
Method Article
Chromatin Extraction from Frozen Chimeric Liver Tissue for Chromatin Immunoprecipitation Analysis
In This Article
Summary
This protocol focuses on chromatin preparation from snap frozen tissues and it is suitable for Crosslinking Chromatin Immunoprecipitation (X-ChIP) followed by either quantitative PCR analysis (X-ChIP-qPCR) or next generation sequencing approaches (X-ChIP-seq).
Abstract
Crosslinking Chromatin Immunoprecipitation (X-ChIP) is a widely used technique to assess levels of histone marks and occupancy of transcription factors on host and/or pathogen chromatin. Chromatin preparation from tissues creates additional challenges that need to be overcome to obtain reproducible and reliable protocols comparable to those used for cell culture. Tissue disruption and fixation are critical steps to achieve efficient shearing of chromatin. Coexistence of different cell types and clusters may also require different shearing times to reach optimal fragment size and hinders shearing reproducibility. The purpose of this method is to achieve reliable and reproducible host chromatin preparations from frozen tissue (liver) suitable for both ChIP-qPCR and next generation sequencing (NGS) applications. We observed that the combination of liquid nitrogen tissue pulverization followed by homogenization leads to increased reproducibility compared to homogenization only, since it provides a suspension consisting mostly of dissociated single cells that can be efficiently sheared. Moreover, the fixation step should be performed under mild rotation to provide homogeneous crosslinking. The fixed material is then suitable for buffer-based nuclei isolation, to reduce contamination of cytoplasmic protein and pathogen DNAs and RNAs (when applicable), avoiding time-consuming centrifugation gradients. Subsequent sonication will complete nuclear lysis and shear the chromatin, producing a specific size range according to the chosen shearing conditions. The size range should fall between 100 and 300 nt for NGS applications, while it could be higher (300-700 nt) for ChIP-qPCR analysis. Such protocol adaptations can greatly improve chromatin analyses from frozen tissue specimens.
Introduction
Since its discovery, epigenetic regulation in mammalian cells has gained increasing recognition1, considering that the understanding of such mechanisms would provide key insights not only in cell biology, but also in disease and tumor biology. Moreover, infectious agents may also cause host epigenetic changes2 whereas the host cell machinery may also affect the chromatin of pathogens, such as persisting DNA viruses3,4. This host-pathogen interplay seems to play a role in infection persistence.2
Through a reversible association with DNA, histone proteins form a complex called nucleosome. Nucleosomes reach in turn a higher level of organization known as chromatin. Chromatin remodeling is known to tightly regulate gene expression, granting or denying access to transcription factors (TFs)5. These factors can either trigger or block the recruitment of the RNA polymerase II (PolII) onto gene promoters, influencing mRNA synthesis from the DNA template6. Histone proteins contain tails7, flanking both ends of the histone fold, which can be subject to post-translational modifications (PTMs), allowing a tight regulation of the gene transcription by structural chromatin changes. Most of the histone PTMs are located at the tail N-terminus, with acetylation and methylation being the best studied PTMs, although phosphorylation8, ubiquitination9 and ribosylation10 have also been reported. Characterizing and studying such proteins is then essential to get a deep insight into gene regulation.
Currently, there is a handful of well-established methods and tools available to study direct DNA-protein interactions: Electrophoretic mobility shift assay (EMSA), Yeast one-hybrid assay (Y1H) and DNA footprinting11. However, these methods focus per se on single DNA-protein interactions and are not applicable for genome-wide studies. Another limitation of those techniques is the lack of histone association with the DNA segments investigated. Thus, such approaches are not meant to reflect the complexity of the transcriptional machinery in vivo and they do not take into account important structural changes12 or other required enzymes/cofactors13 that could influence (either promoting or inhibiting) protein binding to the DNA.
The idea that fixing cells with agents like formaldehyde (FA) could provide an in vivo snapshot of protein-DNA interactions, created the basis for the development of chromatin immunoprecipitation assays (ChIP)14. This, together with the availability of the quantitative PCR (qPCR) technology and of highly specific antibodies, allowed the development of ChIP-qPCR assays. Subsequently, the advent of next-generation sequencing techniques (NGS), whose costs are getting more affordable, conceded to couple ChIP experiments with NGS approaches (ChIP-seq), thus providing researchers with new powerful tools enabling investigation of chromatin regulation. In these assays, isolated or cultured cells are fixed with disuccinimidyl glutarate (DSG) and/or FA, nuclei are isolated, chromatin is then fragmented and precipitated by the antibody of interest. Hereafter, DNA is purified and analyzed by PCR or NGS approaches. In contrast to EMSA, Y1H and DNA footprinting, ChIP assays have the ability to provide a global snapshot of protein-DNA interaction within the cell. This offers flexibility and allows the analysis of multiple loci within the same sample. However, due to the nature of the assay, ChIP may, eventually, detect not only direct interactions, but also indirect ones, not offering the precision of the above-mentioned methods, when interested in direct protein-DNA interactions.
Chromatin preparation protocols from cell culture material are well-established15 and highly reproducible, allowing the user to obtain chromatin suitable both for qPCR and NGS approaches in 1-2 work days. However, obtaining high quality chromatin from whole tissues still represents a challenge because of the need to dissociate the cells within the tissue while achieving optimal fixation and shearing of the chromatin. In addition, composition and morphology of distinct type of tissues vary, thus requiring adjustment of existing protocols16,17. The use of cryopreserved tissue offers additional challenges in comparison to fresh samples. This is due to the difficulty of obtaining a single cell suspension without extensive material loss. This leads to improper shearing, hindering downstream applications. Nonetheless, accessing frozen tissue specimens rather than the fresh counterpart not only increases work flexibility but it may also represent the only option for researchers working with specimens that originate from longitudinal or comparative studies. A handful of chromatin preparation protocols for frozen tissue have been published. These are mostly based on specimen thawing followed by mincing, manual/machine-based dissociation or liquid nitrogen pulverization steps18,19,20.
Here we describe an optimized chromatin preparation method15 for frozen unfixed liver specimens, which combines tissue pulverization in liquid nitrogen with pestle homogenization, to achieve a reproducible chromatin shearing suitable for X-ChIP approaches aiming at analyzing both viral and host genomes.
Protocol
Tissue sampling from human liver chimeric mice21 was performed in accordance with the European Union directive 86/609/EEC and approved by the ethical committee of the city and state of Hamburg in accordance with the principles of the Declaration of Helsinki.
1. Reagents preparation
- Prepare 1.25 M glycine solution in deionized water. Sterile filter with 0.22 µm pore-sized filter. Store at 4 °C.
- Prepare 5 M sodium chloride (NaCl) solution. Store at room temperature.
- Prepare CaCl2 solution: 300 mM CaCl2 and 10 mM Tris-HCl pH 8 in deionized water. Sterile filter with 0.22 µm pore-sized filter and store at RT.
- Prepare a 10% Triton X-100 dilution in deionized water. Store at RT.
- Prepare Tris-EDTA buffer: 1 mM EDTA and 10 mM Tris pH 8 in deionized water. Store at 4 °C.
- Prepare the following buffers according to the amount required:
- Prepare buffer A: 50 mM Hepes-KOH pH 7.5, 140 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 10% Glycerol, 0.5% NP-40 and 0.25% Triton X-100 in deionized water. Sterile filter with 0.22 µm pore-sized filter. Store at 4 °C.
- Prepare buffer B: 10 mM Tris-HCl pH 8, 200 mM NaCl, 1mM EDTA, 0.5 mM egtazic acid (EGTA). Sterile filter with 0.22 µm pore-sized filter. Store at 4 °C.
- Prepare buffer C: 1% SDS, 10 mM EDTA and 50 mM Tris-HCl pH 8 in deionized water. Sterile filter with 0.22 µm pore-sized filter. Store at RT.
- Prepare chromatin dilution buffer: 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.6 mM Tris-HCl pH 8 and 166 mM NaCl in deionized water. Sterile filter with 0.22 µm pore-sized filter. Store at 4 °C.
2. Material preparation
- Collect dry ice, ice, and liquid nitrogen.
CAUTION: Handle dry ice and liquid nitrogen with the necessary care to avoid burns. - Pre-cool the centrifuge at 4 °C.
NOTE: This step is important to avoid protein degradation and de-crosslinking during the washing steps as this will reduce the quality of the chromatin. - Put a sterile plate on dry ice and let it chill.
NOTE: Make sure that the plate is big enough to facilitate the cutting process. A 100 mm Petri plate/cell culture dish is recommended. - Take out the necessary aliquot of glycine 1.25 M and let it reach RT.
- Take out the necessary aliquots of buffer A, B and PBS. Add protease and/or deacetylase and phosphatase inhibitors to reach 1-fold concentration and leave them on ice.
- Take out the necessary aliquot of buffer C leaving it at RT. Do not add the protease and/or deacetylase and phosphatase inhibitors until specified.
- Take out the necessary aliquot of RT PBS.
CAUTION: Buffer C contains sodium dodecyl sulfate (SDS). Adopt the appropriate safety measures when preparing the buffer.
NOTE: SDS precipitates on ice, and protease and deacetylase inhibitors are not stable at RT. - Pre-cool the mortar pouring liquid nitrogen in its chamber, strictly following supplier instructions. Cool down the metal pestle in dry ice for at least 5 min.
NOTE: It is possible to use an alternative mortar to the one proposed. However, the device used in this protocol, due to its peculiar construction, allows to work with small amount of tissue without substantial loss during the pulverization process. - Pre-chill the Dounce homogenizer with the associated pestle A on ice.
NOTE: Pestle A has a loose fit with the homogenizer. This allows to obtain a single cell suspension without significant cell lysis.
3. Tissue crosslinking
- Cut about 50 mg of frozen tissue directly on the dish on dry ice with the help of a scalpel and tweezers.
NOTE: It is suggested to keep the scalpel at RT, as this will ease the cutting process. Avoid applying too much pressure on the scalpel, as this will increase the risk to scatter tissue pieces outside of the cutting area. To note, 50 mg of tissue (liver in this case) should yield around 5 million cells. Note that the warm blade will thaw the cutting edge. Considering the relatively large size of the tissue piece, however, this should have a limited effect. When smaller pieces are cut, it could be beneficial to use a cold scalpel paying attention to avoid scattering of the tissue. - Put the cut tissue in a 1.5 mL tube already chilled on dry ice. Avoid tissue thawing.
- Move the tube containing the tissue to the mortar, letting it sit there for 5 min.
NOTE: Letting the sample rest in the mortar decreases its temperature (from -80 °C to -196 °C). This increases its toughness and makes the pulverization step easier. - Apply pressure to the sample with the help of the pre-chilled pestle until no more solid crumbles are visible.
NOTE: It is important to avoid pestle heating by excessive rotational forces, as this will thaw the sample. After every sample pulverization clean the pestle with 70% ethanol (EtOH) and let it chill again on dry ice. - Remove the tube containing the sample from the mortar and add 950 µL of ice-cold PBS with the required inhibitors. Pipette gently up and down until the sample is completely resuspended. Proceed immediately to step 3.6.
- Transfer the tissue suspension to the homogenizer and apply 20-30 strokes with pestle A to obtain a finer suspension. Avoid foaming.
NOTE: The strokes amount should be optimized according to the tissue consistence. This step dissociates further the small clusters of cells obtained after pulverization. An improper homogenization can affect shearing efficiency. - Transfer the homogenate to a new 1.5 mL tube, already prechilled on ice.
- Centrifuge for 5 min at 1,300 x g at 4 °C and carefully remove the supernatant.
- Resuspend the pellet completely in 950 µL of RT PBS by gentle pipetting and add 63.6 µL of 16% MeOH-free FA to have a 1% final concentration. Proceed immediately to step 3.10.
CAUTION: FA is a toxic chemical. Handle it under a fume hood with proper safety measures.
NOTE: Incomplete resuspension can provoke cell aggregation during the fixation step. This hinders the lysis and the shearing process. - Rotate 10 min at RT. Proceed then immediately to step 3.11
NOTE: Rotation is necessary to avoid aggregates. The amount of time required for fixation should be optimized, according to the target of interest and the sample type. It is important to note that excessive fixation times could hinder proper shearing. - Add 113 µL of 1.25 M glycine at RT to obtain a 125 mM final concentration and rotate for 5 min.
NOTE: Glycine quenches the fixative reaction avoiding over-crosslinking. - Centrifuge at 1,300 x g for 3 min at 4 °C.
- Discard the supernatant and resuspend the pellet carefully by pipetting in 950 µL of ice-cold PBS with the required inhibitors.
- Centrifuge at 1,300 x g for 3 min at 4 °C.
- Repeat steps 3.13-3.14 and proceed immediately to the chromatin isolation steps.
4. Chromatin isolation
- Add 950 µL of Buffer A with the required inhibitors to the pellet. Mix gently by pipetting until the pellet is completely resuspended and rotate 10 min at 4 °C.
NOTE: This step lyses the fixed single cell suspension, without nuclei lysis. This allows for ridding the sample of cytosolic proteins and RNAs. Prolonging lysis time could be beneficial for tough-to-lyse cells, increasing, though, the handling time of the tissue. At this point it is possible to check the preparation under the microscope after trypan blue/DAPI staining to check the size of the clusters and the presence of single cells. However, the single nuclei may not be easy to appreciate because of the fixed tissue material. - Centrifuge at 2,000 x g for 5 min at 4 °C and carefully remove the supernatant.
- Add 950 µL of Buffer B with the required inhibitors to the pellet. Mix gently by pipetting until the pellet is completely resuspended and rotate 10 min at 4 °C.
NOTE: This step washes out the lysis buffer from the nuclei preparation to avoid further undesired lysis. - Centrifuge at 2000 x g for 5 min at 4 °C. Meanwhile, add the required inhibitors (same as step 2.5) to buffer C.
- Carefully remove the supernatant.
- Add 300 µL of RT buffer C to the pellet and pipette vigorously.
- Vortex the sample for 15-30 s and spin the tube briefly to collect the drops on the lid.
NOTE: This step is important to liberate and lyse the fixed nuclei. To preserve the sample integrity and at the same time avoid SDS precipitation keep the sample prior to sonication in a plastic rack kept on ice to maintain a temperature of 9-11 °C.
5. Chromatin fragmentation
- Transfer the sample to three clean 0.65 mL sonication-certified tubes ensuring 100 µL of lysed nuclei suspension per tube.
NOTE: It is possible to use 1.5 mL sonication-certified tubes with a maximum volume of 300 µL. A specific holder for those tubes is needed. 0.65 mL should offer more homogeneous shearing due to the smaller volume of the sample per tube. - Sonicate the chromatin for 28 cycles at high intensity with the 30 s ON and 30 s OFF setting. Ensure that the sonicator bath is properly cooled (ice or cooling device).
NOTE: This step needs optimization in almost every case. The user should keep in mind that increasing shearing time will provide smaller and more homogeneous fragments; however, this may increase the chance to lower chromatin quality. Choose the lowest number of cycles that provides the required fragment size. During the optimization of this step, it is useful to perform nuclear staining to check whether the number of cycles was sufficient to lyse the majority of nuclei. - Transfer the sonicated chromatin to a new 1.5 mL tube previously chilled on ice.
- Add 30 µL of Triton X-100 10% solution and vortex for 5-10 s.
NOTE: Triton X-100 binds the SDS preventing further precipitation at 4 °C. The final amount of Triton X-100 should always be 1%. - Centrifuge at 16,000 x g for 15 min at 4 °C.
- Transfer the supernatant to a clean 1.5 mL tube pre-chilled on ice.
- NOTE: The supernatant contains the sheared chromatin and should appear clear. The pellet contains "not shearable" rests and it should remain quite small (mostly brown in case of liver tissue). Look for indication of unsuccessful shearing: chromatin solution that did not become clearer and similar pellet dimensions to the ones from step 4.5.
6. DNA purification
- Transfer 10-25 µL of sheared chromatin to a new tube and add Buffer C to reach a final volume of 200 µL. Store the rest of the chromatin at -80 °C until further use. If needed, the procedure can be interrupted at this step and the sample stored at -20 °C.
- Add 8 µL of 5 M NaCl and incubate at least 6 h at 65 °C in a heating block under shaking at 1000 rpm.
NOTE: This step de-crosslinks the chromatin. It is safer to extend the de-crosslinking overnight when possible. The presence of NaCl makes the process more efficient. - Let the samples cool at RT for 5 min and add 2 µL of RNase A.
- Incubate for 1 h at 37 °C under shaking at 1000 rpm.
- Remove the samples from the heating block and add 7 µL of 300 mM CaCl2 and 2 µL of Proteinase K.
- Set the heating block at 56 °C and incubate for 30 min under shaking at 1000 rpm. Meanwhile prepare one phase-separation tube for every sample by centrifuging them down at 16,000 x g for 1 min at 4 °C.
NOTE: These special tubes make the phase separation during nucleic acid phenol-chloroform extraction easier. - Remove the tubes from the heating block and let them equilibrate at RT for 3 min.
- Transfer 400 µL of the sample to a previously centrifuged phase-separation tube.
- Add 400 µL of phenol-chloroform-isoamyl alcohol solution (PCI) and vortex for 5 s.
CAUTION: PCI is a highly volatile and toxic compound. Please handle it with the necessary safety measures under a fume hood. - Centrifuge at 16,000 x g for 5 min at 4 °C.
- Add 400 µL of Chloroform and vortex for 5 s.
CAUTION: Chloroform is a highly volatile and toxic compound. Please handle it with the necessary safety measures under a fume hood.
NOTE: This step washes out possible residues of phenol, that could interfere with downstream PCR applications. - Centrifuge at 16,000 x g for 5 min at 4 °C.
- Transfer 400 µL of the upper phase to a new 1.5 mL tube where 24 µL of 5 M NaCl and 0.75 µL glycogen were added. Briefly vortex.
- Add 1,055 µL of 100% EtOH and vortex thoroughly. Ensure proper mixing.
- Incubate at -80 °C for 1 h or at -20 °C overnight (ON).
NOTE: This step precipitates the sheared DNA; to maximize the yield it is suggested to choose the ON incubation. - Centrifuge at 16,000 x g for 30 min at 4 °C.
- Carefully remove the supernatant paying attention to not dislocate the pellet.
- Add 500 µL of cold 70% EtOH. Tilt the tube gently to ensure the pellet is washed.
NOTE: This step is essential to remove salt residues that could have co-precipitated with the nucleic acids. Salts can interfere with other downstream applications. - Centrifuge at 16,000 x g for 15 min at 4 °C.
- Remove carefully the whole supernatant and let the pellet dry at RT.
NOTE: Incubating the tube on a heating block at 37 °C will reduce the time required for drying. - Add 50 µL of Tris-EDTA solution (TE-Buffer) and put the tube on the heating block at 37 °C for 5-10 min under shaking at 300 rpm.
NOTE: This step ensures the pellet dissolution. The protocol can be paused here, and the sample can be stored at 4 °C for up to 1 week or at -20 °C for longer storage. - Perform DNA analysis on 1% agarose gel.
7. DNA size analysis
- Prepare a 1% agarose gel by mixing 1 g of agarose per 100 mL running buffer (i.e., Tris-acetate-EDTA (TAE) or Tris-borate-EDTA (TBE)). Heat the suspension until the agarose is completely dissolved. Add 10 µL of EtBr for every 100 mL of agarose solution before pouring the gel solution.
CAUTION: EtBr is a DNA intercalating agent known to be carcinogenic. Please handle it with the necessary safety measures under a fume hood.
NOTE: EtBr staining (directly in gel or after the run) is strongly suggested. Other DNA-intercalating dyes did not perform well in our hands when working with DNA smears. Narrow loading wells provide a better resolution when compared to wider ones. - Mix 10 µL of the sample with 2 µL of 6x Loading Dye. Next, load 10 µL of the sample in the gel and run it until the last band of the loading dye ran for 2/3 of the gel. Make sure to add a DNA ladder.
- Image the gel and verify if the smear size falls in the range for the desired application.
If the chromatin passes the quality control, it can be used for downstream applications.
Results
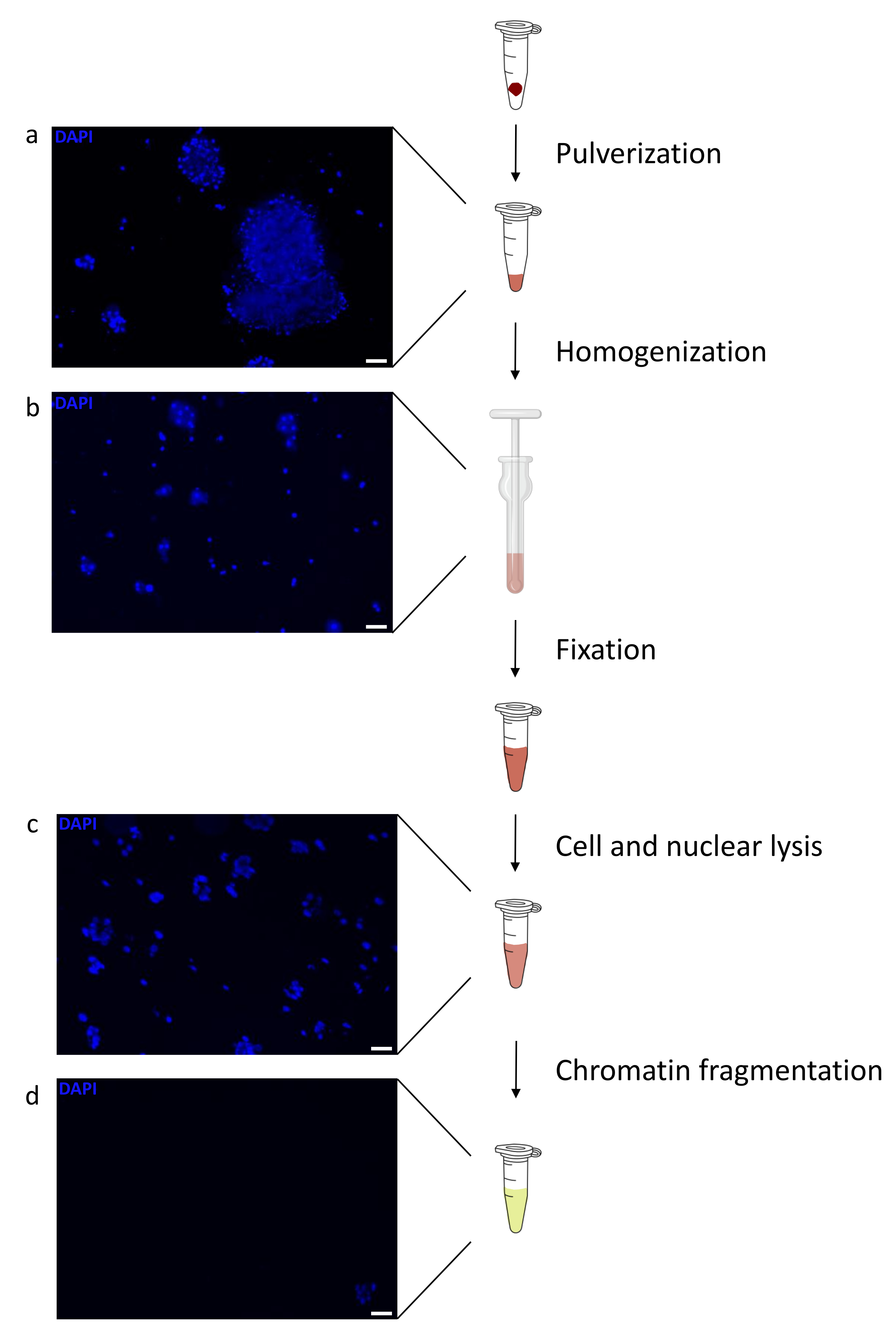
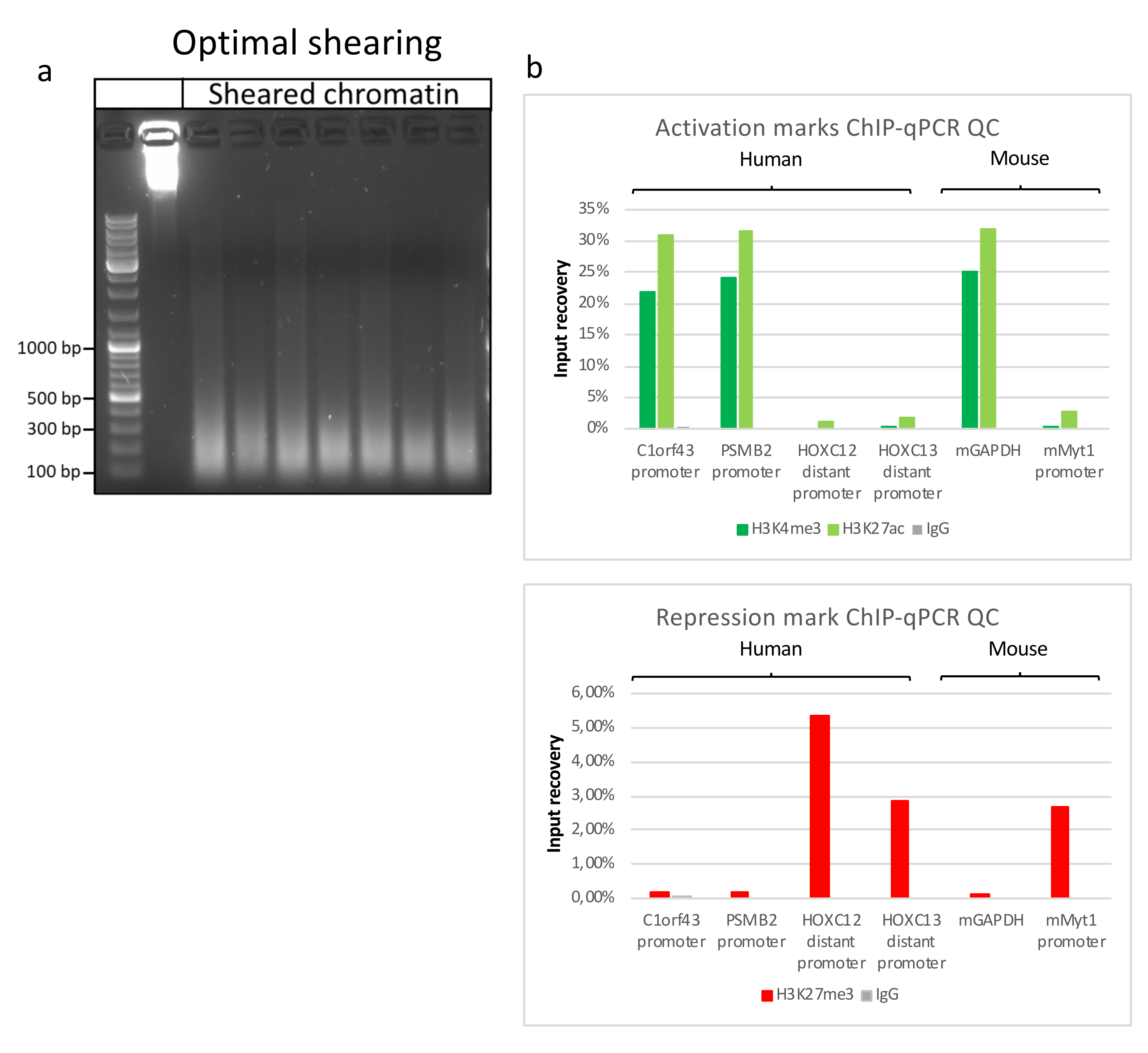
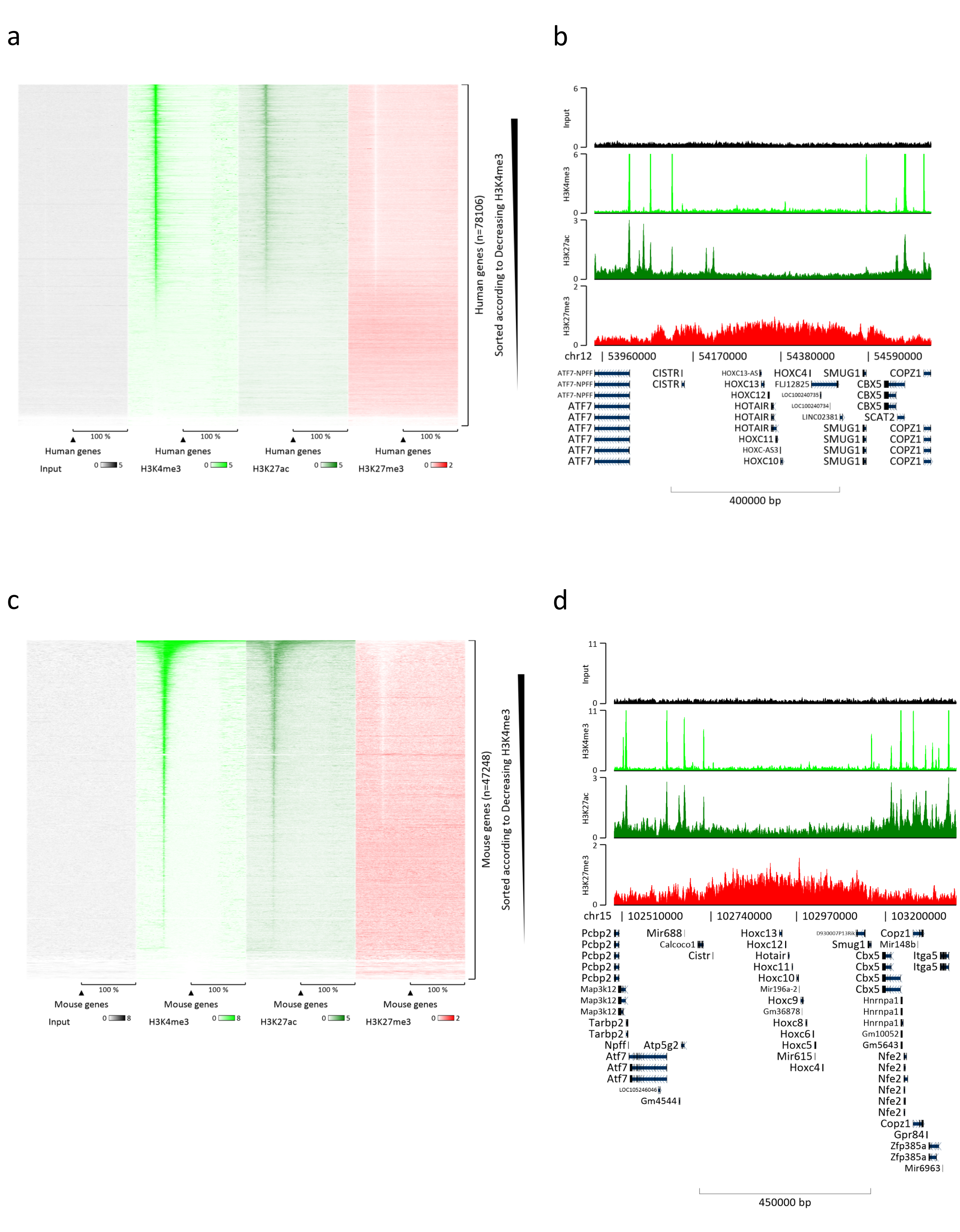
Preparing chromatin is a crucial step in achieving a successful ChIP. In order to prepare good quality chromatin from frozen specimens, we should ensure efficient tissue disruption before fixation to avoid the presence of tissue clumps that could hinder efficient shearing. Figure 1 shows a summarized pipeline of the protocol. Pulverization alone is not sufficient to completely dissociate the tissue since it produces cell clusters of variable size and few single cells (Figure 1a). Associating the first pulverization step with Dounce homogenization, the amount of tissue-clumps is strongly reduced and the remaining ones are smaller (Figure 1b). After the fixation and lysis steps, the number of visible single nuclei (Figure 1c) increases, while the typical spherical appearance is lost. After sonication for 28 cycles, the nuclear staining (Hoechst 33258/DAPI) is mostly not visible anymore. This is indeed a sign of successful shearing (Figure 1d). After de-crosslinking of a chromatin aliquot and visualization of the DNA on agarose gel, successful shearing can be recognized by the presence of fragments in the range of 100-300 bp. (Figure 2a) The amount of DNA can vary according to the composition of the tissue piece prepared. Such chromatin can be successfully used for ChIP-qPCR. As shown in Figure 2b the chromatin could be successfully precipitated with H3K4me3, H3K27ac (active genes related modifications) and H3K27me3 (silenced genes related modification) antibodies. Chromosome 1 Open Reading Frame 43 (C1orf43), Proteasome 20S Subunit Beta 2 (PSMB2) and Glyceraldehyde 3-phosphate dehydrogenase (mGapdh) promoter regions resulted enriched in H3K4me3 and H3K27ac in comparison with Homeobox C13 (HOXC13), Homeobox C12 (HOXC12) and the mouse Myelin Transcription Factor 1 (mMyt1) promoter regions (Table 1). This is because C1orf43, PSMB2 and mGapdh are constitutively transcribed in the liver, while HOXC13, HOXC12 and mMyt1 are silenced. H3K27me3 shows the opposite behavior confirming the success of the ChIP assay. The fact that the liver of these mice is a chimera, allowed us to analyze both murine and human chromatin. In addition, the same chromatin could be successfully used for ChIP-seq experiments. After the sequencing step, the reads were aligned to an index composed of both murine and human genomes to reduce the amount of unaligned fragments. Subsequently, the reads where separated according to species and further analyzed with EaSeq22 .The signal intensity was then measured at the transcription start site (TSS) of every gene and the result sorted for H3K4me3 signal intensity. Figure 3a and Figure 3c show both a marked presence of H3K4me3 and H3K27ac at the TSS for a considerable portion of the genes within both mouse and human chromatin. In addition to that, H3K27me3 anticorrelates with H3K4me3/H3K27ac. H3K27me3 is present on the entire length of the gene and not only at the TSS, as expected from this PTM. Figure 3b and Figure3d show the HOXC/HoxC cluster known for being enriched for H3K27me3 and transcriptionally inactive in both mouse and human livers. The profiling of H3K4me3 and H3K27ac shows peaks for this two PTMs while the signal intensity of H3K27me3 tends to be lower and more distributed.
Due to the complexity of chromatin preparation, over-fixation may happen, lysis or sonication time may be sub-optimal, big cell clumps may persist, or inadequate handling of the sample could be inadequate. These are all events affecting the quality of the preparation. In some cases, the enrichment of chromatin fragments within the correct size will still be present or will be shifted to a higher size. In other cases, there may be a loss of material due to premature lysis or unsuccessful shearing. Figure 4 shows some examples of such negative and suboptimal results. Lane 3 and 4 show an enrichment of the fragment size between 200 bp and 800 bp. However, it is clear that the fragment size spans from 100 bp to >10,000 bp. In lane 5 and 6 an enrichment in the 100-250 bp range is present with a clear loss of material during the preparation. This could explain why the sonication produced smaller fragments. Lane 7 shows a slightly sub-optimal preparation with the fragment range increased, while lane 8 shows an almost complete loss of material. This can be caused by premature nuclear lysis or insufficient tissue dissociation with consequent loss after step 5.5.

Figure 1: Chromatin preparation protocol overview. Pictures were taken after tissue pulverization (a), additional manual homogenization (b), after nuclear lysis (c) and after sonication (before centrifugation) (d). Nuclear staining was performed with Hoechst 33258/DAPI. Scale bar = 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative chromatin shearing, and its quality assessed by ChIP-qPCR. 1% agarose gel with fragmented chromatin samples according to protocols from different chromatin preparations. A control of unsheared chromatin is added to ensure no chromatin/DNA degradation beforehand (a). Sheared chromatin has been tested for quality performing a ChIP-qPCR assay. H3K4me3, H3K27ac and H3K27me3 antibodies were used to precipitate the freshly prepared chromatin. (b) qPCR analysis was performed on human (C1orf43 and PSMB2), murine (Gapdh) active promoters and human (HOXC13, HOXC12), murine (Myt1) inactive promoters. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative ChIP-seq analysis. Reads have been aligned to an index created with both human and mouse genomes (hg19 and mm10). After alignment human and murine reads were separated and further analyzed. Heatmap of human genes where the signal was quantified at the TSS and showed in descending order for H3K4me3 intensity (a). Example of human gene cluster of suppressed genes (HOX cluster) surrounded by active genes (b). Heatmap of murine genes where the signal was quantified at the TSS and shown in descending order for H3K4me3 intensity (c). Example of a murine gene cluster of suppressed genes (Hox cluster) surrounded by active genes (d). All the data shown has been normalized by EaSeq per million reads. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Suboptimal and failed chromatin preparations. 1% Agarose gel with fragmented chromatin samples according to protocol. The figure contains unsheared chromatin used as a control (Lane 2), not optimal shearing (Lane 3-4), optimal shearing with clear material loss (Lane 5-6), suboptimal shearing (Lane 7) and extensive material loss (Lane 8). Please click here to view a larger version of this figure.
{kind=link}
| Primer name | Sequence | |
| C1orf43 promoter | Forward | AGTGGGTGGAGAATGCAGAC |
| Reverse | GAGATTACCCCACCCCATTC | |
| PSMB2 promoter | Forward | CTTATTCAACCCCCGACAAA |
| Reverse | GATGAAGGACGGTGAGAGGA | |
| HOXC13 distal promoter | Forward | GAGCCCGAGATTCACTCAAC |
| Reverse | TTATGCCCAGTTTTGGGGTA | |
| HOXC12 distal promoter | Forward | AAAGCTTCCCACTGCAAAGA |
| Reverse | AAATCTGGGGGCGAACTACT | |
| mGAPDH promoter | Forward | GGTCCAAAGAGAGGGAGGAG |
| Reverse | GCCCTGCTTATCCAGTCCTA | |
| mMYT promoter | Forward | CAGCCCAATTCTAGCCACAT |
| Reverse | CCAAAGCAGGGGAGTAGGAG |
Table 1: qPCR primers list for active and inactive genes used for ChIP-qPCR assays.
Discussion
Chromatin preparation from snap frozen tissue remains a challenge because of the number of steps that need to be optimized in order to achieve reproducible and reliable results. Most of the already published protocols16,23 require tissue mincing before the manual dissociation (douncing). We tried to avoid steps that could provoke protein degradation before the fixation of the sample as much as possible. The pulverization step is already used in frozen liver preparations24 and makes the manual dissociation easier and reproducible (see Figure 2a). With the use of a mortar specifically designed for 1.5 mL tubes (see Protocols), the specimen loss during the pulverization process is reduced, allowing to process small amounts of tissue such as liver biopsy specimens. In principle it is possible to use direct tissue homogenization without any grinding steps; however, tissue homogenization without previous pulverization has a worse reproducibility in our experience and the appearance of problems for downstream applications was higher (data not shown).
Most of the problems encountered by preparing chromatin from tissues derives from the nature of these samples and the incapability to check properly whether the cell clusters are small enough for fixation without losing quality. Moreover, checking each aliquot at each step would be time consuming increasing the chance of protein degradation.
Fixation (step 3.9) is a fundamental and crucial part of the chromatin preparation. Due to the nature of the tissue, the fixation step has been delayed until the tissue was homogenized. Such postponed fixation step has the advantage to produce a more homogeneous cell suspension. However, we recognize, that in case of particularly manipulation-sensitive targets, it may be necessary to perform the fixation just before step 3.6. This would help protect extremely sensitive proteins or PTMs, although it may increase the size of the cell clusters, that when fixed may result in nonhomogeneous shearing. The concentration of the FA solution used in the protocol is standard, however, it can be modified to try to improve the overall fixation. The fixation time chosen here also reflects standard conditions commonly used in the field. In case of higher concentration of the fixating solution, the fixation time may be reduced, while in case of a lower amount it should be increased. The operator should consider that a change of the fixation time may either lead to over-fixation of the sample or give room for protein degradation. In case of aiming at precipitating big complexes (or part of it) and TFs, it would be advantageous to perform a double step fixation using a DSG solution followed by a FA one25,26. DSG in this case would stabilize protein-protein interactions, while formaldehyde acts mostly on direct DNA-protein interactions27.
The operator should take into account the possibility of implementing a column-based kit for DNA purification starting at step 6.7 which is faster and does not use toxic compounds. However, there will always be a certain amount of unbound DNA that will be lost. For this reason, we suggest using the classical phenol-chloroform extraction followed by EtOH precipitation. Moreover, before running the agarose gel (step 7.2) it could be beneficial to measure the DNA concentration and load the same amount for every well to have a clearer picture.
A limitation of this protocol stems from the fact that we explored and utilized this protocol only using liver specimens derived from human-liver chimeric mice28. Per se the liver consists of epithelial and connective tissue29. In case of disease, fibrotic tissue and fat tissue may be present30,31 creating additional challenges during tissue disruption. However, we recognize that our protocol may not be used on bone, muscle and adipose tissue without optimization of the dissociation and sonication steps. To note is that every tissue requires some kind of optimization due to the absence of a protocol suitable for all of them like for cell culture samples15. We believe, though, that with little or no optimization at all, this protocol could be successfully applied to other tissues that share similarities with the liver in composition, like lung, intestine, stomach, pancreas or kidney tissues.
Our protocol has also been successfully used to analyze TFs and histone modifications on the HBV covalently closed DNA episome (cccDNA)32. This opens the chance to apply such an approach for other viral genomes affecting the liver such as human Cytomegalovirus33 (hCMV) and human Adenoviruses34 (HAdV). It is not excluded that it would be possible to analyze other DNA viruses that establish a persistent infection in other tissues like Kaposi Sarcoma Herpes Virus35 (KHSV), Herpes Simplex Virus36 (HSV1/2) Polyoma viruses, Epstein-Barr Virus37 (EBV).
Disclosures
The authors have nothing to disclose.
Acknowledgements
The study was supported by the German Research Foundation (DFG) by a grant to Maura Dandri (SFB 841 A5) and by the State of Hamburg with the Research Program (LFF-FV44: EPILOG).
We would like to thank Dr. Tassilo Volz, Yvonne Ladiges and Annika Volmari for the technical help and for critically reading the manuscript. Dr. Thomas Günther and Prof. Adam Grundhoff for providing very helpful suggestions and the primer sets for the ChIP-qPCR analysis.
Materials
| Name | Company | Catalog Number | Comments |
| 0.22µm sterile syringe filter | Labsolute | 7699822 | |
| 1.5 mL Safeseal tubes | Sarstedt | 7,27,06,400 | |
| 6x orange loading dye | Thermofisher | R0631 | |
| Benchtop refrigerated centrifuge | |||
| Bioruptor NGS | Diagenode | ||
| Blade or Scalpel | |||
| Calcium chloride dihydrate | Carl Roth | HN04 | |
| Chloroform | Sigma Aldrich (Merck) | C2432 | |
| cOmplete Protease Inhibitor Cocktail | Roche | 11697498001 | |
| Deacetylase Inhibitor | Active Motif | 37494 | |
| Dounce tissue grinder set | Sigma Aldrich (Merck) | DWK885300-0001-1EA | |
| EDTA 500 mM solution | PanReac AppliChem | A4892 | |
| EGTA | Sigma Aldrich (Merck) | E4378 | |
| EtBr | Carl Roth | 2218 | Concentration 10mg/mL |
| Ethanol absolute | CHEMSOLUTE | 2273 | |
| Glycerol | Sigma Aldrich (Merck) | G9012 | |
| Glycin | Carl Roth | 0079 | |
| Glycogen | Roche | 10901393001 | Concentration: 20mg/mL |
| Heating block | |||
| HEPES | Sigma Aldrich (Merck) | H4034 | |
| LE Agarose | Biozym | 840000 | |
| Liquid nitrogen cooled mini mortar | Bel-Art | H37260-0100 | |
| MeOH free Formaldehyde 16% | Thermofisher | 28908 | |
| NP-40 | Roche | 11332473001 | |
| PBS 1x | Thermofisher | 10010015 | |
| Pefabloc SC-Protease-Inhibitor | Sigma Aldrich (Merck) | 11429868001 | |
| Phase Lock Gel - Heavy | QuantaBio | 2302830 | |
| Phenol:Chloroform:Isoamyl alcohol 25:24:1 | Sigma Aldrich (Merck) | P3803 | |
| Potassium chloride | Carl Roth | 6781 | |
| Potassium hydroxyde | Merck | 105033 | |
| Proteinase K | Lucigen | MPRK092 | Concentration: 50 µg/µL |
| RNAse A | Lucigen | MRNA092 | Concentration: 5 mg/mL |
| SDS 10% solution | PanReac AppliChem | A3950 | |
| Sodium carbonate anhydrous | Carl Roth | A135 | |
| Sodium chloride | Sigma Aldrich (Merck) | S7653 | |
| Sterile Petri dishes | Sarstedt | 83,39,02,500 | |
| Tris-HCl solution | Sigma Aldrich (Merck) | T2694 | |
| Triton-X100 | Sigma Aldrich (Merck) | X100 |
References
- Waddington, C. H., Pantelouris, E. M. Transplantation of nuclei in newt's eggs. Nature. 172 (4388), 1050-1051 (1953).
- Silmon de Monerri, N. C., Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. American Journal of Pathology. 184 (4), 897-911 (2014).
- Knipe, D. M., et al. Snapshots: chromatin control of viral infection. Virology. 435 (1), 141-156 (2013).
- Tropberger, P., et al. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proceedings of the National Academy of Sciences of the United States of America. 112 (42), 5715-5724 (2015).
- Sproul, D., Gilbert, N., Bickmore, W. A. The role of chromatin structure in regulating the expression of clustered genes. Nature Reviews Genetics. 6 (10), 775-781 (2005).
- Fischle, W., Wang, Y., Allis, C. D. Histone and chromatin cross-talk. Current Opinion in Cell Biology. 15 (2), 172-183 (2003).
- Ling, X., Harkness, T. A., Schultz, M. C., Fisher-Adams, G., Grunstein, M. Yeast histone H3 and H4 amino termini are important for nucleosome assembly in vivo and in vitro: redundant and position-independent functions in assembly but not in gene regulation. Genes & Development. 10 (6), 686-699 (1996).
- Zhang, L., Eugeni, E. E., Parthun, M. R., Freitas, M. A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 112 (2), 77-86 (2003).
- Wang, H., et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 431 (7010), 873-878 (2004).
- Hassa, P. O., Haenni, S. S., Elser, M., Hottiger, M. O. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going. Microbiology and Molecular Biology Reviews. 70 (3), 789-829 (2006).
- Dey, B., et al. DNA-protein interactions: methods for detection and analysis. Molecular and Cellular Biochemistry. 365 (1-2), 279-299 (2012).
- Hager, G. L., McNally, J. G., Misteli, T. Transcription dynamics. Molecular Cell. 35 (6), 741-753 (2009).
- Nagy, Z., Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 26 (37), 5341-5357 (2007).
- Solomon, M. J., Larsen, P. L., Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 53 (6), 937-947 (1988).
- Gunther, T., Theiss, J. M., Fischer, N., Grundhoff, A. Investigation of viral and host chromatin by ChIP-PCR or ChIP-Seq analysis. Current Protocols in Microbiology. 40, 11-21 (2016).
- Cotney, J. L., Noonan, J. P. Chromatin immunoprecipitation with fixed animal tissues and preparation for high-throughput sequencing. Cold Spring Harbor Protocols. 2015 (4), 419 (2015).
- Haim, Y., Tarnovscki, T., Bashari, D., Rudich, A. A chromatin immunoprecipitation (ChIP) protocol for use in whole human adipose tissue. American Journal of Physiology-Endocrinology and Metabolism. 305 (9), 1172-1177 (2013).
- Castellano-Castillo, D., et al. Chromatin immunoprecipitation improvements for the processing of small frozen pieces of adipose tissue. PLoS One. 13 (2), 0192314 (2018).
- Savic, D., Gertz, J., Jain, P., Cooper, G. M., Myers, R. M. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenetics Chromatin. 6 (1), 30 (2013).
- Perna, A., Alberi, L. A. TF-ChIP method for tissue-specific gene targets. Frontiers Cell Neuroscience. 13, 95 (2019).
- Allweiss, L., et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut. 67 (3), 542-552 (2018).
- Lerdrup, M., Johansen, J. V., Agrawal-Singh, S., Hansen, K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nature Structural & Molecular Biology. 23 (4), 349-357 (2016).
- Perna, A., Alberi, L. A. TF-ChIP Method for Tissue-Specific Gene Targets. Frontiers in Cellular Neuroscience. 13, 95 (2019).
- Liang, N., Fan, R., Goni, S., Treuter, E. Preparation of Frozen Liver Tissues for Integrated Omics Analysis. Methods in Molecular Biology. 1951, 167-178 (2019).
- Liu, Z., et al. Proteomic and network analysis of human serum albuminome by integrated use of quick crosslinking and two-step precipitation. Scientific Reports. 7 (1), 9856 (2017).
- Singh, A. A., et al. Optimized ChIP-seq method facilitates transcription factor profiling in human tumors. Life Science Alliance. 2 (1), 201800115 (2019).
- Aoki, T., et al. Bi-functional cross-linking reagents efficiently capture protein-DNA complexes in Drosophila embryos. Fly. 8 (1), 43-51 (2014).
- Allweiss, L., Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. Journal of Hepatology. 64, 17-31 (2016).
- Krishna, M. Microscopic anatomy of the liver. Clinics in Liver Disease. 2, 4-7 (2013).
- Tannapfel, A., et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Archiv. 458 (5), 511-523 (2011).
- Schuppan, D., Afdhal, N. H. Liver cirrhosis. Lancet. 371 (9615), 838-851 (2008).
- Allweiss, L., et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut. , 322571 (2021).
- Gerna, G., Kabanova, A., Lilleri, D. Human cytomegalovirus cell tropism and host cell receptors. Vaccines. 7 (3), (2019).
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clinical Microbiology Reviews. 21 (4), 704-715 (2008).
- Frohlich, J., Grundhoff, A. Epigenetic control in Kaposi sarcoma-associated herpesvirus infection and associated disease. Seminars in Immunopathology. 42 (2), 143-157 (2020).
- Nicoll, M. P., Proenca, J. T., Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiology Reviews. 36 (3), 684-705 (2012).
- Thorley-Lawson, D. A., Hawkins, J. B., Tracy, S. I., Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Current Opinion in Virology. 3 (3), 227-232 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved