
Method Article
A Fluorogenic Peptide Cleavage Assay to Screen for Proteolytic Activity: Applications for coronavirus spike protein activation
* These authors contributed equally
In This Article
Summary
We present a fluorogenic peptide cleavage assay that allows a rapid screening of the proteolytic activity of proteases on peptides representing the cleavage site of viral fusion peptides. This method can also be used on any other amino acid motif within a protein sequence to test for the protease activity.
Abstract
Enveloped viruses such as coronaviruses or influenza virus require proteolytic cleavage of their fusion protein to be able to infect the host cell. Often viruses exhibit cell and tissue tropism and are adapted to specific cell or tissue proteases. Moreover, these viruses can introduce mutations or insertions into their genome during replication that may affect the cleavage, and thus can contribute to adaptations to a new host. Here, we present a fluorogenic peptide cleavage assay that allows a rapid screening of peptides mimicking the cleavage site of viral fusion proteins. The technique is very flexible and can be used to investigate the proteolytic activity of a single protease on many different substrates, and in addition, it also allows exploration of the activity of multiple proteases on one or more peptide substrates. In this study, we used peptides mimicking the cleavage site motifs of the coronavirus spike protein. We tested human and camel derived Middle East Respiratory Syndrome coronaviruses (MERS-CoV) to demonstrate that single and double substitutions in the cleavage site can alter the activity of furin and dramatically change cleavage efficiency. We also used this method in combination with bioinformatics to test furin cleavage activity of feline coronavirus spike proteins from different serotypes and strains. This peptide-based method is less labor- and time intensive than conventional methods used for the analysis of proteolytic activity for viruses, and results can be obtained within a single day.
Introduction
Viral fusion with the host cell membrane is a crucial step in the life cycle of enveloped viruses and facilitates entry into the cell and replication of the virus. Typically, viruses possess a specialized protein (or proteins) that binds to receptors on the host cell membrane and triggers the virus-cell membrane fusion1. Viral fusion proteins have been grouped into three main classes (I-III)1. A number of viruses that are currently a major concern for public health, such as influenza virus (representing a long-standing zoonotic emergence from birds) and Middle East Respiratory Syndrome-coronavirus (MERS-CoV) (representing a recent zoonotic emergence from camels), utilize the so-called class I fusion proteins, which require proteolytic processing by host proteases, to exert their fusogenic activity2. Similarly, feline coronavirus (FCoV), which represents a major disease threat for wild and domestic cats, also possess a class I fusion protein. Class I viral fusion proteins are typically synthesized as an uncleaved precursor, and generally consist of two domains that are responsible for receptor binding and triggering the fusion event respectively. To date, the influenza hemagglutinin (HA) is the best understood fusion protein and numerous studies have described its mechanistic role in host cell entry and fusion3. In this case cleavage of the fusion protein occurs at a specific sequence or cleavage site within HA, and in combination with pH-dependent conformational changes, results in the exposure of the viral fusion peptide1,2.
The fusion peptide and its preceding protease recognition sequence are critical for the pathogenicity and the host adaptation of the virus. Changes in the protease recognition sequence can alter cleavage significantly with potentially dramatic consequences for the virus and the host4. On one hand, it can abrogate cleavage and thus eliminate the life cycle of the virus. But on the other hand, a mutation can increase the substrate specificity for a given protease and/or allow a "new" protease to cleave the fusion protein. Subsequently, this can also expand the cell and tissue tropism as observed, e.g., with influenza virus subtypes5,6. Usually low pathogenicity avian influenza (LPAI) viruses and most human influenza viruses are confined to the gastrointestinal or respiratory tract because of the limited localization of the proteases that activate them. Typically, the fusion peptide of such HA proteins is preceded by a four-amino acid sequence that consists of 1-2 non-consecutive basic amino acids, which is recognized by trypsin-like serine proteases like trypsin, matriptase or TMPRSS27,8. When the virus acquires insertions or mutations that change the cleavage site to a polybasic site, it allows furin to activate the HA and to potentially cause a much more severe systemic infection6,9. These viruses are referred to as high pathogenicity avian influenza virus (HPAI), typified by H5N1 strains.
In contrast to influenza HA, many coronaviruses, such as MERS-CoV, have two distinct cleavage sites within their spike protein. The S1/S2 site separates the N-terminal receptor binding domain (S1) from the C-terminal fusion domain (S2), with a second cleavage site, called S2', downstream of the S1/S2 site, in proximity to the fusion peptide10. It was suggested that the sites are cleaved sequentially, at S1/S2 followed by S2'. In contrast to the HA of most influenza virus strains, the MERS-CoV S protein S1/S2 and S2' sites can be recognized by proteases of the proprotein convertase (PC) family, such as furin. Members of this family cleave at paired basic residues with the motif R/K-(X)0,2,4,6-R/K(X, any amino acid)2. Generally, the amino acids upstream of the cleavage site are referred to as P1, P2, P3, etc. counting from the cleavage site and the amino acids downstream are designated as P1', P2', P3', etc.11. FCoV strains can either have a single or a dual cleavage site. Like MERS-CoV, some FCoV strains also possess two cleavage sites (S1/S2 and S2') in their S protein. However, this characteristic is exclusive of serotype I FCoVs (clade A). In contrast, members of the serotype II (clade B) grouping only have a single cleavage site (S2')2,12. Several proteases have been suggested to cleave the FCoV cleavage sites, including furin, trypsin-like proteases and cathepsin. It has been proposed that the S protein of enteric FCoV (also known as feline enteric coronavirus or FECV) is likely to be cleaved by furin in the S1/S2 site, and mutations in this site (as well as at S2') leads to changes in protease requirements. These mutations have been associated with changes in the tropism and pathogenicity of these viruses, allowing the virus to become systemic and macrophage-tropic (also known as feline infectious peritonitis virus or FIPV)13.
Viruses naturally introduce mutations into their genomes during each replication cycle and frequently new subtypes and strains of influenza, MERS-CoV and FCoV are described14,15,16. As a part of a rapid evaluation to assess the public health threat of emerging viruses, it is critical to investigate changes in the cleavage site and how it affects the range of proteases activating these viruses. Here, we describe a peptide-based assay that allows a very quick assessment of how the cleavage site changes in MERS-CoV S protein affect the substrate specificity of a given protease and to rapidly screen various proteases for their ability to cleave a given or multiple sequences4. In a second set of experiments, we used the technique to determine the furin cleavage activity over different FCoV serotypes and strains.
The peptides used in this assay are modified with the fluorescence resonance energy transfer (FRET) pair, 7-methoxycoumarin-4-yl acetyl (MCA) at the N-terminus and N-2,4-dinitrophenyl (DNP) at the C-terminus. During the assay, MCA is excited and emits light energy that is quenched by DNP as long as the pair is in close vicinity to each other. If cleavage occurs, however, DNP is not able to quench the emission anymore and it can be read by the fluorescence plate reader. The changes in the fluorescence is measured during the experiment to determine the peptide cleavage rate, and to calculate the velocity at which the protease cleaves the specific peptide (also known as Vmax)17.
In order to design the peptides, the gene of the respective fusion protein must have been sequenced and/or made available in a database. However, the method is less labor-intense and costly than conventional methods that usually require the cloning of the fusion protein gene into expression vectors to express it in mammalian cells to analyze the cleavage. From start to end, this may take several days up to a few weeks while the peptide assays presented here can be done within one day as soon as peptides and proteases are available. The setup of the assay takes between 5 and 30 min depending on the number of samples and the runtime in the fluorescence plate reader is 1 h. Analysis of the data may take up to 2 h again depending on the sample size.
Here, we chose two different examples to present the assay. In the first example, we present data that compares the furin-mediated cleavage of human and camel-derived MERS-CoV, to assess the potential of the camel-derived strains of being activated in humans if they cross the species barrier. In the second example, we used a fluorogenic peptide assay to determine the furin-mediated cleavage of the S1/S2 and S2' sites or S2' site of two serotype I and two serotype II FCoV strains, respectively. For these experiments, we used trypsin cleavage as a positive control.
Protocol
1. Designing and Preparing the Peptides
- Acquire the sequence of the fusion protein of interest from a public database such as NCBI (https://www.ncbi.nlm.nih.gov/) or the virus pathogen database (https://www.viprbrc.org). Choose the protease recognition site preceding the fusion peptide and include 2-3 amino acids upstream and downstream of this sequence.
- When ordering the peptides, modify them with the FRET pair 7-methoxycoumarin-4-yl acetyl (MCA) at the N-terminus and N-2,4-dinitrophenyl (DNP) at the C-terminus.
NOTE: Several suppliers provide these modifications. Price and delivery time depend on the supplier of choice. However, alternative modifications exist which vary in sensitivity and require adjustments of the plate reader in terms of wavelengths. - Resuspend the peptide according to the manufacturers recommendations by gently pipetting up and down. For example, resuspend peptides in 70% ethanol to the final concentration of 1 mM.
- Optionally, add the tube containing the peptide and the solvent into a sonication bath until it is fully resuspended if the peptide does not resuspend very well by pipetting.
- Aliquot the peptide into light dampening or resistant tubes to protect the peptide from bleaching. Keep aliquot sizes small to avoid multiple freeze/thaw cycles (e.g., 100 µL). Store aliquots at -20° C.
2. Preparing the Fluorescence Plate Reader
- Turn on the plate reader and wait until the self-test is finished.
- Open the operating software on the attached computer and make sure it is connected with the plate reader.
- Open the temperature setting and set to 30 °C (or the required temperature for optimal performance for the protease).
- Click on Control | Instrument Setup to set up the experiment.
- Choose Kinetic.
- Select Fluorescence.
- Enter Excitation and Emission wavelengths: 330 nm and 390 nm respectively.
- Unselect Auto Cut-off.
- Select Medium, normal sensitivity.
- Choose a runtime of 1 h for the assay and select one measurement every 60 s.
- Set 5 s mixing before first measurement and 3 s before each measurement
- Select wells to read and start the assay.
3. Preparing the Assay
- Prepare the assay buffer by calculating the appropriate quantities for each ingredient and adding it. For furin, make buffer consisting of 100 mM HEPES, 1 mM CaCl2, 1 mM 2-mercaptoethanol, 5% Triton X-100. For trypsin, use standard phosphate buffered saline (PBS) solution.
NOTE: Volumes of 10 mL or less are sufficient. Depending on the protease(s) used in the assay the buffer varies. - Chill the buffer on ice.
- Place the assay plate on ice with a thin metal plate underneath to support cooling and stability. Use a solid black polystyrene 96-well plate with a flat bottom and non-treated.
NOTE: It is essential to use black plates to prevent fluorescent "leakage" from adjacent wells. - Calculate the appropriate amount of protease to add for each reaction based on previous publications or experimental data. For furin, use 1 unit per reaction which corresponds to 0.5 µL. For trypsin, use 0.5 µL of 160 nM TPCK trypsin.
- Per sample prepare 3 technical replicates per assay in a total volume of 100 µL per sample.
NOTE: It was experimentally evaluated that it does not make a difference whether the pipetting is carried out under normal light conditions or dimmed light. However, the peptides should not be exposed to light for an extended period of time. - Pipette the appropriate amount of assay buffer (94.5 µL as described under Step 3.1) into each well.
- Add the protease to each well. For both, furin and TPCK trypsin, use 0.5 µL per sample. To 6 wells (3 wells for a blank control and 3 wells for a peptide control), add 0.5 µL buffer instead of the respective protease.
- Add the peptide to a final concentration of 50 µM to each well except the blank control. In this case, pipette 5 µL peptide prepared as described under Step 1.3. Add 5 µL of buffer to the blank control instead of peptide.
NOTE: Several concentrations of the peptide were initially tested with the described enzyme concentrations to ensure that the enzymes were saturated. - Insert the plate into the fluorescence plate reader and click start.
4. Data Analysis
- Save the experiment file.
- Click on Export and export the file as a .txt.
- Import the .txt file into a spreadsheet.
- For each technical replicate per sample, create a graph plotting the relative fluorescent units (RFU) on the y-axis against the time on the x-axis (Supplemental Figure 1).
- Select the data range where the graph is in a linear range and the closest to the start of the fluorescence increase.
- Plot the selected data on a second graph and add a linear trendline.
- Select in the trendline options Display equation on chart. The equation will show the Vmax.
- Calculate the average Vmax for each sample from the 3 technical replicates.
- Repeat the experiment twice more to obtain 3 biological replicates.
- Calculate the standard deviation based on the data from 3 independent biological replicates.
Results
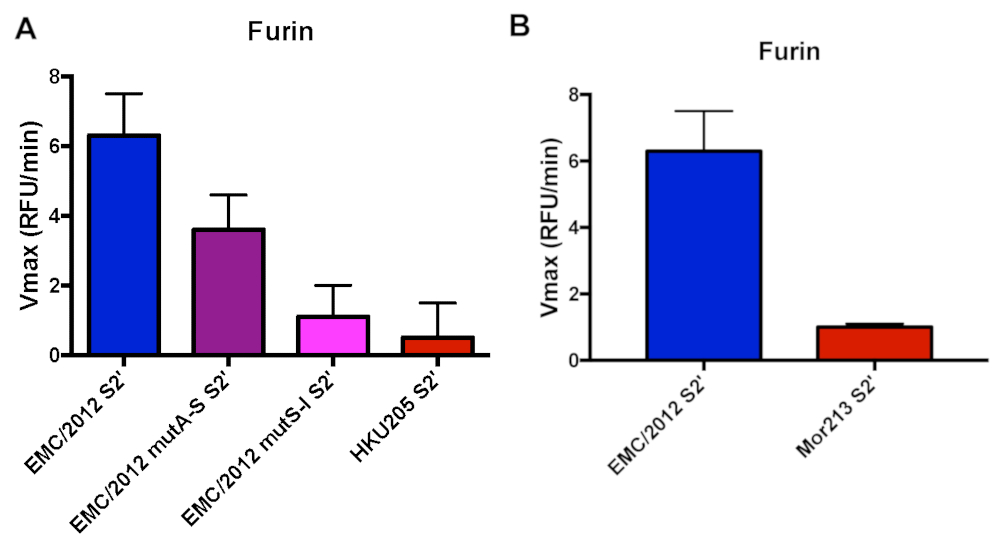
In the first part of this study, we used peptides that represent the S2' cleavage site of three distinct MERS-CoV S proteins: from the prototypical EMC/2012 human strain (Genbank AFS88936.1) and from two selected camel-derived MERS-CoVs, NRCE-HKU205 (Genbank AHL18090.1) and Mor215 (Genbank AVN89324.1) strains, isolated in Egypt and Morocco, respectively (Table 1)16,18.
Compared to the EMC/2012 strain, the HKU205 S2' cleavage site has two mutations in the P2 and P1' positions that could potentially impact its recognition by furin and alter the cleavage efficiency (Table 1). In addition, we were interested to investigate if and how each individual mutation found in the HKU205 strain affects furin cleavage. We, therefore, included two peptides where the individual substitutions were introduced into the EMC/2012 peptide sequence (EMC/2012 mutA-S S2' and EMC/2012 mutS-I S2') (Table 1). Furin was able to cleave the EMC/2012 peptide efficiently with a Vmax of 6.3 ± 1.2 RFU/min, while we detected almost no cleavage when using the S2' peptide of the HKU205 strain (Vmax 0.5 ± 0.1 RFU/min) (Figure 1a). When investigating the EMC/2012 mutA-S S2' peptide, we found that the cleavage was strongly reduced compared to the wild type sequence (Vmax 3.6 ± 1 RFU/min). There was almost no cleavage when testing the EMC/2012 mutS-I S2' peptide (Vmax 1.1 ± 0.9 RFU/min) (Figure 1a).
Recently, MERS-CoV isolates from Western Africa were reported that are phylogenetically distinct from MERS-CoV found in the Arabian Peninsula16. One of the recently described isolates is the Mor213 strain, which carries a leucine instead of an arginine in the P4 position of the S2' site (Table 1), and which could potentially impact its recognition by furin and alter the cleavage efficiency. Our peptide assay showed that there is only minimal cleavage of the Mor213 S2' site as compared to the EMC/2012 site (Figure 1b). The Vmax for the Mor213 peptide is 1.0 ± 0.1.
For the second part of this study, we used the same peptide assay to evaluate furin-mediated cleavage of FCoV S protein cleavage sites. We used peptides mimicking the S1/S2 cleavage site of two FCoV serotype I viruses: FECV I And-4 (from Whittaker lab) and FIPV I Black (Genbank AB088223.1). We also used peptides mimicking the S2' site of these viruses, as well as two FCoV serotype II: FECV II 1683 (Genbank AFH58021.1) and FIPV II 1146 (Genbank AAY32596.1) strains (Table 1). Typically, furin cleavage occurs when basic residues (arginine and lysine) are present in the P1 and P4 positions of the cleavage site. Mutations in the P1, P4 and P1' positions are suggested to interfere with furin cleavability, therefore altering the protease requirements of the protein. (Table 1). Furin cleavage was observed for the prototypical S1/S2 peptide FECV I And-4 S1/S2 (Vmax 100.36 ± 7.61 RFU/min) but not in the mutated S1/S2 peptide FIPV I Black S1/S2 (Vmax -1.37 ± 1.29 RFU/min) (Figure 2A). Similarly, furin was able to cleave the prototypical S2' peptide FECV II 1683 S2' (Vmax 5.46 ± 0.97 RFU/min), but not the mutated S2' peptides FECV I And-4 S2' (Vmax 0.26 ± 0.11 RFU/min), FIPV I Black S2' (Vmax 0.30 ± 0.14 RFU/min) and FIPV II 1146 S2' (Vmax -0.36 ± 0.11 RFU/min) (Figure 2B). We used trypsin as a positive control for peptide cleavage and we observed trypsin cleavage in all the peptides used (Supplemental Figure 2).

Table 1. Peptides used and corresponding amino acid sequences. The peptides used in the assays contain the (7-methoxycoumarin-4-yl) acetyl/2,4-dinitrophenyl (MCA/DNP) FRET pair. The P4 and P1 cleavage site positioned arginine (R) residues, which are recognized by furin, are in bold. Residues in red correspond to mutations compared to reference sequences.

Figure 1. Furin-mediated proteolytic cleavage of human and camel-derived MERS-CoV S2' sites fluorogenic peptides. A. Furin cleavage assay of human MERS-CoV EMC/2012 S2' site and camel-derived strain HKU205 (double mutant), along with single mutant variants of EMC/2012 (mutA-S and mutS-I). B. Furin cleavage assay of human MERS-CoV EMC/2012 S2' site and camel-derived strain Mor213. For panels A and B, peptides were incubated with recombinant furin and the increase in fluorescence due to proteolytic processing was measured using a fluorometer. The assays were performed in triplicates with results representing averages of Vmax from three independent experiments (n=3). Error bars indicate SD. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Furin-mediated proteolytic cleavage of FCoV S1/S2 and S2' sites fluorogenic peptides. A. Furin cleavage assay of FECV I And-4 and FIPV I Black S1/S2 sites. Peptides were incubated with recombinant furin. B. Furin cleavage assay of FECV I And-4, FIPV I Black, FECV II 1683 and FIPV II 1146 S2' sites. The increase in fluorescence due to proteolytic processing was measured using a fluorometer. The assays were performed in triplicates with results representing averages of Vmax from three independent experiments (n=3). Error bars indicate SD. Please click here to view a larger version of this figure.
{kind=link}
Supplemental Table 1: Vmax values used for Figure 1 and Figure 2. The Vmax values were calculated from graphs as illustrated in Supplemental Figure 1 and as described in Protocol section 4. Please click here to download this table.
Supplemental Figure 1: Illustration of raw data derived from the plate reader software. Increase of fluorescence over time graphs that were used for determination of the Vmax. Please click here to download this figure.
Supplemental Figure 2: Trypsin-mediated proteolytic cleavage of FCoV S1/S2 and S2' sites fluorogenic peptides. Trypsin cleavage assay of FECV I And-4 and FIPV I Black S1/S2 sites and of FECV I And-4, FIPV I Black, FECV II 1683 and FIPV II 1146 S2' sites. Please click here to download this figure.
Discussion
We present here a fluorogenic peptide assay that allows for a fast screening of protein sequences for their proteolytic cleavage by proteases. In our first example we chose different cleavage site motifs of the MERS-CoV spike (S) protein to illustrate one application for this assay. Several enveloped viruses that possess class I fusion proteins such as MERS-CoV, SARS-CoV (Severe Acute Respiratory Syndrome) and influenza are major concerns for public health and new subtypes are constantly evolving that have a potential to cross species barriers from their natural hosts to humans1,15,16. Isolating the viruses and cultivating them in the lab may not always be feasible or requires special laboratories that are not available in every research facility. Hence, there is a need for methods to assess the potential public health threat of emerging viruses that can be conducted under regular laboratory conditions. In addition to viruses targeting humans, some animal viruses like FCoV also possess the same class I fusion proteins, therefore, sharing the similar characteristics than their human counterparts. Isolating these viruses can also be a challenge, making the use of innovative tools to study them necessary.
A crucial step in the life cycle of the above-mentioned viruses is host cell entry and fusion mediated by proteolytic processing of the respective fusion protein by host proteases1,2. A standard way to investigate this part of the viral life cycle is to clone the fusion protein gene into a mammalian expression vector, transfect mammalian cells that allow for protein expression, incubate or co-transfect with the protease of interest, isolate the protein and perform a western blot analysis. This method comes with several limitations: availability of viral DNA/RNA for the cloning, costs for synthesizing the gene if no DNA or RNA is available, availability of an antibody to detect the fusion protein and its cleavage product(s), and it can take up to several weeks until the entire study is performed. It even becomes more time-consuming, money and labor intensive if the interest of the study is to investigate several mutations in the cleavage site because it requires site-directed mutagenesis. The fluorogenic peptide assay we describe here does not have these limitations. The peptides of interest can be designed based on publicly available sequences, there are several suppliers that provide a wide range of recombinant proteases that are relevant for these kinds of studies and the assay including the analysis can be performed within a single day.
In our example, we examined furin-mediated cleavage of the S2' protease recognition site of the MERS-CoV S protein derived from humans and camels from Egypt and Morocco. Both the human EMC/2012 and the camel HKU205 S2' sites contain a typical RXXR furin cleavage motif. However, according to the PiTou 2.0 cleavage scoring algorithm the HKU205 S2' site is not predicted to be cleaved by furin, which we experimentally confirmed19. The reason why HKU5 S2' is not processed by furin might be due to the isoleucine in the P1' position. When we tested two peptides that carried the individual mutations in the EMC/2012 S2' sequence, we were able to confirm that the isoleucine in the P1' largely abrogates cleavage whereas the alanine to serine substitution resulted in reduced cleavage. The Mor213 S2' site does not contain a furin cleavage motif and it was not surprising to detect almost no cleavage. We also examined how furin cleaves the S1/S2 and the S2' protease recognition sites of the FCoV S protein. We were able to observe furin cleavage of both FECV peptides (FECV I And-4 S1/S2 and FECV II 1683 S2'), while mutated sequences from FIPV viruses were not cleaved by furin.
When comparing furin-mediated cleavage of the EMC/2012 S2' peptide (Figure 1A) with the FECV I And-4 S1/2 peptides (Figure 2A), we found that there was an approximate 26-fold difference in the Vmax. This can be explained by the furin cleavage motif in both sequences (Table 1). While the minimum requirement for furin cleavage is a RXXR motif, a RXRR sequence is much more favorable2. Therefore, the EMC/2012 S2' peptide, which has a RSAR motif, is cleaved with less specificity compared to the FECV I And-4 S1/2 peptide that contains a RSRR furin cleavage site (Table 1).
However, one major limitation of the assay is that the peptides do not reflect the tertiary structure of the protein they are usually embedded in. Cleavage of the full length fusion protein exposes the fusion peptide and triggers host cell entry1. The interaction between protease and fusion protein might also be impacted by the tertiary structure of the fusion protein while we assume that the peptides are more or less linear. Hence, cleavage detected in the peptide assay may be artificial and may not reflect the in vivo situation. This was reported for cleavage of the influenza H3N2 HA subtype by matriptase. While several groups described cleavage of the H3N2 HA by matriptase in peptide assays, it was also shown that incubation of full length H3N2 HA protein and matriptase in cell culture did not result in a fusogenic HA protein4,20,21. There are other examples that show that the biologically important regulation of the proteolytic cleavage is on the level of protein conformation. It has been described that fusion proteins sometimes need a series of pre-fusion events to be able to expose the cleavage sites upon fusion. The Semliki Forest virus fusion protein, for example, requires structural rearrangements during a number of prefusion events in order to expose its fusion protein and make it available for proteolytic activation22. Therefore, the results obtained in a fluorogenic peptide assay need to be validated by cell fusion assays or similar experiments. However, the peptide assay still allows a fast screening of several protease/peptide combinations and to reduce the labor and time of follow up experiments since combinations that do not show cleavage are very likely to exhibit the same result in vivo.
The second major limitation of the assay concerns the proteases. So far it is only possible to test soluble proteases but not transmembrane proteases. Depending on the intent of the experiment, this can be a problem. For example, influenza HAs are activated by a number of transmembrane proteases located at cell membranes8. To a certain extent, this problem can be circumvented by using the soluble proteolytic active domains, but the results have to be interpreted with caution and should be validated with conventional methods. We want to refer to the beforementioned example with matriptase and its activity towards H3N2 HA. In vivo matriptase is located in the cell membrane but the commercially available enzyme is the soluble catalytic domain. As discussed with respect to the fusion proteins, it may be possible that it also requires the full-length protease to interact with the full-length protein substrate to obtain cleavage and that testing only single domains may result in false positives.
This methodology has shown to be efficient to study the cleavage of specific amino acid sequences by furin. However, the applications of the technique extents to any available protease (e.g., cathepsins, proprotein convertase), making it a useful method to screen peptide candidates for protease cleavage. Moreover, it has been shown that this assay can also be used to test the efficacy of protease inhibitors and hence provides a fast and easy to tool to screen protein inhibitors23.
The fluorogenic peptide cleavage assay described here represents an addition to bioinformatic cleavage scoring algorithms, which predicts the peptide cleavage by a determined protease, based on the amino acid properties and sequence. These two techniques, in combination with tradition western blotting techniques can be used as an efficient method to study protease cleavage in vitro.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Research funding for the MERS project presented in this manuscript was provided by NIH grant R21 AI111085. The feline coronavirus study was supported by research grants from Morris Animal Foundation, Winn Feline Foundation and the Cornell Feline Health Center. We also would like to thank Malik Peiris for providing us with Mor213 sequence before it was publicly accessible.
Materials
| Name | Company | Catalog Number | Comments |
| Peptides | Biomatik | N/A | |
| Furin | NEB | P8077S | |
| Trypsin, TPCK-treated | Sigma-Aldrich | 4352157-1KT | |
| HEPES | Sigma-Aldrich | H3375 | |
| CaCl2 | Sigma-Aldrich | C1016 | |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250 | |
| Triton-X100 | Sigma-Aldrich | X100 | |
| PBS | Corning | 21-040-CV | |
| SpectraMax Gemini XPS | Molecular Devices | XPS | |
| SoftMax Pro 6.5.1 | Molecular Devices | N/A | |
| 96-well plate (solid black polystyrene with a flat bottom and non-treated) | Costar | 3915 | |
| Light damping tubes | Watson Lab | 131-915BL or 131-915BR | |
| Microsoft Excel | Microsoft | N/A | |
| Prism 7 | GraphPad | N/A |
References
- Harrison, S. C. Viral membrane fusion. Virology. 479-480, 498-507 (2015).
- Millet, J. K., Whittaker, G. R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Research. , (2014).
- Hamilton, B. S., Whittaker, G. R., Daniel, S. Influenza virus-mediated membrane fusion: Determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses. 4 (7), 1144-1168 (2012).
- Straus, M. R., Whittaker, G. R. A peptide-based approach to evaluate the adaptability of influenza A virus to humans based on its hemagglutinin proteolytic cleavage site. PloS one. 12 (3), e0174827(2017).
- Kawaoka, Y., Webster, R. G. Sequence requirements for cleavage activation of influenza virus hemagglutinin expressed in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 85 (2), 324-328 (1988).
- Abdelwhab, E. S. M., et al. A Unique Multibasic Proteolytic Cleavage Site and Three Mutations in the HA2 Domain Confer High Virulence of H7N1 Avian Influenza Virus in Chickens. Journal of Virology. 90 (1), 400-411 (2016).
- Steinhauer, D. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 258 (1), 1-20 (1999).
- Böttcher-Friebertshäuser, E., Klenk, H. D., Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathogens and disease. 69 (2), 87-100 (2013).
- Horimoto, T., Nakayama, K., Smeekens, S. P., Kawaoka, Y. Proprotein-processing endoproteases PC6 and furin both activate hemagglutinin of virulent avian influenza viruses. Journal of Virology. 68 (9), 6074-6078 (1994).
- Belouzard, S., Chu, V. C., Whittaker, G. R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proceedings of the National Academy of Sciences. 106 (14), 5871-5876 (2009).
- Polgar, L. General Aspects of Proteases. Mechanisms of Protease Action. , 43-76 (1989).
- Whittaker, G. R., André, N. M., Millet, J. K. Improving Virus Taxonomy by Recontextualizing Sequence-Based Classification with Biologically Relevant Data: the Case of the Alphacoronavirus 1 Species. mSphere. 3 (1), 1-8 (2018).
- Licitra, B. N., et al. Mutation in spike protein cleavage site and pathogenesis of feline coronavirus. Emerging Infectious Diseases. 19 (7), 1066-1073 (2013).
- Sanjuan, R., Nebot, M. R., Chirico, N., Mansky, L. M., Belshaw, R. Viral Mutation Rates. Journal of Virology. 84 (19), 9733-9748 (2010).
- Trombetta, C., Piccirella, S., Perini, D., Kistner, O., Montomoli, E. Emerging Influenza Strains in the Last Two Decades: A Threat of a New Pandemic? Vaccines. 3 (1), 172-185 (2015).
- Chu, D. K. W., et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proceedings of the National Academy of Sciences. , 1-6 (2018).
- Caprioli, R. M., Smith, L. Determination of Km and Vmax for Tryptic Peptide Hydrolysis Using Fast Atom Bombardment Mass Spectrometry. Analytical Chemistry. 58 (6), 1080-1083 (1986).
- Millet, J. K., Goldstein, M. E., Labitt, R. N., Hsu, H., Daniel, S., Whittaker, G. R. A camel-derived MERS-CoV with a variant spike protein cleavage site and distinct fusion activation properties. Emerging microbes and infections. 5 (12), e126-e129 (2016).
- Tian, S., Huajun, W., Wu, J. Computational prediction of furin cleavage sites by a hybrid method and understanding mechanism underlying diseases. Scientific Reports. 2, (2012).
- Hamilton, B. S., Gludish, D. W. J., Whittaker, G. R. Cleavage Activation of the Human-Adapted Influenza Virus Subtypes by Matriptase Reveals both Subtype and Strain Specificities. Journal of Virology. 86 (19), 10579-10586 (2012).
- Beaulieu, A., et al. Matriptase Proteolytically Activates Influenza Virus and Promotes Multicycle Epithelium Replication in the Human Airway. Journal of virology. 30 (878), 4237-4251 (2013).
- Hammar, L., Markarian, S., Haag, L., Lankinen, H., Salmi, A., Holland Cheng, R. Prefusion rearrangements resulting in fusion peptide exposure in Semliki Forest virus. Journal of Biological Chemistry. 278 (9), 7189-7198 (2003).
- Hamilton, B. S., Chung, C., Cyphers, S. Y., Rinaldi, V. D., Marcano, V. C., Whittaker, G. R. Inhibition of influenza virus infection and hemagglutinin cleavage by the protease inhibitor HAI-2. Biochemical and Biophysical Research Communications. 450 (2), 1070-1075 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved