Design to Implementation Study for Development and Patient Validation of Paper-Based Toehold Switch Diagnostics
* These authors contributed equally
In This Article
Summary
Access to decentralized, low-cost, and high-capacity diagnostics that can be deployed into the community for decentralized testing is critical for combating global health crises. This manuscript describes how to build paper-based diagnostics for viral RNA sequences that can be detected with a portable optical reader.
Abstract
Access to low-burden molecular diagnostics that can be deployed into the community for testing is increasingly important and has meaningful wider implications for the well-being of societies and economic stability. Recent years have seen several new isothermal diagnostic modalities emerge to meet the need for rapid, low-cost molecular diagnostics. We have contributed to this effort through the development and patient validation of toehold switch-based diagnostics, including diagnostics for the mosquito-borne Zika and chikungunya viruses, which provided performance comparable to gold-standard reverse transcription-quantitative polymerase chain reaction (RT-qPCR) based assays. These diagnostics are inexpensive to develop and manufacture, and they have the potential to provide diagnostic capacity to low-resource environments. Here the protocol provides all the steps necessary for the development of a switch-based assay for Zika virus detection. The article takes readers through the stepwise diagnostic development process. First, genomic sequences of Zika virus serve as inputs for the computational design of candidate switches using open-source software. Next, the assembly of the sensors for empirical screening with synthetic RNA sequences and optimization of diagnostic sensitivity is shown. Once complete, validation is performed with patient samples in parallel with RT-qPCR, and a purpose-built optical reader, PLUM. This work provides a technical roadmap to researchers for the development of low-cost toehold switch-based sensors for applications in human health, agriculture, and environmental monitoring.
Introduction
RT-qPCR has remained the gold standard technology for clinical diagnostics due to its excellent sensitivity and specificity. Although highly robust, the method depends on expensive, specialized equipment and reagents that require temperature-controlled distribution and storage. This presents significant barriers to the accessibility of quality diagnostics globally, particularly in times of disease outbreaks and in regions where access to well-equipped labs is limited1,2. This was observed during the 2015/2016 Zika virus outbreak in Brazil. With only five centralized laboratories available to provide RT-qPCR testing, significant bottlenecks resulted, limiting access to diagnostics. This was especially challenging for individuals in peri-urban settings, who were more severely impacted by the outbreak3,4. In an effort to improve access to diagnostics, the protocol shows a platform that has been developed with the potential to provide decentralized, low-cost, and high-capacity diagnostics in low-resource settings. As a part of this, a diagnostic discovery pipeline was established, coupling isothermal amplification and synthetic RNA switch-based sensors with paper-based cell-free expression systems5,6.
Cell-free protein synthesis (CFPS) systems, in particular E. coli based cell-free systems, are an attractive platform for a wide range of biosensing applications from environmental monitoring7,8 to pathogen diagnostics5,6,9,10,11,12. Comprising the components necessary for transcription and translation, CFPS systems have significant advantages over whole-cell biosensors. Specifically, sensing is not limited by a cell wall and, in general, they are modular in design, biosafe, inexpensive, and can be freeze-dried for portable use. The ability to freeze-dry gene circuit-based reactions onto substrates such as paper or textiles, enables transport, long-term storage at room temperature5, and even incorporation into wearable technology13.
Previous work has demonstrated that E. coli cell-free systems can be used to detect numerous analytes, for example, toxic metals such as mercury, antibiotics such as tetracycline7,14, endocrine-disrupting chemicals15,16, biomarkers such as hippuric acid17, pathogen-associated quorum sensing molecules9,18 and illicit substances such as cocaine17, and gamma hydroxybutyrate (GHB)19. For the sequence-specific detection of nucleic acids, strategies have for the most part relied on the use of switch-based biosensors coupled to isothermal amplification techniques. Toehold switches are synthetic riboregulators (also referred to as simply 'switches' in the rest of the text) that contain a hairpin structure that blocks downstream translation by sequestering the ribosomal binding site (RBS) and the start codon. Upon interaction with their target trigger RNA, the hairpin structure is relieved and subsequent translation of a reporter open reading frame is enabled20.
Isothermal amplification can also be used as molecular diagnostics21; however, these methods are prone to nonspecific amplification, which can reduce the specificity and thereby the accuracy of the test to below that of RT-qPCR 22. In the work reported here, isothermal amplification upstream of the switch-based sensors was used to provide combined signal amplification that enables clinically relevant detection of nucleic acids (femtomolar to attomolar). This pairing of the two methods also provides two sequence-specific checkpoints that, in combination, provide a high level of specificity. Using this approach, previous work has demonstrated the detection of viruses such as Zika6, Ebola5, Norovirus10, as well as pathogenic bacteria such as C. difficile23 and antibiotic resistant Typhoid12. Most recently, cell-free toehold switches have been demonstrated for SARS-CoV-2 detection in efforts to provide accessible diagnostics for the COVID-19 pandemic11,12,13.
The following protocol outlines the development and validation of cell-free, paper-based synthetic toehold switch assay for Zika virus detection, from in silico biosensor design, through the assembly and optimization steps, to field validation with patient samples. The protocol begins with the in silico design of RNA toehold switch-based sensors and isothermal amplification primers specific for the Zika viral RNA. Although numerous isothermal amplification methods exist, here the use of nucleic acid sequence-based amplification (NASBA) to increase the concentration of viral RNA target present in the reaction, enabling clinically significant sensitivity was demonstrated. Practically, isothermal amplification methods have the advantage of operating at a constant temperature, eliminating the need for specialized equipment, such as thermal cyclers, which are generally limited to centralized locations.
Next, the process of assembling the synthetic toehold switch sensors with reporter coding sequences through overlap extension PCR, and screening the synthetic toehold switch sensors for optimal performance in cell-free systems using synthetic RNA is described. For this set of Zika virus sensors, we have selected the lacZ gene encoding the β-galactosidase enzyme, which is able to cleave a colorimetric substrate, chlorophenol red-β-D-galactopyranoside (CPRG), to produce a yellow to purple color change that can be detected by eye or with a plate reader. Once top-performing synthetic switches are identified, the process for screening primers for nucleic acid sequence-based isothermal amplification of the corresponding target sequence using synthetic RNA to find sets that provide the best sensitivity is described.
Finally, the performance of the diagnostic platform is validated on-site in Latin America (Figure 1). To determine the clinical diagnostic accuracy, the paper-based cell-free assay is performed using Zika virus samples from patients; in parallel a gold standard RT-qPCR assay is performed for comparison. To monitor colorimetric cell-free assays, we enable on-site quantification of results in regions where thermal cyclers are not available. The hand-assembled plate reader called Portable, Low-Cost, User-friendly, Multimodal (PLUM; hereafter referred to as portable plate reader) is also introduced here24. Initially developed as a companion device for cell-free synthetic toehold switch diagnostics, the portable plate reader offers an accessible way to incubate and read results in a high-throughput manner, providing integrated, computer vision-based software analysis for users.

Figure 1: Workflow for testing patient samples using paper-based cell-free toehold switch reactions. Please click here to view a larger version of this figure.
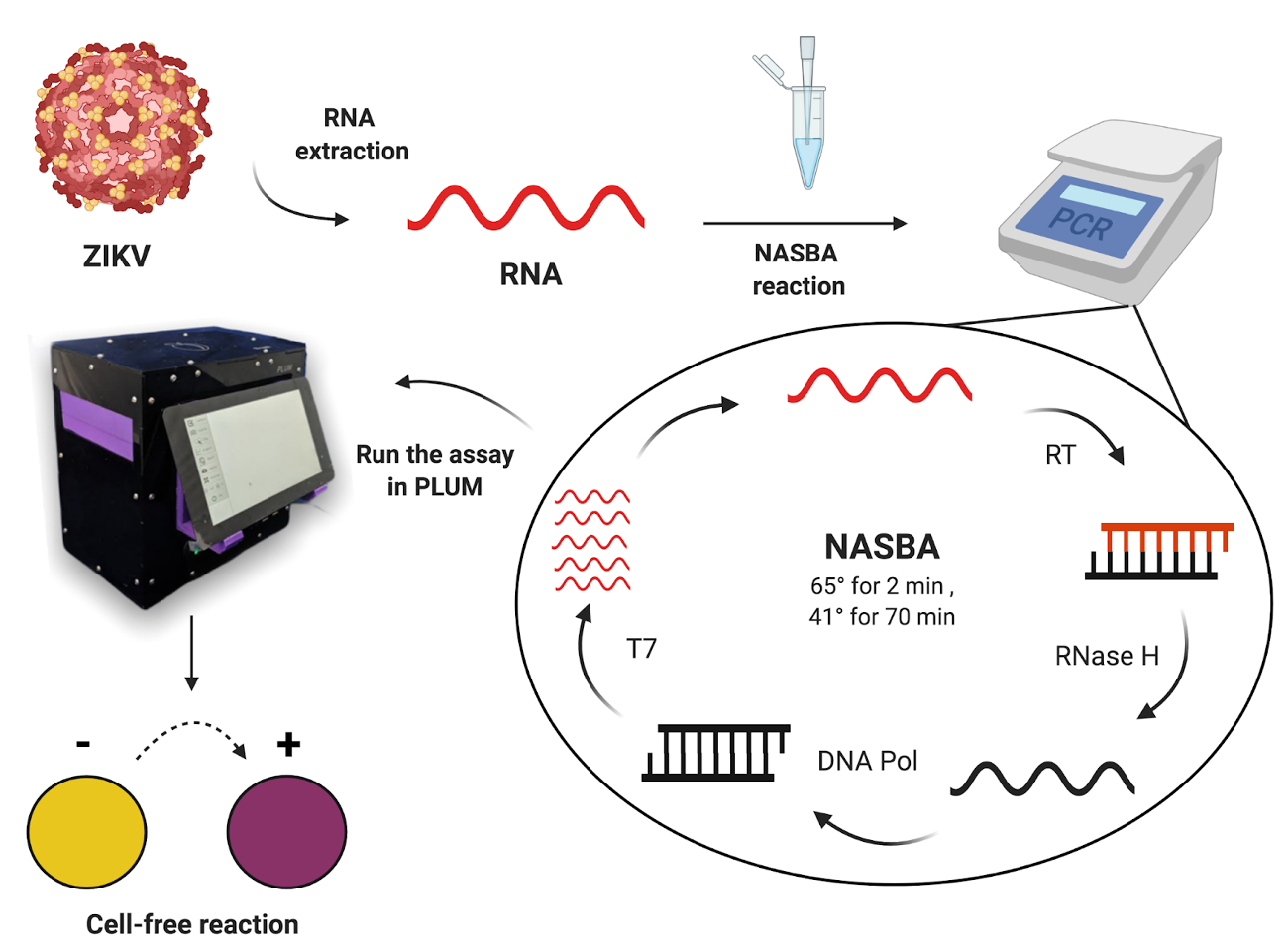{kind=link}
Protocol
All the procedures involving human participants are to be conducted in accordance with ethical standards and relevant guidelines, including the ethical principles for medical research involving human subjects established by the World Medical Association Declaration of Helsinki. This study was approved by the human research ethics committee under license protocol number CAAE: 80247417.4.0000.5190. Informed consent of all patients included in this study was waived by the Fiocruz-PE Institutional Review Board (IRB) for diagnostic samples.
NOTE: The PLUM device will be hereafter referred to as a 'portable plate reader'.
1. Computational design of nucleic acid sequence-based amplification primers
- Scan the Zika genome to identify a set of candidate regions for amplification that are approximately 200 nucleotides (nt) to 300 nt in length by putting the genomic sequence into NCBI/Nucleotide BLAST25,26. Compare the genome against reference RNA sequences (refseq_rna) and search for sites that remain highly conserved and do not have homology with the human genome (other BLAST parameters remain unchanged). Select at least two sites to design the synthetic toehold switch.
- Use the selection criteria of Deiman et al.27 to generate pairs of forward and reverse nucleic acid sequence-based amplification primers for each candidate amplification region. In brief, follow these criterias for designing primers: (1) GC content between 40%-60%; (2) 20 to 24 nt in length with DNA melting temperature above 41 °C; (3) no runs of four or more identical nucleotides; (4) the final 3' nucleotide is an A base; (5) low primer secondary structure and dimer formation probability. Screen 8-10 primer pairs for each target region (recommended).
- Append the prefix sequence containing the T7 promoter sequence AATTCTAATACGACTCACTATAGGGAGAAGG (T7 promoter sequence underlined) to the 5' end of the forward primers to allow for transcription of the amplicons using T7 RNA polymerase (RNAP). Order the primers as DNA oligos from a DNA synthesis company.
2. Computational design of toehold switches
- Install NUPACK28 version 3.2 (nucleic acid design suite) based on the instructions on the website29. Use a UNIX operating system and MATLAB (numeric computing platform) as the programming language (recommended).
- Identify the target sequences (e.g., viral genome) specific to the pathogen of interest for the toehold switch designs as in step 1.1. Choose the Zika target sequences from the nucleic acid sequence-based amplicons generated in step 1.2.
NOTE: In practice, either the nucleic acid sequence-based amplification primers or the synthetic toehold switches can be designed and tested first, depending on the needs of the users. If particular target regions of interest have already been established (e.g., from existing publications or diagnostic protocols), toehold switch design and screening may occur first, followed by design and screening for nucleic acid sequence-based amplification primers. If there are no established target regions, first screen nucleic acid sequence-based amplification primers against the full genome to narrow down the targets of choice while selecting for primer amplification efficiency. If multiple sequences for the pathogen are available, it is best to design nucleic acid sequence-based amplification primers that focus on highly conserved regions (rather than screening the entire genome). - Download and install the required MATLAB software on the computer.
- Set up the environment variables to allow the nucleic acid design suite functions to be called in the software. To do this, open the software, and then open (or create) a startup.m file in the default work folder. Next, copy the following lines of code into the startup.m file and finally, add the folder containing the nucleic acid design suite binaries to the PATH:
NUPACKINSTALL = '/Users/[user_name]/.../nupack3.2.2';
setenv('NUPACKINSTALL',NUPACKINSTALL);
setenv('PATH',[getenv('PATH'),sprintf(':%s/build/bin',NUPACKINSTALL)]); - Open the numeric computing platform software and navigate to the design software folder. Run the design algorithms accessible at https://github.com/AlexGreenLab/TSGEN.
- Input the target sequences into the design_input_file.csv located in the input subfolder. The inputs include Name, Outer sequence, Inner sequence, Temperature, Output name, and Output sequence as defined in Table 1. If no priming sites are determined yet for the target, keep inner and outer sequences the same. Only the first 29 nt of the reporter will be considered during the design process. When finished, save and close the updated spreadsheet.
NOTE: The output sequence is the sequence of the reporter protein planned to be used for the assay (e.g., lacZ). Specifying the inner and outer sequences ensures that the algorithm will not generate synthetic toehold switches that overlap with either of the primers. It also ensures that the assay recognizes three unique sites in the Zika genome to improve its specificity. - Select the parameters to use for the design function: toehold_switch_design_run(num_designs,input_file, options). Fill the parameter values as:
- num_designs - the number of top designs for each target output by the software. The default is 6 and can be changed as needed (a minimum of six designs for each target is recommended).
- input_file - this parameter specifies the name of the file that provides the input sequence information. The default value is 'design_input_file.csv' and can be changed as needed.
- Choose from the following options - (1) SeriesA and SeriesB: the version(s) of toehold switch that the code will generate. Set SeriesB to 1 and SeriesA to 0 to generate series B toehold switch, which is the format used in the Zika virus diagnostic6; (2) Parallel: set to 1 to harness multiple cores to compute the designs; otherwise, set to 0; (3) Antisense: set to 1 to generate toehold switches that hybridize the antisense sequence (i.e., the reverse complement) of the target input; otherwise, set to 0.
- Run the design function using the selected parameters: toehold_switch_design_run(num_designs,input_file,options). The algorithm will then start to generate the toehold switch designs for the targets of interest.
- Upon design completion, locate the top toehold switch design sequences and the corresponding target sequences in the final_designs folder in the form of .csv format spreadsheets. The toehold switch DNA sequences generated by the algorithm will contain the T7 promoter sequence at the 5' end and the conserved 21 nt linker sequence AACCTGGCGGCAGCGCAAAAG at the 3' end.
- Screen the resulting top toehold switch design sequences against other common viruses by putting them on NCBI-BLAST and checking for sequence homology. Accept sequences with 40% homology or less.
- Order the toehold switch hairpin sequences as DNA oligos and later assemble them with the reporter gene into fully functional switches. Order the necessary PCR primers for subsequent sensor assembly depending on the reporter protein of choice.
| Parameter | Definition |
| Name | The desired names of the output toehold switch sequences. |
| Outer sequence | Full NASBA transcript produced from amplification. |
| Inner sequence | The outer sequence excluding the primer binding sites. It matches the outer sequence but excludes the portions of the transcripts that bind to the forward and reverse primers. |
| Temperature | The temperature used by the algorithms to compute the RNA structures. |
| Output name | The name of the output gene (e.g. lacZ, gfp). |
| Output sequence | The sequence of the output gene. |
Table 1: The definition of each parameter used in the toehold switchdesign software.
3. Construction of toehold switches by PCR
NOTE: These steps describe the construction of LacZ toehold switches by overlap extension PCR. Here, the DNA oligo is used as a forward primer and the T7 terminator is used as a reverse primer. We use the pCOLADuet-LacZ plasmid as a template for the lacZ gene (addgene: 75006). Any other DNA templates that contain the corresponding sequence can be used as templates, provided that the T7 terminator is included in the final construct.
- Once synthetic DNA oligos have been received from the commercial provider, prepare solutions of the synthetic DNA and reverse amplification primer at a concentration of 10 µM in nuclease-free water. Assemble reactions in PCR tubes on ice according to Table 2.
NOTE: PCR volumes can be scaled as needed. Use a minimal amount (0.1-1 ng) of pCOLADuet-LacZ DNA template to avoid the need for an additional plasmid removal step or background LacZ signal. For higher DNA template amounts, follow the PCR with a DpnI restriction enzyme digest to remove the residual plasmid template. - Place the reactions in a thermal cycler, following the cycling conditions listed in Table 3. Analyze the PCR products on an agarose gel (Figure 2; see Supplementary Protocol and30).
- Purify the PCR products using a spin column-based PCR purification kit and elute the DNA in 15-30 µL of nuclease-free water, according to the manufacturer's instructions. Quantify the DNA using a spectrophotometer.
| Component | Volume | Concentration |
| 5X Q5 Reaction Buffer | 10 µL | 1x |
| 10 mM dNTPs | 1 µL | 200 µM |
| 10 mM Forward Primer (Synthetic Switch DNA FW) | 2.5 µL | 0.5 µM |
| 10 mM Reverse Primer (T7 terminator RV) | 2.5 µL | 0.5 µM |
| Template DNA (pCOLADuet-LacZ) | variable | <1 ng |
| Q5 High-Fidelity DNA Polymerase | 0.5 µL | 0.02 U/µL |
| Nuclease-free water | to 50 µL | - |
Table 2: The PCR components used to construct toehold switches.
| Step | Temperature | Time | |
| Initial Denaturation | 98 °C | 30 s | |
| 35 Cycles | Denaturation | 98 °C | 10 s |
| Annealing | 60 °C | 20 s | |
| Extension | 72 °C | 1.45 min | |
| Final extension | 72 °C | 5 min | |
| Hold | 4 °C | - | |
Table 3: Cycling conditions used during construction of toehold switches by PCR.
4. Preparation of synthetic RNA target (Trigger)
- Using a molecular biology design software, modify the synthetic trigger RNA templates (selected in step 1.1) to include an upstream T7 promoter sequence, ensuring that the full template remains short (<200 bp). Design forward and reverse primers to amplify the trigger sequence primer (for oligo sequences please see Supplementary Protocol).
- Order the sequences as DNA oligos. Once received, reconstitute the synthetic trigger DNA and amplification primers to 10 µM in nuclease-free water. Assemble the corresponding PCRs in thin-wall tubes on ice according to Table 2 and use 0.5 µL of the trigger DNA ultramer (final concentration of 0.1 µM) as the template DNA.
- Place the reactions in a thermal cycler and use the cycling conditions listed in Table 3. Use an extension time of 15 s. Assess the quality and purify trigger PCR products as described in step 3.3 and Supplementary Protocol.
NOTE: To expedite the process, column-purified trigger PCR products can also be directly used for initial toehold switch screening.
5. In vitro transcription of selected trigger sequences
- Assemble reaction components on ice according to Table 4.
- Incubate in vitro transcription (IVT) reactions at 37 °C for 4 h, followed by DNase I treatment to remove the template DNA. For the DNase I step, add 70 µL of nuclease-free water, 10 µL of 10x DNase I Buffer, and 2 µL of DNase I (RNase-free), and incubate at 37 °C for 15 min. If not proceeding to step 5.4 immediately, inactivate DNase I with the addition of 50 mM EDTA and heat inactivation at 65 °C for 10 min.
- Optional Step: Perform denaturing polyacrylamide gel electrophoresis (e.g., urea-PAGE) analysis on the IVT products to assess RNA quality as described in the Supplementary Protocol.
- Purify the trigger IVT products using a column-based RNA cleanup kit according to the manufacturer's instructions, and then quantify the RNA concentration and purity using spectrophotometry. See Supplementary Protocol for instructions on how to determine the molarity and copy number/µL of the RNA.
| Component | Volume | Concentration |
| 10X Reaction Buffer | 1.5 µL | 0.75x |
| 25 mM NTP mix | 6 µL | 7.5 mM |
| Template trigger DNA | X µL | 1 µg |
| T7 RNA Polymerase Mix | 1.5 µL | - |
| Nuclease-free water | X µL | To 20 µL |
Table 4: In vitro transcription (IVT) of selected trigger sequences.
6. Initial screening of the switches
NOTE: This section describes the steps associated with setting up cell-free, paper-based toehold switch reactions, and how to screen for high-performing toehold switches. The BSA blocked filter paper used in step 6.10 should be prepared in advance as described in the Supplementary Protocol.
- Prepare a CPRG stock solution by dissolving 25 mg of the powder in 1 mL of nuclease-free water. Keep the solution on ice at all times and store at -20 °C for long-term use.
- Determine the number of reactions to set up. For each toehold switch assessed, include a no template control (no switch and no target RNA-otherwise known as cell-free alone control) to account for any background signal that may arise from the master mix. Include a switch only control to assess for the background switch leakiness or OFF rate in the absence of target RNA.
- Assemble a cell-free reaction master mix of solution A, solution B, RNase inhibitor, and CPRG on ice according to the standard protocol listed in Table 5.
NOTE: The steps described here are for a master mix that will be sufficient for a triplicate set of 1.8 µL reactions with added 10% volume to account for pipetting error. The master mix volume should be adjusted as needed depending on the number of toehold switches screened. - For each triplicate cell-free reaction, dispense the master mix into a PCR tube, keeping all the tubes on ice. For the tube set aside as the cell-free alone control, add nuclease-free water to a final volume of 5.84 µL, mix by pipetting up and down, and centrifuge briefly. For the remainder of the tubes, add PCR purified toehold switch DNA to a final concentration of 33 nM.
- For tubes set aside as switch alone controls, add nuclease-free water to a final volume of 5.84 µL. For tubes set aside to test the toehold switch and target RNA combination, add in vitro transcribed target RNA to a final concentration of 1 µM. Then, add nuclease-free water to a final volume of 5.84 µL. Mix all reactions thoroughly by pipetting up and down, and centrifuge briefly.
- Add 30 µL of nuclease-free water to the wells surrounding the reaction wells on a 384-well black, clear bottomed plate. This minimizes evaporation over the course of the experiment and improves reproducibility.
NOTE: If using the portable plate reader device, place a PCR foil over the plate, and use a precision knife to cut out the wells intended for your reaction, as well as each of the four corner wells. The PCR foil prevents the portable plate reader camera overexposure from empty wells. - Using a 2 mm biopsy punch and tweezers, cut BSA-blocked filter paper disks and place them in the reaction wells either by ejecting the punch or using tweezers (see Supplementary Protocol for preparing BSA-blocked filter paper). Dispense triplicate 1.8 µL volumes from each reaction tube onto the filter paper discs in the 384-well plate, keeping all the samples, as well as the 384-well plate, on ice at all times.
NOTE: If using the portable plate reader device, add 1.8 µL of CPRG (0.6 mg/mL) to each of the four corners of the 384-well plate. The yellow color from the CPRG provides pattern recognition to the portable plate reader for alignment of the digital multiwell plate template in image analysis. Cover the wells of the plate with PCR film to prevent evaporation. - Place the plate in a plate reader, read OD570 at 37 °C every min for 130 min.
| Component | Volume | Final Concentration Per Reaction |
| Solution A | 2.38 µL | 40% |
| Solution B | 1.78 µL | 30% |
| RNase Inhibitor | 0.03 µL | 0.5% v/v |
| CPRG (25 mg/mL) | 0.14 µL | 0.6 mg/mL |
| Toehold Switch | X µL | 33 nM |
| Target RNA | X µL | 1 µM |
| Nuclease-free water | to 5.94 µL | - |
| Total volume: | 5.94 µL | |
Table 5: PURExpress cell-free transcription-translation reaction components.
7. Identifying high-performing toehold switches
NOTE: This section describes how to analyze data from step 6 in order to select the best performing toehold switches to move forward with.
- Begin data analysis by first normalizing to the background OD570 absorbance. To do this, subtract the OD570 measurements of reactions that do not have any switch or target RNA (i.e., cell-free alone wells) from the other OD570 measurements.
- Smooth the normalized data using a three-point moving average and adjust the minimum value of each well to zero. See Figure 3 for an example of normalized time-course data collected for the toehold switches 27B, 33B, and 47B.
- Using this processed data, calculate the fold change (or ON/OFF ratio) by determining the difference in the rate of color change (i.e., change in OD570 over time) between the toehold switch and target and switch alone wells (see Supplementary Protocol for calculating the ratio; Figure 4).
- Select switches with the highest ON/OFF fold change for further characterization. Omit the poorly performing toehold switches that display the lowest fold change.
8. Nucleic acid sequence-based amplification primer screening and sensitivity
NOTE: In the following steps, first a screen for functional isothermal amplification primers is done, and then their sensitivity is assessed by determining the number of target RNA copies per µL of synthetic RNA that a given toehold switch can reliably detect when coupled with isothermal amplification. Following isothermal amplification, perform cell-free reactions to identify successful nucleic acid sequence-based amplification primer sets. However, it may be more cost-effective to run polyacrylamide or agarose gels on nucleic acid sequence-based amplification reactions to first narrow the pool of candidate primer sets. In that case, nucleic acid sequence-based amplification primer sets that generate a band on the gel at the appropriate amplicon size can be shortlisted for subsequent cell-free screening.
- Make a 25 µM stock solution of all forward and reverse primer sets in nuclease-free water (see Supplementary Protocol). Determine the number of nucleic acid sequence-based amplification reactions and subsequent cell-free reactions that need to be set up. This will depend on the number of toehold switches and respective primer sets.
NOTE: When screening the primers, it is important to always include a no template control for each primer set to account for any background artifacts that may arise due to primer-associated nonspecific amplification. - Set up one 5 µL reaction as follows (scale this as needed). Thaw all the reagents on ice. Assemble a reaction master mix of the reaction buffer, nucleotide mix, and RNase inhibitor according to Table 6. Mix thoroughly by pipetting up and down until the white precipitate is solubilized, and then dispense aliquots into PCR tubes.
- If the master mix is for screening nucleic acid sequence-based amplification primers, exclude the primers from the master mix at first (dispense 2.55 µL of master mix). Otherwise, if performing primer sensitivity analysis, the primers can be included in the master mix (in which case dispense 2.75 µL aliquots). Add the enzyme mix after the denaturation and equilibration stages.
- If screening nucleic acid sequence-based amplification primers, add the forward and reverse primers to the appropriate tubes. On a thermal cycler, set up the incubation protocol as described in Table 7.
- Add 1 µL of either nuclease-free water or 1 µL of target trigger RNA at 2 pM or approximately 106 copies/µL, making sure to label the tubes accordingly. Mix by gentle pipetting and briefly spin down the tubes.
NOTE: For primer set screening, use a concentration of target trigger RNA that is high enough not to impinge on the lower limit of detection of the isothermal amplification method, yet not high enough to provide activation of the toehold switch if no amplification were to occur. - Move the tubes to the thermal cycler and start the incubation. After 12 min, remove the tubes and add 1.25 µL of the enzyme mix to each tube. Mix by pipetting up and down and briefly centrifuge the tubes.
NOTE: At this point, the tubes may be kept at room temperature; however, the enzyme mix should be kept on ice until added to the tubes. - Return the tubes to the thermal cycler, skipping the 41 °C hold step to start the 1 h reaction incubation.
NOTE: Following incubation, the reactions can be placed on ice for same-day processing or frozen at -20 °C for later processing. - Follow steps 6.2-6.7 and Table 8 to assemble paper-based cell-free toehold switch reactions to assess primer performance. Analyze data as described in steps 7.1-7.2 and Figure 3.
NOTE: In addition to the controls previously mentioned, it is important to also include the negative control to account for any background signal that might arise from the reaction. Additionally, it is important to also include a control with the switch and 2 pM (final concentration) of target RNA, as a no-NASBA amplification control. - Identify candidate primer pairs to move forward with sensitivity analysis. Primer pairs are selected based on whether toehold switch activation occurs following the addition of the incubated reaction. If needed, repeat the primer screen with more primer combinations.
- Keeping RNA samples on ice at all times, make the following serial dilution set of target RNAs in nuclease-free water: 104, 103, 102, 101, and 100 RNA copies/µL. Repeat nucleic acid sequence-based amplification and cell-free reactions with the selected primer sets (steps 8.2-8.7), testing the serial dilution series in order to identify the primer sets that provide the best sensitivity. See Figure 5 for an example of results for determining the primer set sensitivity.
NOTE: Ideally, the selected toehold switch and the primer pair should detect as few as 1-100 target RNA copies per µL of the RNA (stock solution concentration). - Repeat the nucleic acid sequence-based amplification sensitivity experiment in biological triplicate. This step serves to confirm the reproducibility of the results before moving to experiments with patient samples.
- Optional: To have a reliable source of the switch DNA for long-term use and for transport, clone the selected switch sequence(s) into a plasmid using any conventional cloning method. Similarly, the target trigger RNA sequence may also be cloned into a plasmid of choice for storage.
| Component | Volume per reaction | Final Concentration |
| NASBA Reaction Buffer | 1.67 µL | 1x |
| NASBA Nucleotide Mix | 0.833 µL | 1x |
| 25 µM Forward Primer | 0.1 µL | 0.5 µM |
| 25 µM Reverse Primer | 0.1 µL | 0.5 µM |
| RNase Inhibitor (40 U/µL) | 0.05 µL | 0.4 U/µL |
| Target RNA | 1 µL | |
| NASBA Enzyme Mix | 1.25 µL | 1x |
| Total volume | 5 µL |
Table 6: NASBA reaction components.
| Step | Temperature | Time |
| Denaturation | 65 °C | 2 min |
| Equilibration | 41 °C | 10 min |
| Hold | 41 °C | ∞ |
| Incubation | 41 °C | 1 h |
| Hold | 4 °C | - |
Table 7: Reaction conditions for the NASBA.
| Component | Volume | Final Concentration Per Reaction |
| Solution A | 2.38 µL | 40% |
| Solution B | 1.78 µL | 30% |
| RNase Inhibitor | 0.03 µL | 0.5% v/v |
| CPRG (25 mg/mL) | 0.14 µL | 0.6 mg/mL |
| Toehold Switch | X µL | 33 nM |
| Target RNA (if applicable) | X µL | 1 µM |
| NASBA (if applicable) | 0.85 µL | 1:7 |
| Nuclease-free water | to 5.94 µL | - |
| Total volume: | 5.94 µL | |
Table 8: Paper-based cell-free reaction components.
9. Patient samples collection and viral RNA extraction
NOTE: This section describes the protocol to collect patient samples and to extract the RNA using an RNA purification kit. The protocol below is used to obtain serum from peripheral blood. The samples used in this study were collected from patients presenting fever, exanthema, arthralgia, or other related symptoms of arbovirus infection in Pernambuco state, Brazil.
- Collect peripheral blood samples from suspected arbovirus-infected patients. Perform venipuncture (whole blood tube) using a standard aseptic technique. Label the sample tubes with an anonymous code and the date of sample collection.
- Centrifuge blood to separate the serum at 2,300 x g for 10 min. Using a pipette, prepare aliquots of serum (200 µL) that will be used for RNA extraction in 1.5 mL tubes.
NOTE: If it is not possible to perform the RNA extraction shortly after sample collection, serum samples should be stored at -80 °C prior to downstream applications. - Pipette 560 µL of prepared Buffer AVL (viral lysis buffer) containing the carrier RNA into a 1.5 mL tube. Add 140 µL of serum to the Buffer AVL/carrier RNA mixture in the tube. Mix by pulse-vortexing for 15 s.
- Incubate for 10 min at room temperature, and then briefly centrifuge the sample to collect the contents at the bottom of the tube. Add 560 µL of ethanol (96%-100%) to the sample and mix by pulse-vortexing for 15 s. After mixing, centrifuge the sample to collect the contents at the bottom of the tube.
- Carefully add 630 µL of the solution from step 9.4 to the spin-column without wetting the rim. Following this step, close the cap, and centrifuge at 6,000 x g for 1 min. Place the spin-column into a clean 2 mL collection tube and discard the used collection tube containing the filtrate.
- Carefully open the spin-column and repeat step 9.5. Carefully open the spin-column and add 500 µL of Buffer AW1 (wash buffer 1). Close the cap, and centrifuge at 6,000 x g for 1 min. Place the spin-column into a clean 2 mL collection tube and discard the used collection tube containing the filtrate.
- Carefully open the spin-column and add 500 µL of Buffer AW2 (wash buffer 2). Close the cap and centrifuge at 20,000 x g for 3 min. Place the spin-column into a clean 2 mL collection tube, and discard the used collection tube containing the filtrate.
- To avoid contamination by residual buffer AW2, which could cause problems in downstream enzymatic reactions, place the spin-column into a clean 2 mL collection tube and discard the used collection tube with the filtrate. Centrifuge at 20,000 x g for 1 min.
- Place the spin-column in a clean 1.5 mL tube and discard the used collection tube with the filtrate. Carefully open the spin-column and add 60 µL of Buffer AVE (elution buffer) equilibrated to room temperature. Following this step, close the cap, and incubate at room temperature for 1 min.
- Centrifuge at 6,000 x g for 1 min. The eluted RNA can then be used as an input for the NASBA and RT-qPCRs in parallel.
- Follow steps 8.3 to 8.11 to perform the nucleic acid sequence-based amplification and paper-based cell-free reactions using 1 µL of extracted patient RNA, ensuring to include negative and positive control reactions. Following the reactions, prepare the 384-well reaction plate and run the assay in the portable plate reader device at 37 °C (see details in the Supplementary Protocol).
NOTE: Two RNA concentrations of 104 and 102 PFU/mL, extracted from cultured Zika virus are used as positive controls for all experiments during the clinical validation. In addition to the controls previously mentioned, it is important to also include the trigger (10 ng/µL) as a positive control. - At the end of every experiment, the raw data (.csv file) from the prtable plate reader can be uploaded to a secure database online. Additionally, the portable plate reader generates a report indicating whether samples are positive or negative for Zika virus (Figure 6); see the representative results section for examples of all the graphs obtained during the incubation (Figure 7).
NOTE: Users can also transfer files from the portable plate reader to their own computer via the USB.
10. Portable plate reader device
- The portable plate reader device (Figure 8) can be used as an alternative to a conventional plate reader24. For the protocol and representative results, see Supplementary Protocol.
11. RT-qPCR for Zika virus detection
NOTE: This section outlines the steps to perform the RT-qPCR for Zika virus detection from patient samples (see Supplementary Protocol).
- To avoid contamination, assemble the RT-qPCR components in an area isolated from the amplification process (e.g., clean-hood). Place the RT-qPCR buffer, enzymes, and primer/probes on ice or a cold-block. Following this, add the reactions to a 384-well plate on ice according to Table 9.
NOTE: Negative (extraction control and non-template control [NTC]) and positive control (104 PFU/mL) are recommended for all experiments. - After adding the reagents to a 1.5 mL microcentrifuge tube, mix the reaction by pipetting up and down (do not vortex), and dispense 6.5 µL into each well. Add 3.5 µL of each RNA template in triplicate.
- Place a PCR film over the top of the plate. Centrifuge the 384-well plate at 600 x g for 2 min.
- Place the plate in a RT-qPCR machine using the following cycling conditions outlined in Table 10. To analyze the results, use the RT-qPCR machine design and analysis software and consider the automatic threshold and baseline (see details in the Supplementary Protocol).
| Component | Volume | Concentration |
| 2X QuantiNova Probe RT-PCR Master Mix | 5 µL | 1 X |
| 100 µM Forward Primer | 0.08 µL | 0.8 µM |
| 100 µM Reverse Primer | 0.08 µL | 0.8 µM |
| 25 µM Probe | 0.04 µL | 0.1 µM |
| QuantiNova ROX Reference Dye | 0.05 µL | 1 X |
| QuantiNova Probe RT Mix | 0.1 µL | 1 X |
| Template RNA | 3.5 µL | - |
| Nuclease-free water | to 10 µL | - |
Table 9: RT-qPCR components to amplify Zika virus RNA based on Centers for Disease Control and Prevention-CDC USA protocol to detect Zika virus from patient samples31.
| Step | Temperature | Time | |
| Reverse transcription | 45 °C | 15 min | |
| PCR initial activation step | 95 °C | 5 min | |
| 45 Cycles | Denaturation | 95 °C | 5 s |
| Combined annealing/extension | 60 °C | 45 s | |
Table 10: Cycling conditions for RT-qPCR.
Representative Results
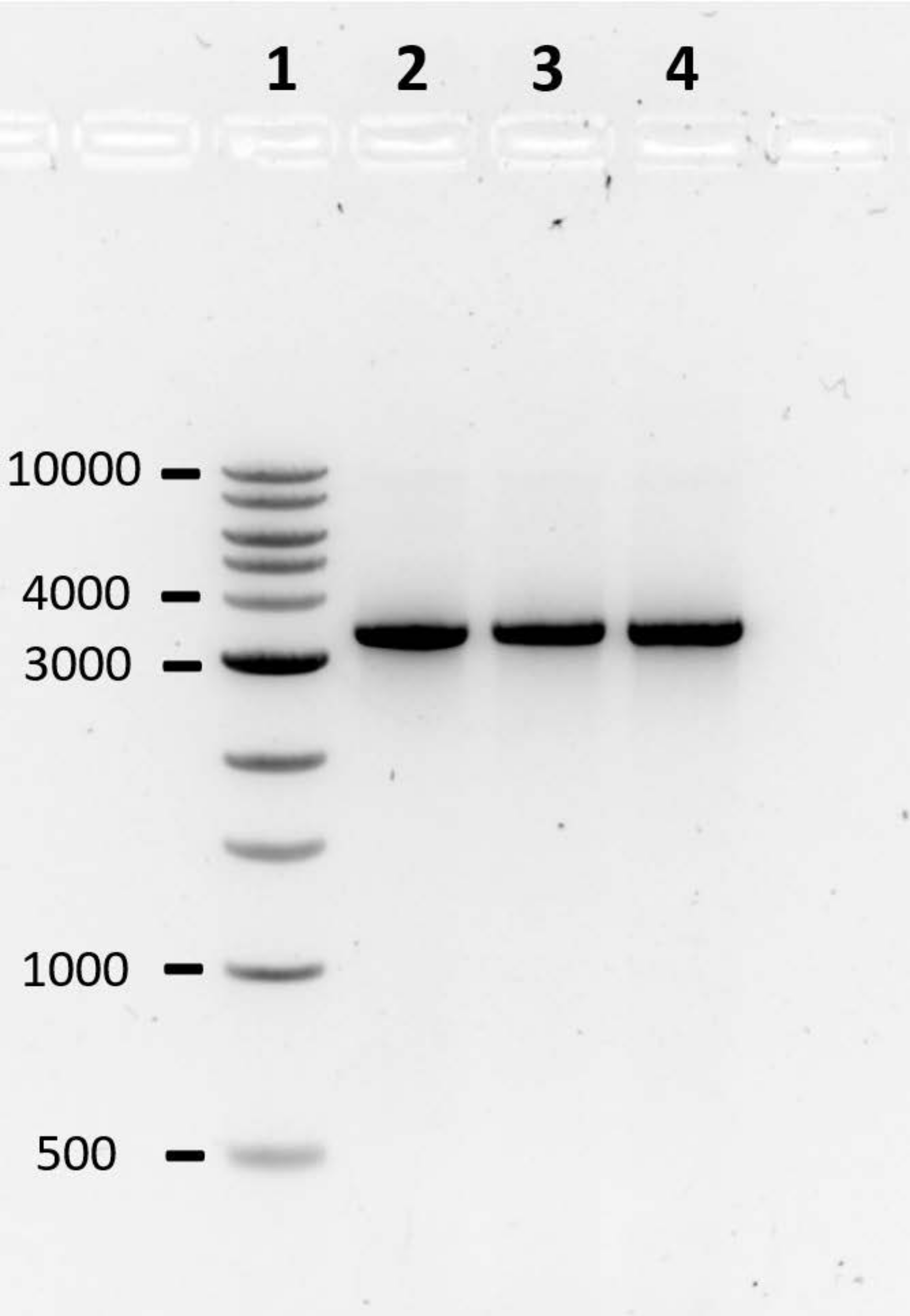
Following the computational design pipeline, the construction of three toehold switches was performed by PCR. The PCR products were analyzed using agarose gel electrophoresis (Figure 2). The presence of a clear band around 3,000 bp, roughly the size of the lacZ gene coupled to a toehold switch, typically indicates a successful reaction. Alternatively, a lane without a band, multiple bands, or a band of the incorrect size, indicates a failed PCR. In the case of a failed PCR, the reaction conditions and/or primer sequences should be optimized.
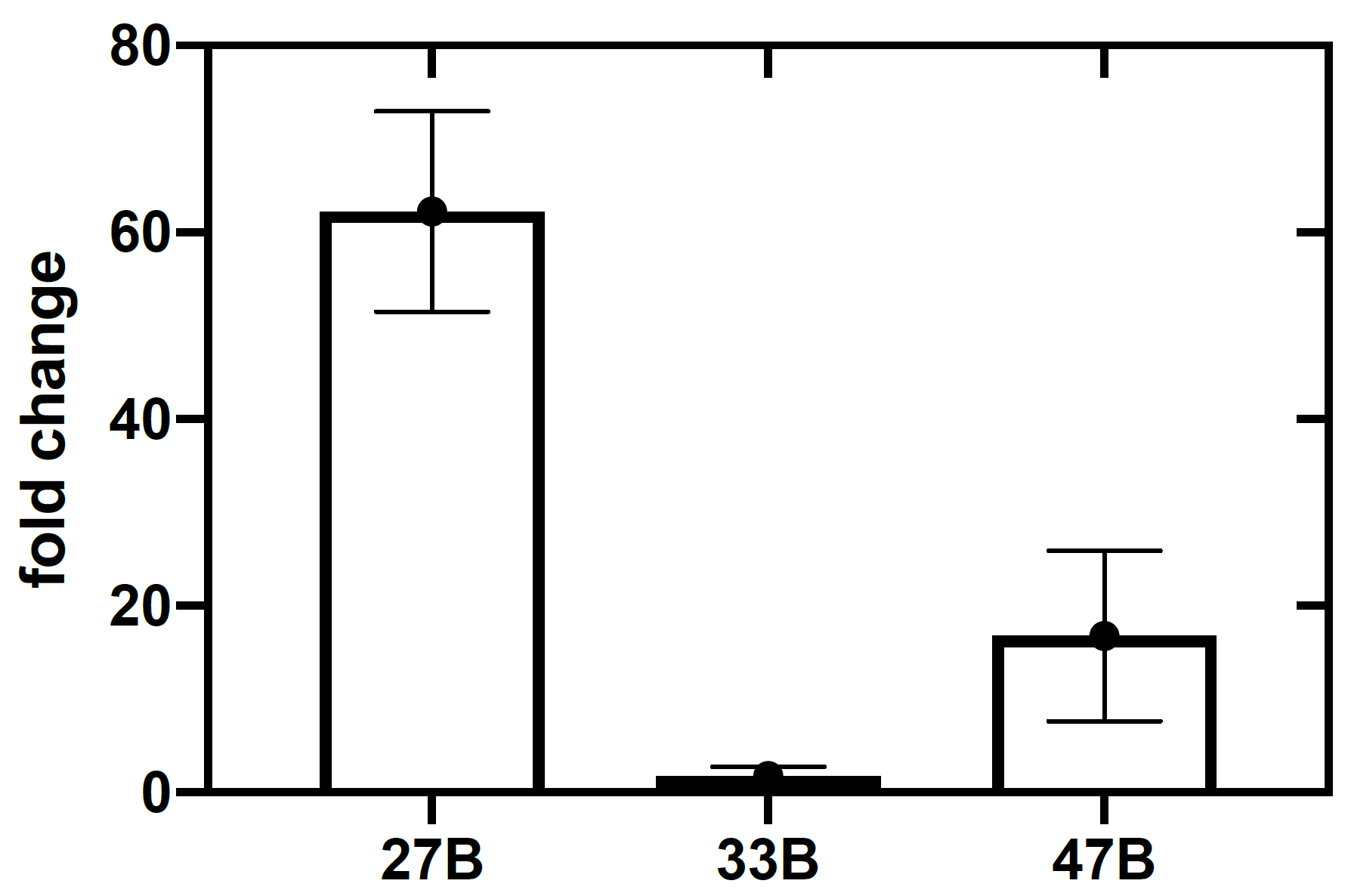
The assembled toehold switches were screened to assess each sensor against its respective in vitro transcribed trigger RNA (Figure 3). While all three sensors displayed an increased OD570 absorbance, switch 27B (Figure 3A) had the most rapid on-rate. Switches 33B and, to a lesser extent, 47B showed an increased OD570 absorbance in the absence of target trigger RNA, indicating that these switches possess some background activity or leakiness (Figure 3B,C)-a trait not wanted in a candidate sensor as it can reduce specificity. To more clearly identify the sensor with the highest ON/OFF signal ratio, the fold change of OD570 absorbance was calculated (see supplemental information section 5) and plotted (Figure 4). From this analysis, it is clear that switch 27B is the sensor that has the best performance with an ON/OFF ratio of around 60.
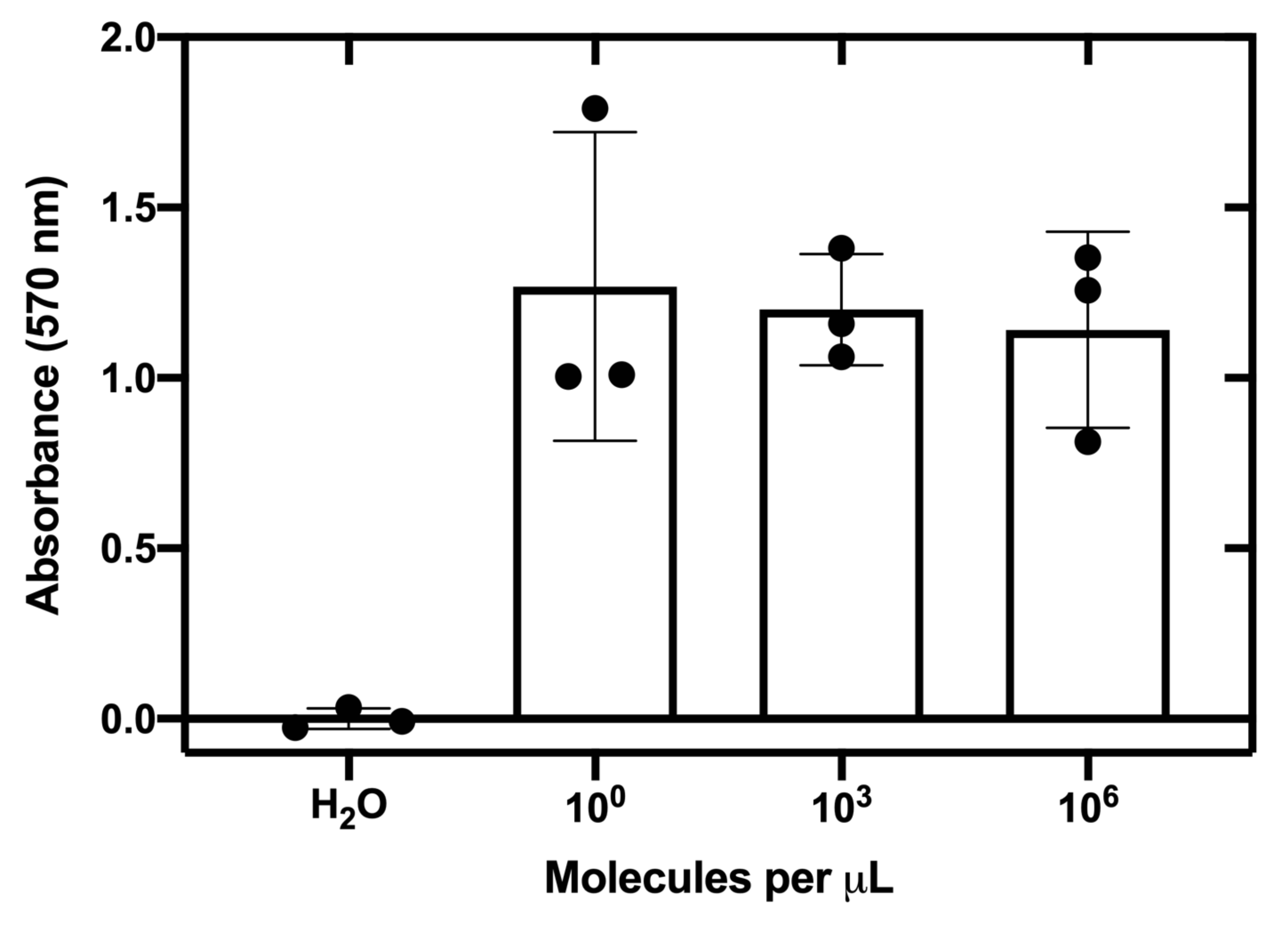
The sensitivity of the top-performing toehold switch (27B) was then evaluated by determining the lowest RNA concentration required to activate the toehold switch when coupled with a NASBA reaction. The graph illustrates that the top-performing Zika sensors can detect RNA at concentrations as low as 1.24 molecules per µL (equivalent to ~2 aM; Figure 5).
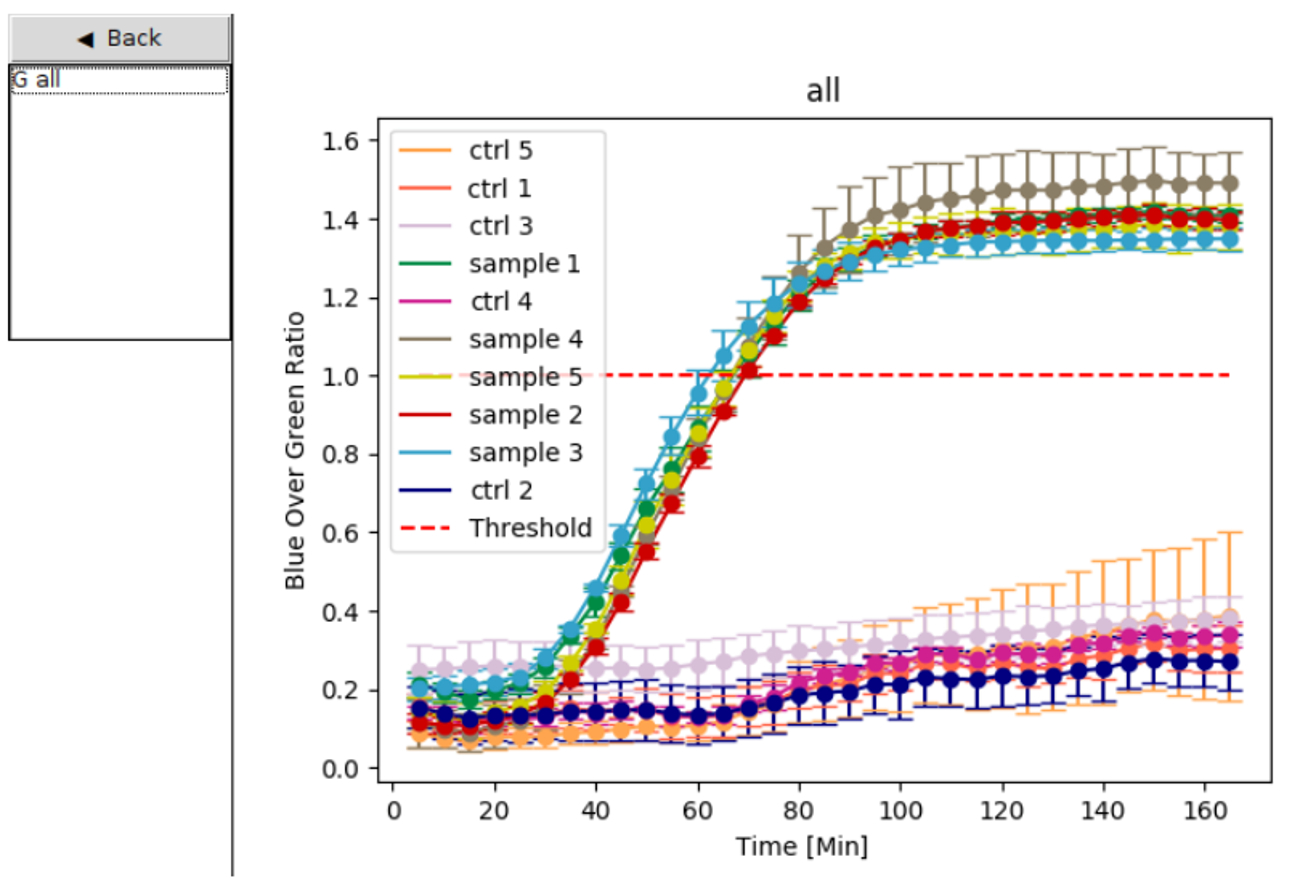
After switch 27B was identified and validated, the sensor materials were distributed to the team members in Recife in the Pernambuco state of Brazil. In Brazil, the clinical diagnostic accuracy of the Zika virus diagnostic platform was assessed using Zika virus patient samples, in parallel with RT-qPCR for comparison. To validate the paper-based Zika diagnostic platform, the portable plate reader was used, which is capable of incubating and reading the colorimetric output of paper-based sensors. The color change from yellow to purple is used to identify a positive sample, while a negative sample remains yellow (Figure 6). An additional option for visualizing the results generated by the portable plate reader (Figure 8) is to plot the colorimetric response for each paper-based reaction over time. Samples were tested in triplicate and those that exceeded the threshold (red line set to 1) were considered positive, while samples below the threshold were considered negative (Figure 6 and Figure 7).
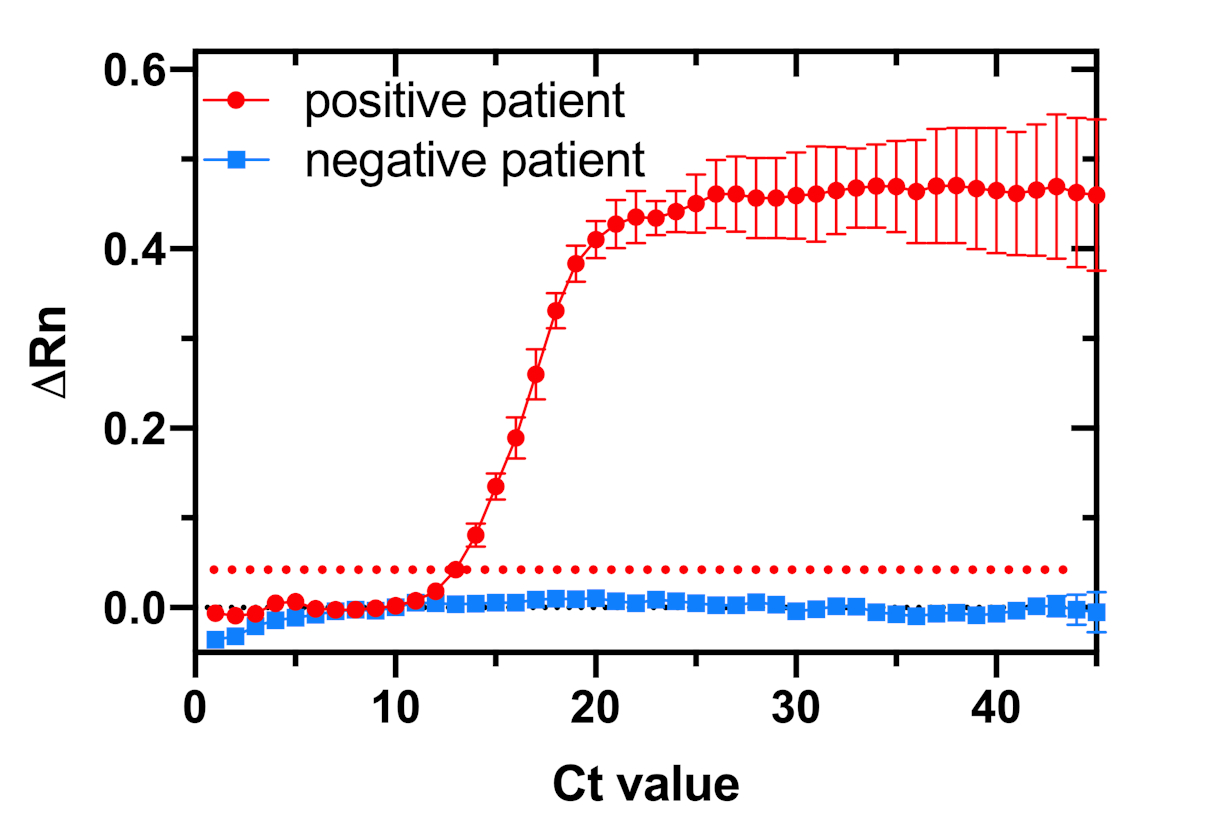
Finally, to evaluate and compare the clinical performance of the Zika toehold switch sensor with the current gold standard method for diagnosing Zika virus infection, all patient samples were tested in parallel with RT-qPCR. The amplification plot of two representative patient samples was tested in triplicate for the detection of Zika virus by RT-qPCR (Figure 9). The samples are considered positive when the cycle threshold (Ct) value is ≤38; the red line indicates a positive sample and the blue line indicates a negative sample for Zika virus.

Figure 2: Agarose gel electrophoresis to assess the quality of PCR products. PCR products are analyzed on a 1% agarose gel in 1X TAE, run at 80 V for 90 min. A single clear band typically indicates a successful reaction. Lane 1: 1 kb DNA ladder; Lanes 2-4: 27B switch DNA, 33B trigger DNA, and 47B trigger DNA, respectively.The numbers on the left-hand side represent band size in bp. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Prototyping three toehold switches for paper-based Zika sensors. The performance of three paper-based RNA toehold switch sensors was measured at 37 °C for over 130 min. Each graph contains two traces, one represents the switch only control, while the other represents the switch and trigger. The three graphs represent data acquired using toehold switch sensors 27B (A), 33B (B), and 47B (C). Error bars represent the standard error of the mean (SEM) from three replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Top-performing sensors are identified by calculating the fold change in absorbance at 570 nm. Fold change (or maximum ON/OFF ratio) is calculated by measuring the ratio of absorbance (OD570) at 130 min between the switch only control, and the switch plus trigger CFPS assay. Error bars represent SEM from three replicates. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Sensitivity assessment of top-performing switch. In vitro transcribed Zika RNA is titrated into NASBA reactions. After a 1 h incubation, the reactions were added to cell-free PURExpress reactions on paper discs at a ratio of 1:7. The fold change after 130 min at 37 °C is plotted. This figure has been reproduced from24. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Portable plate reader capture page loaded with captured image data. This figure shows a sample picture of the final image captured by the portable plate reader during a data collection run. The original date/time stamp is visible at the top of the image. Yellow color indicates control or negative reactions, and the purple color indicates a positive reaction. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Data analysis mode. On the left, users select the data sets they would like to plot; the graphs are then displayed on the right with unique colors for each sample or control set. The dashed red line serves as a threshold for determining positive and negative samples. Samples tested in triplicate that exceed the threshold are considered positive, while samples below the threshold are deemed negative. Error bars represent standard deviation (SD) from three replicates. Ctrl 1 to Ctrl 5 indicates controls. Please click here to view a larger version of this figure.
{kind=link}

Figure 8. PLUM, a portable plate reader. This portable plate reader acts as a lab-in-a-box and serves as a temperature-controlled plate reader to incubate and monitor colorimetric reactions. This portable device can provide quantitative and high-throughput measurements of the paper-based Zika sensors on-site. Please click here to view a larger version of this figure.
{kind=link}

Figure 9: RT-qPCR plot of the amplification of two patient samples tested in triplicate for the detection of the Zika virus. Samples are considered positive when the cycle threshold (Ct) value is ≤38. The dotted red line serves as a threshold for determining positive and negative samples. The red trace indicates a positive sample, and the blue trace indicates a negative sample. ΔRn (delta Rn) value represents the normalized magnitude of the fluorescence signal detected by the RT-qPCR instrument for all the samples tested. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Protocol File. Please click here to download this File.
Supplemental Figures. Please click here to download these figures.
Discussion
The combined paper-based system described here can bring clinically relevant molecular diagnostics, with performance that is functionally comparable to RT-qPCR, to the point-of-need6. Importantly, for remote settings, the availability of diagnostics on-site can decrease the time to results from days to hours. Highlighting the programmability of this approach, the pipeline that has been described can be used to detect virtually any pathogen target. We have paired the molecular tools with a purpose-built portable plate reader, which is compact and compatible with battery operation (8–9 h) and provides onboard data analysis to enable distributed applications. In other work, we have validated the combined hardware and Zika virus diagnostic platform with 268 patient samples, in parallel with RT-qPCR, and found a diagnostic accuracy of 98.5%24. Taken together, our goal is to enable the technology transfer of this platform to researchers so that it can be repurposed and improved by the community to address unmet diagnostic needs.
The in silico toehold switch design process has been integrated and automated into a pipeline that can be divided into three stages. The first stage generates a pool of toehold switch designs that hybridize to the target sequence in one nucleotide increments. The second stage examines the secondary structure and toehold switch availability and eliminates sensors with in-frame premature stop codons. A scoring function that considers multiple factors (e.g., defect level of the toehold switch, toehold switch availability, and target site accessibility) is then implemented to select top toehold switch designs based on overall scores. In the final stage, a list of sequences is generated for the top toehold switch designs and their corresponding target triggers. The top sensor sequences should be screened for specificity against the human transcriptome and closely related viral genomes using NCBI/Primer-BLAST25. It is also best practice to screen the sensor target sites for sequence conservation in the Zika viral genome to ensure that the sensors will provide broad and robust detection. Several versions of toehold switch design software have been developed and the design algorithm allows users to generate two versions, either series A6 or series B toehold switches6. In this article, the focus has been on the series B toehold switch design.
Following commercial DNA synthesis, the toehold switches can be rapidly assembled, and then tested by performing an initial screen against a synthetic target trigger sequence that corresponds to short regions (200-300 nt) of the target genome. For screening the performance of toehold switch-based sensors, it is ideal to add the target sequence in the form of RNA. In this article, the steps required to add in vitro transcribed trigger RNA have been outlined. However, if available, full-length genome templates such as quantified viral RNA extracts or commercial synthetic RNA genomes or standards can be used. Using full-length RNA genomes for initial toehold switch screening is beneficial as it can inform whether additional factors, such as RNA secondary structure, will affect sensor performance. To optimize the ON/OFF ratio of candidate switches, the toehold switch DNA can be titrated into the cell-free reaction. This step can also serve to identify high-performing toehold switches (fold amplification, or high ON/OFF ratio) and omit leaky toehold switches (high signal in the absence of the target RNA) from downstream characterization steps.
To improve the limit of detection of the top-performing toehold switch candidates, NASBA is used to increase clinically-relevant concentrations of target Zika viral RNA to a level that can be detected by toehold switches6. Different combinations of forward and reverse primer sets are screened to determine the best NASBA primer and toehold switch combinations to enable detection at clinically relevant concentrations. Once an ideal primer set and toehold switch combination has been identified, the assay is taken forward to clinical validation. It is important to note that the toehold switch and NASBA screening stages can be labor and resource-intensive and therefore test development is best suited to well-resourced research sites. Although we have not applied process automation, it is likely that this could accelerate the iterative design, build, and test cycle32. Fortunately, the turnaround time from sensor design and testing to deployment can be remarkably short (less than a week), making this strategy ideal for time-critical situations, such as epidemic outbreaks6.
Even after a biosensor with clinically relevant sensitivity has been developed, there are technical challenges that need to be addressed. Since this protocol involves manual operation and is a multi-step procedure, there is a risk of cross contamination between samples. We do our best to abate this risk through careful laboratory practice. In a recent clinical trial of 268 patient samples, we did not encounter any contamination issues; however, it is an important consideration24. With this in mind, the protocol remains a laboratory assay and requires a skilled user with command of proper molecular biology techniques. An additional consideration for deployment is the RNA isolation from patient samples. Here we describe RNA isolation using column-based nucleic acid extraction kits. However, in other work, we have demonstrated an effective and simple boiling lysis method (95 °C for 2 min) for low-burden patient sample processing6. This strategy nearly eliminates the cost associated with RNA extraction and avoids the use of column-based nucleic acid extraction kits, which can pose a logistics challenge in low-resource settings or supply chain issues during crises, such as the COVID-19 pandemic33.
As we have seen during the COVID-19 pandemic, the instruments used to perform RT-qPCR can themselves serve as a bottleneck and limit patient access to testing. This factor, which is also largely financial, leads to a centralized testing mode that can limit diagnostic access. For example, during the 2015/2016 Zika outbreak, only five national reference labs were available in Brazil, which caused delays in patient testing. Without considering the potential benefit of economies of scale, the current cost of goods for the portable plate reader is ~$500 USD, which even if increased five-fold to account for labor and commercial margin, still provides an affordable instrument. This compares well to RT-qPCR instruments that range in cost from $15,000-$90,000 USD34. Furthermore, the estimated cost per test for the cell-free assay in Latin America is around $5.48 USD, while the cost per test of RT-qPCR in Brazil was ~$10-11 USD at the time of the Zika outbreak36. Beyond the cost of equipment, the portable plate reader has a small footprint (20 cm3), automatic analysis, data upload to the cloud via internet, and can be run on battery power. These features dramatically expand the potential settings where testing can be deployed and concomitantly expands the patient population that can be served.
To date the most common commercial E. coli CFPS platforms are the S30 and PURE systems37; however, a key consideration in improving access to diagnostics in low- and middle-income countries is the limited domestic availability of these reagents. An important step toward resolving this challenge is the development of local CFPS production. The Federici lab has recently made significant progress toward developing a non-commercial platform to implement toehold switch-based sensors in lysate-based cell-free systems, reaching a 2.7 fM LOD with Zika virus RNA14. Not only does this achievement allow the reagents to be made in the country of use, avoiding import tariffs and delays, but labor costs also scale to local rates and thus the overall cost can be significantly lowered. In the work outlined by the Federici group, the cost of producing the CFPS expression reaction (5 µL) in Chile was 6.9 cents (USD)35,38, providing a dual incentive (improved logistics and cost) for implementing lysate-based systems35,38.
The placement of RT-qPCR-comparable testing into distributed diagnostic networks could bring significant advantages over current practices that are dependent on the transportation of samples to centralized RT-qPCR facilities. In peri-urban settings, where Zika cases were concentrated, the physical distance between a patient and the diagnostic facility slows diagnosis and increases the risk that results will not reach the patient at a clinically relevant time. It is our hope that the work presented here can contribute to enabling the research community, through the transfer of knowledge, to create decentralized biotechnologies and portable hardware for human health, agriculture and environmental monitoring.
Acknowledgements
The authors thank all members of the Green, Pardee and Pena labs as well as all co-authors of previous manuscripts pertaining to the work disclosed in this manuscript. S.J.R.d.S. was supported by a PhD fellowship sponsored by the Foundation for Science and Technology of Pernambuco (FACEPE), Brazil, reference number IBPG-1321-2.12/18, and currently is supported by a Postdoctoral Fellowship sponsored by the University of Toronto, Canada. P.B. is supported by the William Knapp Buckley Award from the Faculty of Pharmacy, University of Toronto. M.K. was supported by the Precision Medicine Initiative (PRiME) at the University of Toronto internal fellowship number PRMF2019-002. This work was supported by funds to K.P from the Canada Research Chairs Program (Files 950-231075 and 950-233107), the University of Toronto's Major Research Project Management Fund, the CIHR Foundation Grant Program (201610FDN-375469), and to L.P, A.A.G., and K.P through the CIHR/IDRC Team Grant: Canada-Latin/America-Caribbean Zika Virus Program (FRN: 149783), as well as by funds to K.P. from the Canada's International Development Research Centre (grant 109434-001) through the Canadian 2019 Novel Coronavirus (COVID-19) Rapid Research Funding Opportunity. This work was also supported by funds to A.A.G. from an Arizona Biomedical Research Commission New Investigator Award (ADHS16-162400), the Gates Foundation (OPP1160667), an NIH Director's New Innovator Award (1DP2GM126892), an NIH R21 award (1R21AI136571-01A1) to K.P./A.A.G, and an Alfred P. Sloan Fellowship (FG-2017-9108). Figure 1 was created with Biorender.com under academic license to K.P.
Materials
| Name | Company | Catalog Number | Comments |
| 384 well plate covers - aluminum | Sarstedt | 95.1995 | Used to cover the 384-well plates before they are inserted into the PLUM reader |
| 384 well plate covers - transparent | Sarstedt | 95.1994 | Used to cover the 384-well plates before they are inserted into the BioTek plate reader |
| 384 well plates | VWR | CA11006-180 | 2 mm paper-based diagnostics are placed into the wells of these plates for quantification |
| Agarose | BioShop Canada | AGA001.500 | Gel electrophoresis |
| BSA | BioShop Canada | ALB001.500 | Blocking agent for the Whatman filter paper |
| Cell free reactions | New England Biolabs | E6800L | PURExpress |
| CPRG | Roche | 10884308001 | Chlorophenol red-b-D-galactopyranoside |
| Disposable Sterile Biopsy Punches | Integra Miltex | 23233-31 | Used to create 2 mm paper discs that fit into a 384-well plate |
| DNAse I | Thermo Scientific | K2981 | Digests template DNA following incubation of the in vitro transcription reaction |
| DNAse I Kit | Thermo Scientific | 74104 | DNase I Kit For removing template DNA from IVT RNA |
| dNTPs | New England Biolabs | N0446S | Used for PCRs |
| electrophoresis system | Bio-Rad | 1704487 | Used to run the agarose gels |
| Gel imaging station | Bio-Rad | 1708265 | ChemiDoc XRS+ Imaging System |
| IVT kit | New England Biolabs | E2040S | Used for in vitro transcribing template (trigger) RNA for switch screening |
| Nanodrop One | Thermo Scientific | ND-ONE-W | Used for determining nucleic acid concentrations |
| NASBA kit | Life Sciences Advanced Technologies | NWK-1 | Isothermal amplification reaction components |
| Nuclease free H20 | invitrogen | 10977015 | Added to reaction mixes |
| PAGE electrophoresis system | Biorad | 1658001FC | Used to cast and run polyacrylamide gels |
| pCOLADuet-LacZ DNA | Addgene | 75006 | https://www.addgene.org/75006/ |
| Phusion polymerase/reactioin buffer | New England Biolabs | M0530L | Used for PCRs |
| Plate reader | BioTek | BioTek NEO2 | Multi Mode Plate Reader, Synergy Neo2 |
| Primers | Integrated DNA Technologies | Custom oligo synthesis | |
| Q5 polymerase/reaction buffer | New England Biolabs | M0491L | Used for PCRs |
| Qiagen PCR Puriifcation Kit | QIAGEN | 27106 | QIAprep Spin Miniprep Kit |
| RNA loading dye | New England Biolabs | B0363S | 2X RNA loading dye |
| RNA Purification Kit | QIAGEN | EN0521 | QIAamp Viral RNA extraction kit |
| RNase inhibitor | New England Biolabs | M0314S | Used to prevent contamination of RNases A, B, and C |
| RT-qPCR kit | QIAGEN | 208352 | QuantiNova Probe RT-PCR Kit |
| SYBR Gold | Invitrogen S11494 | S11494 | PAGE gel stain for nucleic acids |
| TAE Buffer | BioShop Canada | TAE222.4 | Gel electrophoresis buffer |
| Thermal Cycler | Applied Biosystems | 4484073 | Used for temperature cycling and incubating reactions |
| Whatman 42 filter paper | GE Healthcare | 1442-042 | Used to imbed molecular components for paper-based diagnostics |
References
- Urdea, M., et al. Requirements for high impact diagnostics in the developing world. Nature. 444, 73-79 (2006).
- Yager, P., Domingo, G. J., Gerdes, J. Point-of-care diagnostics for global health. Annual Review of Biomedical Engineering. 10, 107-144 (2008).
- Faria, N. R., et al. Mobile real-time surveillance of Zika virus in Brazil. Genome Medicine. 8 (1), 97 (2016).
- Faria, N. R., et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science. 352 (6283), 345-349 (2016).
- Pardee, K., et al. Paper-based synthetic gene networks. Cell. 159 (4), 940-954 (2014).
- Pardee, K., et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Duyen, T. T. M., et al. Paper-based colorimetric biosensor for antibiotics inhibiting bacterial protein synthesis. Journal of Bioscience and Bioengineering. 123 (1), 96-100 (2017).
- Jung, J. K., et al. Cell-free biosensors for rapid detection of water contaminants. Nature Biotechnology. 38 (12), 1451-1459 (2020).
- Yang, Y. H., et al. Cell-free Escherichia coli-based system to screen for quorum-sensing molecules interacting with quorum receptor proteins of streptomyces coelicolor. Applied and Environmental Microbiology. 75 (19), 6367 (2009).
- Ma, D., Shen, L., Wu, K., Diehnelt, C. W., Green, A. A. Low-cost detection of norovirus using paper-based cell-free systems and synbody-based viral enrichment. Synthetic Biology. 3 (1), (2018).
- Park, S., Lee, J. W. Detection of coronaviruses using RNA toehold switch sensors. International Journal of Molecular Sciences. 22 (4), 1-13 (2021).
- Amalfitano, E., et al. A glucose meter interface for point-of-care gene circuit-based diagnostics. Nature Communications. 12 (1), 1-10 (2021).
- Nguyen, P. Q., et al. Wearable materials with embedded synthetic biology sensors for biomolecule detection. Nature Biotechnology. 39 (11), 1366-1374 (2021).
- Pellinen, T., Huovinen, T., Karp, M. A cell-free biosensor for the detection of transcriptional inducers using firefly luciferase as a reporter. Analytical Biochemistry. 330 (1), 52-57 (2004).
- Salehi, A. S. M., et al. Cell-free protein synthesis approach to biosensing hTRβ-specific endocrine disruptors. Analytical Chemistry. 89 (6), 3395-3401 (2017).
- Salehi, A. S. M., et al. Biosensing estrogenic endocrine disruptors in human blood and urine: A RAPID cell-free protein synthesis approach. Toxicology and Applied Pharmacology. 345, 19-25 (2018).
- Voyvodic, P. L., et al. Plug-and-play metabolic transducers expand the chemical detection space of cell-free biosensors. Nature Communications. 10 (1), 1-8 (2019).
- Yang, W. C., Patel, K. G., Wong, H. E., Swartz, J. R. Simplifying and streamlining Escherichia coli-based cell-free protein synthesis. Biotechnology Progress. 28 (2), 413-420 (2012).
- Gräwe, A., et al. A paper-based, cell-free biosensor system for the detection of heavy metals and date rape drugs. PLOS One. 14 (3), 0210940 (2019).
- Green, A. A., Silver, P. A., Collins, J. J., Yin, P. Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell. 159 (4), 925-939 (2014).
- Craw, P., Balachandran, W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab on a Chip. 12 (14), 2469-2486 (2012).
- Zyrina, N. V., Antipova, V. N. Nonspecific Synthesis in the Reactions of Isothermal Nucleic Acid Amplification. Biochemistry (Moscow). 86 (7), 887-897 (2021).
- Takahashi, M. K., et al. A low-cost paper-based synthetic biology platform for analyzing gut microbiota and host biomarkers. Nature Communications. 9 (1), 1-12 (2018).
- Karlikow, M., et al. Field validation of the performance of paper-based tests for the detection of the Zika and chikungunya viruses in serum samples. Nature Biomedical Engineering. 6 (3), 246-256 (2022).
- Agarwala, R., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 44 (1), 7-19 (2016).
- . BLAST: Basic Local Alignment Search Tool Available from: https://blast.ncbi.nlm.nih.gov/Blast.cgi (2022)
- Deiman, B., Van Aarle, P., Sillekens, P. Characteristics and applications of nucleic acid sequence-based amplification (NASBA). Molecular Biotechnology. 20 (2), 163-179 (2002).
- Zadeh, J. N., Wolfe, B. R., Pierce, N. A. Nucleic acid sequence design via efficient ensemble defect optimization. Journal of Computational Chemistry. 32 (3), 439-452 (2011).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. DNA Gel Electrophoresis. Journal of Visualized Experiments: JoVE. , (2021).
- Lanciotti, R. S., et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerging Infectious Diseases. 14 (8), 1232 (2008).
- Walsh, D. I., et al. Standardizing automated DNA assembly: Best practices, metrics, and protocols using robots. SLAS Technology. 24 (3), 282 (2019).
- Silva, S. J. R., de Magalhães, J. J. F., de Pena, L. Simultaneous circulation of DENV, CHIKV, ZIKV and SARS-CoV-2 in Brazil: An inconvenient truth. One Health. 12, 100205 (2021).
- Kimura, S., Fujinaga, K., Sekiya, T. PCR machine. Tanpakushitsu kakusan koso. Protein, Nucleic Acid, Enzyme. 41 (5), 514-517 (1996).
- Arce, A., et al. Decentralizing cell-free RNA sensing with the use of low-cost cell extracts. Frontiers in Bioengineering and Biotechnology. 9, 727584 (2021).
- Silva, S. J. R., Pardee, K., Balasuriya, U. B. R., Pena, L. Development and validation of a one-step reverse transcription loop-mediated isothermal amplification (RT-LAMP) for rapid detection of ZIKV in patient samples from Brazil. Scientific Reports. 11 (1), 1-13 (2021).
- Shimizu, Y. Cell-free translation reconstituted with purified components. Nature Biotechnology. 19 (8), 751-755 (2001).
- Lavickova, B., Maerkl, S. J. A simple, robust, and low-cost method to produce the PURE cell-free system. ACS Synthetic Biology. 8 (2), 455-462 (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved