentration are used for this purpose (Figure 1d, Figure 2a). Usually, 3-5 calibration wells containing NB per plate are sufficient. Before evaluating any HiP-FA experiment, a NB calibration curve should be constructed for the set-up by performing a conventional titration series of NB dissolved in the agarose gel with a competitor DNA of any sequence at different concentrations (Figure 3a), as explained in detail in step 8. In the case of very strong binding (KD < 500 pM), the extrapolation used for determination of low concentrations of competitor DNA becomes limiting, since it is less accurate than a direct measurement. However, for TFs with such low KDs, the HiP-FA setup can be used to perform a conventional competitive titration in binding buffer without the use of an agarose gel matrix (Figure 5). For example, one full titration with 12 different concentrations of competitor DNA can be performed in a single row of a 96-well plate.
The controlled delivery system also requires fast TF-DNA binding kinetics and stable proteins, since the diffusion though the agarose gel is dynamical (although slow). Both properties can be tested directly with the HiP-FA setup by following, over time, the FA of the TFs of interest when bound to their respective fluorescently labeled reference DNA. We measured KON and KOFF rates for the investigated factors and found them to be on the order of milliseconds to seconds20, in accordance with other studies30. This is sufficiently fast to ensure that measurements take place at equilibrium. In the case of other binding reactions with slower kinetics, the diffusivity of the competitor can be tuned by lowering its concentration or reducing gel pore size. In the case of the tested TFs, which all have fast TOFF (~seconds), a total measurement time of about 1-2 h is sufficient to ensure thermodynamic equilibrium at each measurement.
Another potential issue related to the protein is the formation of protein aggregates that may alter FA measurements. The use of other buffer conditions containing different additives (like tensides) can prevent aggregate formation, if necessary.
We have worked under the linearity assumption of the PWM; however, HIP-FA can be scaled to include all possible di-nucleotide mutations of the consensus sequence. Finally, HiP-FA can be adapted to measure other types of binding interactions. The prerequisite is to have available a suitable reference molecule bound by the protein which can be fluorescently labeled. With the controlled delivery system, a concentration gradient can be generated for any kind of ligand; therefore, protein-protein and drug-protein interactions can be measured with similarly high fidelity and throughput.
Disclosures
The authors declare no conflict of interests.
Acknowledgements
We thank J. Müller for cDNA clones and members of the Gaul lab, in particular S. Bergelt, for valuable advice and spirited discussion. This work was supported by SFB 646, Regulatory Networks in Genome Expression and Maintenance (C.J., P.B.), the Center for Integrated Protein Science (U.G.) and the Graduate School for Quantitative Biosciences Munich (M.S.). U.G. acknowledges support by the Deutsche Forschungsgemeinschaft (SFB 646, SFB 1064, CIPSM, QBM), the Bundesministerium für Bildung und Forschung (BMBF: ebio - Innovationswettbewerb Systembiologie), and the Humboldt-Foundation (Alexander von Humboldt, Professorship).
Materials
| Name | Company | Catalog Number | Comments |
| Cy5-labled 16- / 18-bp DNA-oligomers | Eurofins | Custom synthesis | |
| 16- / 18-bp DNA-oligomers | Eurofins | Custom synthesis | |
| Nile Blue A | Sigma | N5632-25G | |
| Sensoplate plus microplate 96- or 384-well, PS | Greiner | 655891 | 175 µm thick glass bottom |
| 384 Well Sensoplate, black | Greiner | 788896 | |
| Agarose, low gelling temperature | Sigma | A9414-50G | |
| Sodium Chloride | Merck | 1.06404.1000 | |
| Tween-20 | Sigma | P1379-1L | |
| Di-Potassium hydrogen phosphate trihydrate | Merck | 1.05099.1000 | |
| Potassium dihydrogen phosphate | Merck | 1.04873.1000 | |
| Q-POD Element | Merck Millipore | ZMQSP0DE1 | |
| Millipak 40 Gamma Gold Filter | Merck Millipore | MPGL04GK2 | |
| Milli-Q Integral 3 Water Purification System | Merck Millipore | ZRXQ003WW | |
| Quantum TIX | Merck Millipore | QTUMOTIX1 | |
| DL-Dithiothreitol | Sigma | 43815-1G | |
| Mastercycler gradient | Eppendorf | Z316083 | |
| SafeSeal tube 1.5 mL | Sarstedt | 72.706.200 | |
| Tube 15 mL | Sarstedt | 62.554.502 | |
| Multiply-Pro cup 0.2 mL PP | Sarstedt | 72.737.002 | |
| MICROSCOPY SETUP: | |||
| Automated widefield microscope | LEICA | DMI6000 | |
| Long distance objective | LEICA | HCX PL FLUOAR L 60x/0.60 N.A. Dry | |
| 638 nm line continuous diode laser | Omicron | PHOxX 638-40, 40mW | |
| Back-illuminated EM-CCD Camera | Andor | iXon DV897 | |
| Dichroic mirror | AHF | 640nm cut-off | |
| Bandpass filter | AHF | ET bandpass 700/75 | |
| Linear polarizer | Thorlabs | LPVISC050-MP2 | |
| Polarizing beam splitter | Thorlabs | BS010 | |
| Achromatic lens | Thorlabs | 200 mm focal length | |
| Multimode optical fiber | Optronis | FVP600660710 | |
| ROBOTIC SYSTEM: | |||
| Our robotic system includes a Biomek NXP workstations with a 96-channel head and with Span-8 pipettors, connected with a servo-shuttle, are used for all liquid transfer steps. In addition, the system is equipped with orbital shakers and a microplate reader (Paradigm, Molecular device) served by the Span-8 gripper | Beckman Coulter | Biomek NXP | |
| SOFTWARE: | |||
| Programming language | National Instruments | Labview 9.0 | |
| Script for the HiP-FA software available at | https://github.com/GeneCenterMunich/HiP-FA |
References
- Berg, O. G., von Hippel, P. H. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. Journal of Molecular Biology. 193, 723-750 (1987).
- Hammar, P., et al. Direct measurement of transcription factor dissociation excludes a simple operator occupancy model for gene regulation. Nature Genetics. 46, 405-408 (2014).
- Chen, J., et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 156, 1274-1285 (2014).
- Segal, E., Raveh-Sadka, T., Schroeder, M., Unnerstall, U., Gaul, U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 451, 535-540 (2008).
- Schroeder, M. D., et al. Transcriptional control in the segmentation gene network of Drosophila. PLoS Biology. 2, 271(2004).
- He, X., Samee, M. A., Blatti, C., Sinha, S. Thermodynamics-based models of transcriptional regulation by enhancers: the roles of synergistic activation, cooperative binding and short-range repression. PLoS Computionnal Biology. 6, (2010).
- Wilson, M. D., et al. Species-Specific Transcription in Mice Carrying Human Chromosome 21. Science. 322, 434-438 (2008).
- Galas, D. J., Schmitz, A. Dnaase footprinting - simple method for detection of protein-DNA binding specificity. Nucleic Acids Research. 5, 3157-3170 (1978).
- Hellman, L. M., Fried, M. G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nature Protocols. 2, 1849-1861 (2007).
- Liedberg, B., Nylander, C., Lundstrom, I. Surface-plasmon resonance for gas-detection and biosensing. Sensors and Actuators. 4, 299-304 (1983).
- Wienken, C. J., Baaske, P., Rothbauer, U., Braun, D., Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nature Communications. 1, (2010).
- Berger, M. F., et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature Biotechnology. 24, 1429-1435 (2006).
- Nitta, K. R., et al. Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife. 4, (2015).
- Jolma, A., et al. DNA-Binding Specificities of Human Transcription Factors. Cell. 152, 327-339 (2013).
- Noyes, M. B., et al. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Research. 36, 2547-2560 (2008).
- Nutiu, R., et al. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nature Biotechnology. 29, (2011).
- Riley, T. R., et al. SELEX.-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods Molecular Biology. 1196, 255-278 (2014).
- Maerkl, S. J., Quake, S. R. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 315, 233-237 (2007).
- Isakova, A., et al. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nature Methods. 14, 316-322 (2017).
- Jung, C., et al. True equilibrium measurement of transcription factor-DNA binding affinities using automated polarization microscopy. Nature Communications. 9, 1605(2018).
- Weber, G. Polarization of the fluorescence of macromolecules: Fluorescent conjugates of ovalbumin and bovine serum albumin. Biochemical Journal. 51, 155-168 (1952).
- Github. , Available from: https://github.com/Reutern/PySite (2018).
- Github. , Available from: https://githum.com/GeneCenterMunich/HiP-FA (2018).
- Roehrl, M. H., Wang, J. Y., Wagner, G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization. Biochemistry. 43, 16056-16066 (2004).
- St Johnston, D., Nuesslein-Volhard, C. The origin of pattern and polarity in the Drosophila embryo. Cell. 68, 201-220 (1992).
- Pankratz, M., Jäckle, H. The Development of Drosophila melanogaster, Vol. 1. Bate, M., Martinez Arias, A. , Cold Spring Harbor Laboratory Press. Cold Spring Harbor. 467-516 (1993).
- Zhao, Y., Ruan, S. X., Pandey, M., Stormo, G. D. Improved Models for Transcription Factor Binding Site Identification Using Nonindependent Interactions. Genetics. 191, (2012).
- Noureddine, M. A., et al. Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genetics. 5, 1000462(2009).
- Veprintsev, D. B., Fersht, A. R. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Research. 36, 1589-1598 (2008).
- Geertz, M., Shore, D., Maerkl, S. J. Massively parallel measurements of molecular interaction kinetics on a microfluidic platform. Proceedings of the National Academy of Sciences. 109, 16540-16545 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved