
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Engineering Antiviral Agents via Surface Plasmon Resonance
In This Article
Summary
The present protocol describes new tools for SPR binding assays to examine CV-N binding to HA, S glycoprotein, related hybrid-type glycans, and high-mannose oligosaccharides. SPR is used to determine the KD for binding either dimeric or monomeric CV-N to these glycans.
Abstract
Surface plasmon resonance (SPR) is used to measure hemagglutinin (HA) binding to domain-swapped Cyanovirin-N (CV-N) dimer and to monitor interactions between mannosylated peptides and CV-N's high-affinity binding site. Virus envelope spikes gp120, HA, and Ebola glycoprotein (GP) 1,2 have been reported to bind both high- and low-affinity binding sites on dimeric CVN2. Dimannosylated HA peptide is also bound at the two low-affinity binding sites to an engineered molecule of CVN2, which is bearing a high-affinity site for the respective ligand and mutated to replace a stabilizing disulfide bond in the carbohydrate-binding pocket, thus confirming multivalent binding. HA binding is shown to one high-affinity binding site of pseudo-antibody CVN2 at a dissociation constant (KD) of 275 nM that further neutralizes human immunodeficiency virus type 1 (HIV-1) through oligomerization. Correlating the number of disulfide bridges in domain-swapped CVN2, which are decreased from 4 to 2 by substituting cystines into polar residue pairs of glutamic acid and arginine, results in reduced binding affinity to HA. Among the strongest interactions, Ebola GP1,2 is bound by CVN2 with two high-affinity binding sites in the lower nanomolar range using the envelope glycan without a transmembrane domain. In the present study, binding of the multispecific monomeric CV-N to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) glycoprotein is measured at KD = 18.6 µM as compared with nanomolar KD to those other virus spikes, and via its receptor-binding domain in the mid-µ-molar range.
Introduction
Tetherin-associated antiviral activity is induced by interferon-α, and it comprises protein-based tethers, that leads to the retention of fully formed virions on infected cell surfaces1. The necessity for tetherin glycosylation in the inhibition of virus release remains uncertain, implying the importance of glycosylation patterns on recombinantly expressed glycans for in vitro studies1,2, which depends on the conformation of (in the case of influenza virus) surface-expressed influenza hemagglutinin HA3,4. It has been noted that modification of oligosaccharide tethered to N-linked glycosylation is enough for tetherin-mediated restriction of HIV type-1 release2, while dimerization plays an essential role in preventing virus release, thereby involving the transmembrane domain or glycosyl-phosphatidyl-inositol (GPI)-anchor for tethering the budding virions5. Unique features are described for human and murine tetherin to block multiple enveloped viruses, retroviruses, and filoviruses. BST-2/tetherin is an interferon-inducible antiviral protein of the innate immunity1,6, acting with broad-spectrum antiviral activity and is antagonized by envelope glycoproteins5 to either translocate tetherin or disrupt the structure of tetherin6. For example, surface-expressed envelope glycoprotein HA and neuraminidase on influenza A virus are well known for tetherin antagonism in a strain-specific manner7, facilitating the recognition of host receptor binding sites8. Glycan-targeting antibodies are studied in the stoichiometry of their interactions with the rapidly customizing glycan shields on HA, resulting in binding affinity to influenza A H3N2 and H1N1 subtypes4.
To elucidate the binding mechanisms between antiviral agents and virus envelope spikes, i.e., carbohydrate ligands, and complementary immunological and spectroscopic methods, mono-, di- and tri-mannose moieties are chemically synthesized. The mannosylated peptides are created via azido glycosylation of glycosyl {beta}-peracetates to 1,2-trans glycosyl azides transformation9, mimicking the typically found N-acetyl glucosamine and high-mannose oligosaccharides on the surface of life-threatening viruses. Triazole bioisosteres are utilized to mimic linkages that form the mannosylated residue of HA peptide10 and facilitate site-specific interactions with antiviral CV-N derivatives around the second N-linked glycosylation spot on the HA head domain (HA top with 4 N-linked glycans N54, N97, N181, N301)8,11,12. Interactions between glutamic acid (Glu) and arginine (Arg) and the resulting helix dipole manifestated good stability of both model peptides and proteins but are visualized using SPR. If compared with recognizing a single chemically synthesized glycosylation site on HA10 by directly inhibiting receptor binding on the glycan moieties, a higher affinity of a four-site mutated Fc structure to its receptor is shown to elicit effector functions in vivo, revealing the unrelated composition of N-linked glycans attached to Fc mutant to be mechanistically determined13.
CV-N displays antiviral activity against HIV14,15, influenza16, and Ebola virus, which is mediated by nanomolar binding to high-mannose oligosaccharide modifications on envelope spike proteins12,17,18,19. Influenza HA binding to one high-affinity carbohydrate-binding site (H) in CV-N or two Hs in covalently linked dimeric CVN2 is determined to have equilibrium dissociation constants (KD) = 5.7 nM (Figure 1A) and KD = 2.7 nM, respectively. Both CV-N and CVN2 harbor another one or two low-affinity carbohydrate-binding sites (L)s12,17,20,21. Ebola GP1,2 binds to 2H of CVN2 with affinities in the lower nanomolar range (KD = 26 nM). CV-N WT binding to Ebola GP1,2 and HA exhibits affinities from KD = 34 nM to KD = 5.7 nM (A/New York/55/04)12. Lectins, such as CV-N, which specifically target high-mannose glycans on the viral envelopes, further inhibit replication of hepatitis C virus, SARS-CoV, herpesvirus, Marburg virus, and measles virus22.
The small CV-N molecule has been studied thoroughly for more than 20 years as it functionalizes to bind a wide range of viruses to inhibit viral entry16,18. Structural analyses and binding affinity assays indicate cross-linking of two Ls in a domain-swapped CVN2 dimer by bivalent binding in the micromolar range to enhance avidity to viral envelope glycoproteins10,19. Selective binding of Manα1-2Manα on Man(8) D1D3 arms and Man(9) comprises two binding sites of differing affinities located on opposite protein protomers20, thereby reaching nanomolar binding affinities (Figure 1B). Thus, CVN2 is considered a pseudo-antibody concerning its application to bind epitopes on HIV gp120, similar to virus-neutralizing antibodies17. Herein, the author is interested in investigating the potential binding of CVN2 to the SARS-CoV-2 spike via its receptor-binding domain (RBD). Binding curves of immobilized human angiotensin-converting enzyme (ACE)-2 with the SARS-CoV-2 RBD result in KD = 4.7 nM for this biologically relevant binding interaction23.
By contrast, selected immunoglobulin classes recognize specific and consistent structural protein patterns, which impart a substrate for affinity maturation in the membrane-anchored HA regions24. CV-N shows highly potent activity in almost all influenza A and B viruses16, and it is a broadly neutralizing antiviral agent. Our knowledge is incomplete on the location of targeted epitopes on the stem of HA1 and HA2 that possibly involve epitopic structures for glycan-targeting by highly neutralizing antibodies and as compared with lectin binding25.

Figure 1: Schematic representation of the SPR binding assay for CV-N to virus envelope spikes. (A) SPR Assay for CV-N binding to ligand: HA full-length protein (90 kDa). Kinetic data set (5120, 2560, 1280, 640, 320, 160, 80, 40, 20, 10, 5, 2.5, 0 nM) showing real-time double-referenced binding to influenza HA A/New-York/55/04 (H3N2). (B) CVN2L0 variant V2 binding to immobilized ligand DM within a concentration range of 500 nM to 16 µM. Sequence: L residues are highlighted in yellow. H residues are highlighted in gray. E58 and R73 are a replacement for cysteines in the wildtype protein and make V2 a stable protein fold with three instead of four disulfide bonds Please click here to view a larger version of this figure.
{kind=link}
Whereas the glycan shield on the membrane-distal HA top part induces high-affinity binding to CV-N12, CVN2 binding to HA adjacent to a disulfide bridge of the HA top part has further been observed at its low-affinity sites10,12. Various polar interactions and interaction sites are identified in carbohydrate-binding by CV-N. These interactions are verified by generating knock-out variants in the binding site to correlate binding affinities to in silico predicted glycosylation12. Thus, the project aims to compare previously tested chemically mannosylated HA peptides in binding affinity and specificity with short peptide sequences from SARS-related 2019-nCoV spikes and SARS-CoV-2, naturally occurring modified by a small number of different N-linked glycosylation sites and O-linked glycosylation. Using cryo-electron microscopy and binding assays, Pinto and coworkers report a monoclonal antibody, S309, that potentially recognizes an epitope on SARS-CoV-2 spike protein containing a conserved glycan within the Sarbecovirus subgenus, without competing with receptor attachment26. The protocol of this study describes how designing, expressing, and characterizing CV-N variants are important to study how CV-N and CVN2 bind to glycosylated proteins and synthetic mannosylated peptides using the SPR technology10,12.
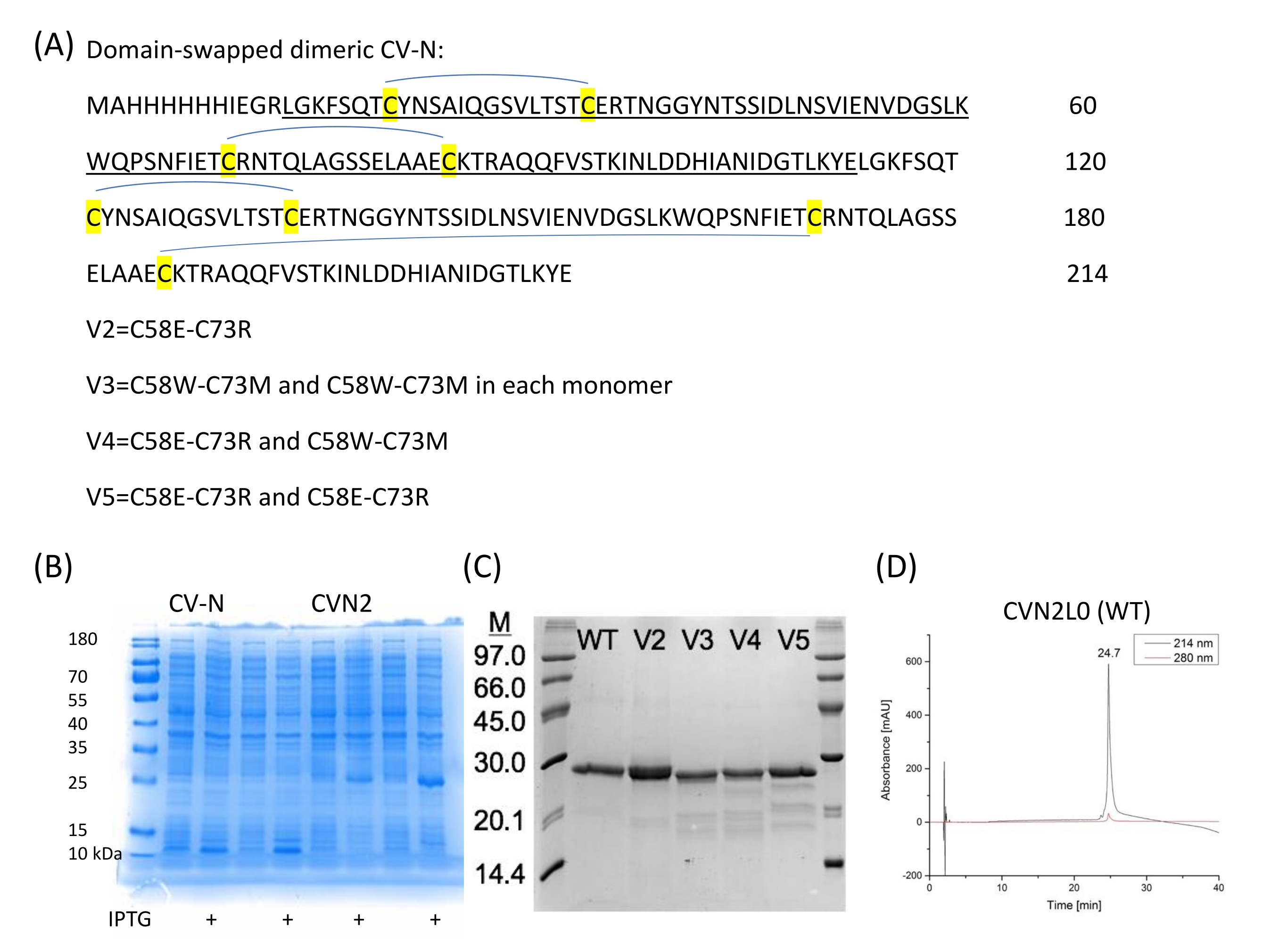
Tandem-linked dimer CVN2L027 and binding-site variants (V2-V5) are recombinantly expressed and variants are with disulfide bond replacements (C58E and C73R) (Figure 2A). Also, a mutant with a single-point mutation E41A is prepared because this position has been seen as an intermolecular cross-contacting residue. This mutant is another interesting molecule for SPR binding measurements between the lectin and high-mannose oligosaccharides deciphering binding domains and allows comparison with the dimeric form. The domain-swapped crystal structure of CVN2 shows a flexible linker, that extends between 49 and 54 residues. The two domains can continue moving around the hinge as rigid bodies, developing either a monomer through intramolecular domain interactions (domain A -residues 1-39;90-101- with domain B -residues 40-89) or a dimer by intermolecular domain swapping [domain A (of the first monomer) with domain B (of the second), and domain B (of the first monomer) with domain A (of the second copy)]. There are no close interactions between the two protomers' A and B domains, except for Glu4128. The gene for CV-N can be developed using a repetitive PCR method with 40-mer synthesized oligos29 and is then subcloned into the NdeI and BamHI sites of pET11a for transformation (electroporation) into electrocompetent cells as described by Keeffe, J.R.27. The protein, which is used for achieving the respective crystal structure (PDB ID 3S3Y), includes an N-terminal 6-histidine purification tag followed by a Factor Xa protease cleavage site. Site-directed mutagenesis is utilized to make point mutations, switch codons, and insert or delete single or multiple bases or codons for amino acid exchange. These transformations provide invaluable insight into protein function and structure. Recombinantly expressed and purified CV-N, CVN2, and CVN3 have been biophysically well studied20,21,27, are cheap to produce, and therefore used to characterize binding assays to glycans immobilized onto SPR sensor chips. Conventional enzyme-linked immunosorbent assay (ELISA) provides less reproducibility concerning the immobilization technique of glycan ligands and transforms real-time binding of various binding-site variants, which is shown for SPR, into endpoint assays.
Binding-affinity variant CVN2L0-V2 (an intact fold of homodimeric CV-N with a disulfide bridge substitution10) is expressed with a His-tag in Escherichia coli (E. coli), purified over Ni-NTA column applying affinity chromatography and tested for binding to HA (H3N2), monomannosylated HA-peptide and dimannosylated HA-peptide using SPR. Chemically mannosylated peptides, or HA and S protein, all are ligands and amine coupled to the hydrophilic chip surface via reactive esters or biotin-streptavidin protein engineering. The same procedure of sequential runs is applied to those ligands, injecting various dilutions of CV-N and variants of CV-N (and CVN2) to obtain kinetic information for the molecular interaction analyses as described below30. RBD-immobilized SPR sensor chip is used for binding studies on CV-N to S peptides, and affinities are compared to SARS-CoV-2 binding with the human ACE2.
Protocol
For the present study, a CVN-small ubiquitin-like modifier (SUMO) fusion protein has been used in enzyme-linked immunosorbent assays instead of CV-N and is suitable for cell-based assays. Recombinant full-length influenza A virus HA H3 protein is obtained commercially (see Table of Materials) or expressed in mammalian HEK293 cell lines and baculovirus-infected insect cells according to standard protocols12. Wuhan-1 spike protein is expressed in mammalian HEK293 cells. The synthesis of monomannosylated peptide (MM) and dimannosylated peptide (DM) allows the detection of homogeneous ligands to CVN2 and monomannosylated small molecule10.
1. Creating CV-N constructs
- For each of the CVN2 variants and CVN2L0 protein (PDB ID 3S3Y), obtain the gene construct with an N-terminal pelB leader sequence and His-tag in pET27b(+) vector from commercial sources (see Table of Materials, Supplementary File 1).
- Obtain CVN2L0 and its variants (V2, V3, V4, and V5; Figure 2A,C) in the background of a CVN2L0 template gene consisting of two distinct DNA sequences for each CV-N repeat.
- Dissolve the lyophilized plasmid DNA in sterile deionized distilled water (ddH2O) to a final concentration of 100 ng/µL.
2. Preparation of LB-agar plates with plasmid DNA transformed cells
- Prepare culture medium LB-Lennox by dissolving 10 g/L of peptone, 5 g/L of yeast extract, and 5 g/L of NaCl in ddH2O (see Table of Materials), and adjust the pH to 7.4. Perform the transformation into competent E. coli BL21 (DE3) for each variant (V2-V5) by chemical method following a previously published report10.
- Split the solution (900 µL and 100 µL), transfer 100 µL on LB-agar plates (50 µg/mL kanamycin), and gently use a sterile cell spreader. Incubate the agar plates overnight at 37 °C.
3. Cloning
- Subclone the gene for CV-N into the NdeI and BamHI sites of pET11a (see Table of Materials) for transformation (electroporation) into electrocompetent cells following reference27.
4. Site-directed mutagenesis
- To generate CVN2L029 and mutant CVN-E41A in the background of a CVN2L0 template gene containing two distinguished DNA sequences for each CV-N repeat27.
- Make mutations using a site-directed mutagenesis kit (see Table of Materials) and specific mutagenic primers 5'-gagaaccgtcaacgtttgcgataacagagttcagg-3' and 5'-cctgaactctgttatcgcaaacgttgacggttctc-3' for running the PCR31.
- Start a series of sample reactions using multiple concentrations of double-stranded DNA (dsDNA) template ranging from 5-50 ng (e.g., 5, 10, 20, and 50 ng of dsDNA template). Keep the primer concentration constant.
NOTE: The PCR mix and thermal cycling protocol are generally used as described in the instruction manual for the site-directed mutagenesis kit32.
- Start a series of sample reactions using multiple concentrations of double-stranded DNA (dsDNA) template ranging from 5-50 ng (e.g., 5, 10, 20, and 50 ng of dsDNA template). Keep the primer concentration constant.
- Add the Dpn I restriction enzyme (1 µL, 10 U/µL, see Table of Materials) below the mineral oil overlay. Thoroughly and gently mix reactions, spin down in a table-top microcentrifuge for 1 min, and incubate immediately at 37 °C for 1 h for digesting the parental supercoiled dsDNA.
5. Transformation of bacterial cells
- Thaw the XL1-Blue supercompetent cells (see Table of Materials) gently on ice. To transform each control and sample reaction, aliquot the supercompetent cells (50 µL) to a prechilled polypropylene round-bottom tube (14 mL).
- Transfer 1 µL of the Dpn I-treated single-stranded DNA (ssDNA) from each control and sample reaction (mutated ssDNA) to separate aliquots of the supercompetent cells, which synthesize the complementary strand. Swirl the transformation reactions carefully to mix and incubate the reactions on ice for 30 min.
NOTE: Before transferring the Dpn I-treated DNA to the transformation reaction, it is recommended to remove any remaining mineral oil carefully from the pipette tip. As an optional control, the transformation efficiency of the XL1-Blue supercompetent cells needs to be checked by mixing 0.1 ng/µL of the pUC18 control plasmid (1 µL) with a 50 µL aliquot of the supercompetent cells.
- Transfer 1 µL of the Dpn I-treated single-stranded DNA (ssDNA) from each control and sample reaction (mutated ssDNA) to separate aliquots of the supercompetent cells, which synthesize the complementary strand. Swirl the transformation reactions carefully to mix and incubate the reactions on ice for 30 min.
- Apply heat pulse to the transformation reactions at 42 °C for 45 s, and then place the reactions on ice for 2 min.
NOTE: The applied heat pulse has already been optimized for the mentioned conditions in polypropylene round-bottom tubes (14 mL). - Add 0.5 mL of NZY+ broth (containing per liter: 10 g of NZ amine (casein hydrolysate), 5 g of yeast extract, 5 g of NaCl, 12.5 mL of 1 M MgCl2, 12.5 mL of 1 M MgSO4, 10 mL of 2 M glucose, pH 7.5, and preheated to 42 °C) and incubate the transformation reactions at 37 °C with shaking at 225-250 rpm for 1 h. Plate the correct volume of each transformation reaction (5 µL from control plasmid transformation; 250 µL from sample transformation) on LB-ampicillin agar plates.
NOTE: For the transformation controls and mutagenesis, spread cells on LB-ampicillin agar plates having 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, 80 µg/mL) and isopropyl-1-thio-β-D-galactopyranoside (IPTG, 20 mM) (see Table of Materials). Inoculate 50 mL of the cell cultures with a single colony of transformed E. coli cells to purify mutated plasmid DNA for analyses. Mutagenesis is confirmed by DNA sequencing at an external facility.
6. Expression and protein purification
- For a large-scale culture, inoculate a small amount of LB (containing ampicillin) with a single colony from the transformed plate.
- Using the overnight culture, inoculate the expression culture with additives, such as 10 mM of MgCl2, 10 mM of MgSO4, and 20 mM of glucose, diluting the seed culture to 1/100.
- Grow cells with vigorous shaking at 37 °C. Grow cells to an Abs 600 nm between 0.4-0.6 (mid-log phase) before cooling the cells to 20 °C. Induce with 1 mM IPTG and grow overnight.
- Then, harvest the cells by centrifuging at 4,000 x g for 15 min at 4 °C, and discard the supernatant with a pipette.
- Resuspend the cell pellet in phosphate-buffered saline (PBS) buffer and re-centrifuge at 4,000 x g for 15 min at 4 °C. Then, discard the supernatant with a pipette. Resuspend the remaining pellet in 10 mL of lysis buffer and incubate the suspension for 1 h at 37 °C.
NOTE: Composition of lysis buffer: (50 mM of NaH2PO4, 300 mM of NaCl, 2% Triton-X100, 500 ng/mL of lysozyme, 1 mM of phenylmethylsulfonylfluoride (PMSF), 1 mM of dithiothreitol, 1 mM of MgCl2, pH 8, see Table of Materials).- Subject the mixture to two freeze-thaw cycles (-80 °C). Separate soluble and insoluble fractions by centrifugation at 4,000 x g for 15 min at 4 °C and analyze them using polyacrylamide gel electrophoresis (PAGE), in particular, sodium dodecyl sulfate (SDS)-PAGE33(Figure 2B,C).
NOTE: Cells can be lysed in several ways, such as freeze-thaw, sonication, homogenization, enzymatic lysis, or a combination of these methods. Purification from inclusion bodies is recommended for collecting high protein yields26.
- Subject the mixture to two freeze-thaw cycles (-80 °C). Separate soluble and insoluble fractions by centrifugation at 4,000 x g for 15 min at 4 °C and analyze them using polyacrylamide gel electrophoresis (PAGE), in particular, sodium dodecyl sulfate (SDS)-PAGE33(Figure 2B,C).
- Purify proteins using Ni-NTA column chromatography (see Table of Materials). Load the soluble fraction onto a regenerated Ni-NTA bead column (1 mL/min). Wash the system with TBS buffer (50 mM of Tris, 150 mM of sodium chloride, pH 7.5) before starting a gradient (0-100% 500 mM imidazole in TBS over 60 min) and collecting fractions (1 mL/min). Dialyze the purified proteins for biochemical characterization against 100 mM PBS (Figure 2C,D).
- Alternatively, use His-Select Ni2+ affinity gel (see Table of Materials) in 14 mL tubes to bind and re-suspend his-tagged recombinantly expressed CV-N in buffer solutions with 20 mM imidazole and 250 mM imidazole, respectively. Incubate in batch for at least 30 min.
NOTE: Apply these semi-purified proteins to a single-use prepacked column for buffer exchange and cleanup of biological samples, for example, carbohydrates and proteins, which can load 1-1.5 mL eluate from immobilized metal affinity chromatography. - Transfer the protein solutions to centrifugation tubes with a 10 kDa cut-off filter (see Table of Materials) and concentrate them by centrifuging for 10 min at 4,500 x g and 4 °C. For SPR measurements, exchange the analyte solutions to 10 mM HEPES, 150 mM sodium chloride, 3 mM ethylenediaminetetraacetic acid (EDTA), and 0.05% Tween20, pH 7.4 (HBS-EP(+), see Table of Materials).
- Add this SPR running buffer to a dilution factor of 1:10 and centrifuge four times to the initial volume for 10 min at 4,500 x g and 4 °C.
- Determine the protein concentration at 280 nm using a NanoDrop UV-Vis spectrophotometer (see Table of Materials) based on the calculated extinction coefficient (20,440 M-1 cm-1) for the main protein CVN2L0 showing a size of 23,474 Da34. Use PBS (100 mM, pH 7.0) or SPR buffer as a blank, and measure the protein concentration at three dilution steps (1:1, 1:10, and 1:100).

Figure 2: CV-N sequences and expression. (A) CVN2 without a linker between each CV-N repeat (101 amino acids each) and four disulfide bridges is expressed in pET11a vector in E. coli. (B) Expressions of two independent colonies for CV-N (monomer) and CVN2 (dimer). (C) Disulfide bond variants are purified and analyzed on SDS-PAGE. A low molecular weight marker (6 µL) is used as a reference. WT = CVN2L0 bearing four disulfide bridges as marked in (A). V2 is a variant with a disulfide bond replacement by polar residues at positions 58 and 73. V3-V5 are variants with two remaining S-S bonds and either polar (C58E-C73R) or non-polar (C58W-C73M) substituting amino acids or a combination of these residue pair substitutions. (D) HPLC chromatograms of purified CVN2L0 are elutated at a flow rate of 1 mL/min with a linear gradient from 5%-65% buffer B in buffer A over 30 min. Buffer A is: 0.1% (v/v) trifluoroacetic acid in ddH2O, buffer B is: 0.08% (v/v) trifluoroacetic acid in acetonitrile. Protein is analyzed on a high-performance silica gel 300-5-C4 (150 x 4.6 mm) column at 214 nm and 280 nm. Please click here to view a larger version of this figure.
{kind=link}
7. SPR spectroscopy
- Use the Dual Channel SPR system (see Table of Materials) with running buffer HBS-EP(+) and 10 mM glycine HCl pH 1.5-1.6 as the regeneration buffer. Turn on the instrument, degasser, auto-sampler, and pump and wash the entire system with ddH20 for 1 h. Place ready-to-use running buffer in a separate bottle.
- Drop emersion oil onto the detector and mount a glass sensor chip (see Table of Materials) coated with a thin gold film and on the upper side functionalized with carboxymethyldextran hydrogel directly onto the detector below the three-port flow cell. Fix the setting by pulling down the handling.
NOTE: C19RBDHC30M 200 nm streptavidin derivatized carboxymethyldextran hydrogel with a medium density of biotinylated severe acute respiratory syndrome coronavirus-2 RBD protein, is a ready-to-use sensorchip with the pre-immobilized ligand.
8. SPR binding assay for CV-N binding to HA, S protein, and RBD
- Immobilize the proteinaceous ligands to sensor chips following the steps below.
- Open a run table by clicking on Form in the menu bar and Run Table Editor in the integrated SPRAutoLink software (see Table of Materials). Choose and click on BASIC_Immobilization from the list of available run tables and follow the steps of the experimental procedure on the computer screen. The respective Sample Editor used is shown in the right upper corner.
- Click on Sample Set Editor in the Form section to fill out the reagents list for two racks placed in the autosampler for further analyses. Click on Autosampler Direct Control as a "Tool" in the menu bar to bring the racks forward or back home. Choose 4 °C as the operating temperature.
NOTE: The SPR software allows for "SPR Instrument Direct Control" and "Pump Direct Control" via selecting the corresponding tools, as well as autosampler-handling, and by clicking on Form; also Run Table Editor, Data-Plot, or Post-Processing can be chosen to perform data analysis. Files are directly saved in the default directory and exported as scrubber.files from the Post-Processing window.
- Start the pump to infuse ddH20 by clicking on Tools and Pump Direct Control and record data by clicking on SPR Instrument Direct Control, and each time Start in the newly appearing windows. Put the coupling reagents (step 8.3) in 300 µL vials, put them into the autosampler racks and start the run table by clicking on Run.
NOTE: Chip surfaces are either conditioned with 10 mM glycine buffer pH 9.0 or may have been flushed with 1 M sodium chloride, 0.1 M sodium borate buffer pH 9.0 to condition carboxyl derivatized chip surface for EDC/NHS activation mix30. - For this simple protein-protein interaction, use the CMD500D chip (see Table of Materials) to generate a micro-refractive index units (µRIU) = 2500 - 3000 flow cell with immobilized HA and µRIU = 400 flow cell with spike protein. At an infuse flow rate of 15 µL/min, inject an aqueous and equal mixture of 0.4 M N-ethyl-N'-(dimethylaminopropyl) carbodiimide hydrochloride (EDC*HCl) and 0.1 M N-hydroxysuccinimide (NHS) by applying the following sequential steps.
- Refill pump refill at 25,000 µL/min, perform baseline adjustment for 30 s, inject 90 µL of sample activation solution (EDC/NHS) over 6 min contact time, and then hold for another 5 min.
- Repeat this cycle after baseline running for 1 min at 10 µL/min on only the left flow cell (blue) to inject and immobilize chemically synthesized peptides10, HA, and spike protein at 20 µg/mL, and allow for the subsequent baseline adjustment with ddH2O for 1.5 min before quenching the activated chip surface with 1 M ethanolamine HCl pH 8.5.
- Switch the tubes from the liquid sampler to the degasser from ddH20 into the bottle with HBS-EP(+) (Supplementary Figure 1).
- Analyze the SPR sensorgrams.
- To perform kinetic studies, use various analyte concentrations (10-5-10-8 M), with a regeneration step after each injection and blank measurements after different analytes. Change the flow rate to 10 µL/min and start injections for 4 min contact time, then 5 min baseline generation, and two regeneration steps of 2 min each with an interval of 30 s.
- Inject the buffer solution for blank measurements whose sensorgrams are subtracted from sample runs to normalize different protein concentrations.
- Click on Form, scroll down, and change to "Post-Processing" by clicking this operation mode. Click on Add to select binding curves generated over time in the Data Plot Form for each flow cell, and export the overlay as a scrubber file (.ovr). Click on File to open the file saving options. Obtain response curves by aligning left and right curves and subtracting signals of the second reference channel from those of the ligand channel.
NOTE: Data is operated in "Post-Processing" by defining sensorgrams computationally. It is put into an overlay of sensorgrams from left and right flow cells, or represented as sensorgrams from the difference of both channels. - Clean the entire fluidics with 50-100 mM glycine buffer pH 9.5, water, and 20% ethanol before and after binding measurements to remove traces of salt or any protein contamination, or more stringent, with 0.5% SDS and glycine.
NOTE: To prevent instrument damage, it is recommended to check the mechanical stability of the glass chip before re-inserting the chip cartridge into the instrument if chips have been stored under buffer or at 100% humidity.
Results
A dimeric domain-swapped CVN2L0 molecule is tested for binding to the HA top region in three separate SPR experiments and binding affinity is presented in KD values. Domain B is assumed to comprise H-binding sites, which are impacted by replacing a disulfide bond into ionic residues, and domain A forms L10,18. Single injections of CVN2L0 and variants V2 (three disulfide bridges) and V5 (two disulfide bridges) are first tested for binding to the HA-coup...
Discussion
CV-N's binding affinity is correlated with the number of functional binding sites [2H on domains B, and 2L on domain(s) A when engineered as domain-swapped dimer]. A variant with an altered binding affinity (CVN2L0-V2, a homodimeric stable fold of CV-N comprising a disulfide bridge knock-out) is expressed in E. coli, purified, and positively tested for binding to HA-protein (H3N2) using SPR10, and shows a conformational change upon binding HA with either H or L carbohydrate-binding si...
Disclosures
The author has nothing to disclose.
Acknowledgements
The author acknowledges Dr. Christian Derntl from the Department for Biotechnology and Microbiology at the TU Wien and the Department of Medicine III, Division of Nephrology and Dialysis at the Medical University of Vienna, especially Dr. Markus Wahrmann for technical and scientific support. Protein expression in mammalian cells was supported by the Department of Biotechnology at the University of Natural Resources and Life Sciences (BOKU) Vienna. The author wants to express her deep acknowledgement to Dr. Nico Dankbar from XanTec bioanalytics in Duesseldorf, Germany, for helpful scientific discussions on performing the SPR binding assays.
Materials
| Name | Company | Catalog Number | Comments |
| Äkta primeplus | Cytiva | ||
| Amicon tubes | Merck | C7715 | |
| Ampillicin | Sigma-Aldrich | A5354 | |
| Beckmann Coulter Cooler Allegra X-30R centrifuge | Beckman Coulter | B06320 | |
| Cell spreader | Sigma-Aldrich | HS86655 | silver stainless steel, bar L 33 mm |
| Custom DNA Oligos | Sigma-Aldrich | OLIGO | |
| Custom Gensynthesis | GenScript | #1390661 | cloning vector: pET27b(+) |
| Cytiva HBS-EP+ Buffer 10, 4x50mL | Thermo Scientific | 50-105-5354 | |
| Dionex UlitMate 3000 | Thermo Scientific | IQLAAAGABHFAPBMBFB | |
| Dpn I restriction enzyme (10 U/μL) | Fisher Scientific | ER1701 | |
| DTT | Merck | DTT-RO | |
| EDC | Merck | 39391 | |
| EDTA | Merck | E9884 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120086 | |
| Eppendorf Safe-Lock Tubes | Eppendorf | 30120094 | |
| Eppendorf Minispin and MiniSpin Plus personal microcentrifuge | Sigma-Aldrich | Z606235 | |
| Ethanol | Merck | 51976 | |
| Ethanolamine HCl | Merck | E6133 | |
| Falcon 50mL Conical Centrifuge Tubes | Fisher Scientific | 14-432-22 | |
| Falcon 14 mL Round Bottom Polystyrene Test Tube, with Snap Cap, Sterile, 25/Pack | Corning | 352057 | |
| Glucose | Merck | G8270 | |
| Glycine HCl | Merck | 55097 | |
| HA H3 protein | Abcam | ab69751 | |
| HEPES | Merck | H3375 | |
| His-select Ni2+ | Merck | H0537 | |
| Imidazole | Merck | I2399 | |
| IPTG | Merck | I6758 | |
| Kanamycin A | Sigma-Aldrich | K1377 | |
| Kromasil 300-5-C4 | Nouryon | ||
| LB agar | Merck | 52062 | |
| LB agar | Merck | 19344 | |
| LB Lennox | Merck | L3022 | |
| Lysozyme | Merck | 10837059001 | |
| Magnesium chloride | Merck | M8266 | |
| Magnesium sulfate | Merck | M7506 | |
| NaH2P04 | Merck | S0751 | |
| NanoDrop UV-Vis2000c spectrophotometer | Thermo Scientific | ND2000CLAPTOP | |
| NaOH | Merck | S5881 | |
| NHS | Merck | 130672 | |
| NZ amine (casein hydrolysate) | Merck | C0626 | |
| PBS | Merck | 806552 | |
| PD MidiTrap G-10 | Sigma-Aldrich | GE28-9180-11 | |
| Peptone | Merck | 70171 | |
| pET11a | Merck Millipore (Novagen) | 69436 | |
| PMSF | Merck | PMSF-RO | |
| QIAprep Spin Miniprep Kit (1000) | Qiagen | 27106X4 | |
| Reichert Software Package Autolink1-1-9 | Reichert | ||
| Reichert SPR SR7500DC Dual Channel System | Reichert | ||
| Scrubber2-2012-09-04 for data analysis | Reichert | ||
| SDS | Merck | 11667289001 | |
| Site-directed mutagenesis kit incl pUC18 control plasmid | Stratagene | #200518 | |
| Sodim chloride | Merck | S9888 | |
| Sodium acetate.Trihydrate | Merck | 236500 | |
| SPR sensor chip C19RBDHC30M | XanTec bioanalytics | SCR C19RBDHC30M | |
| SPR sensor chip CMD500D | XanTec bioanalytics | SCR CMD500D | |
| Sterilin Standard 90mm Petri Dishes | Thermo Scientific | 101R20 | |
| TBS | Merck | T5912 | 10x, solution |
| Triton-X100 | Merck | T8787 | |
| Tryptone | Merck | 93657 | |
| Tween20 | Merck | P1379 | |
| Vortex-Genie 2 Mixer | Merck | Z258423 | |
| X-gal | Merck | XGAL-RO | |
| XL1-Blue Supercompetent Cells | Stratagene | #200236 | |
| Yeast extract | Merck | Y1625 |
References
- Perez-Caballero, D., et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 139 (3), 499-511 (2009).
- Waheed, A. A., Gitzen, A., Swiderski, M., Freed, E. O. High-mannose but not complex-type glycosylation of tetherin is required for restriction of HIV-1 release. Viruses. 10 (1), 26 (2018).
- Wilson, I. A., et al. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 289 (5796), 366-373 (1981).
- Otterstrom, J. J., et al. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proceedings of the National Academy of Sciences of the United States of America. 111 (48), 5143-5148 (2014).
- Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proceedings of the National Academy of Sciences of the United States of America. 106 (8), 2886-2891 (2009).
- Tokarev, A., Skasko, M., Fitzpatrick, K., Guatelli, J. Antiviral activity of the interferon-induced cellular protein BST-2/tetherin. AIDS Research and Human Retroviruses. 25 (12), 1197-1210 (2009).
- Gnirss, K., et al. Tetherin sensitivity of influenza A viruses is strain specific: Role of hemagglutinin and neuraminidase. Journal of Virology. 89 (18), 9178-9188 (2015).
- Fleury, D., et al. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nature Structural Biology. 6 (6), 530-534 (1999).
- Salunke, S. B., et al. Iron(III) chloride as an efficient catalyst for stereoselective synthesis of glycosyl azides and a cocatalyst with Cu(0) for the subsequent click chemistry. Chemical Communication. (Camb). 47 (37), 10440-10442 (2011).
- Schilling, P. E., et al. Mannosylated hemagglutinin peptides bind cyanovirin-N independent of disulfide-bonds in complementary binding sites. RSC Advances. 10 (19), 11079-11087 (2020).
- Fleury, D., Wharton, S. A., Skehel, J. J., Knossow, M., Bizebard, T. Antigen distortion allows influenza virus to escape neutralization. Nature Structural Biology. 5 (2), 119-123 (1998).
- Maier, I., Schiestl, R. H., Kontaxis, G. Cyanovirin-N binds viral envelope proteins at the low-affinity carbohydrate binding site without direct virus neutralization ability. Molecules. 26 (12), 3621 (2021).
- Ahmed, A. J., Keremane, S. R., Vielmetter, J., Bjorkman, P. J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. Journal of Structural Biology. 194 (1), 78-89 (2016).
- Boyd, R. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrobial Agents and Chemotherapy. 41 (7), 1521-1530 (1997).
- Bolmstedt, A. J., O'Keefe, B. R., Shenoy, S. R., McMahon, J. B., Boyd, M. R. Cyanovirin-N defines a new class of antiviral agent targeting N-linked, high-mannose glycans in an oligosaccharide-specific manner. Molecular Pharmacology. 59 (5), 949-954 (2001).
- O'Keefe, B. R., et al. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrobial Agents and Chemotherapy. 47 (8), 2518-2525 (2003).
- Shenoy, S. R., et al. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chemical Biology. 9 (10), 1109-1118 (2002).
- Bewley, C. A., Kiyonaka, S., Hamachi, I. Site-specific discrimination by cyanovirin-N for alpha-linked trisaccharides comprising the three arms of Man(8) and Man(9). Journal of Molecular Biology. 322 (4), 881-889 (2002).
- Barrientos, L. G., Matei, E., Lasala, F., Delgado, R., Gronenborn, A. M. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Engineering Design & Selection. 19 (12), 525-535 (2006).
- Bewley, C. A., Otero-Quintero, S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. Journal of the American Chemical Society. 123 (17), 3892-3902 (2001).
- Bewley, C. A. Solution structure of a cyanovirin-N:Man alpha 1-2Man alpha complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 9 (10), 931-940 (2001).
- Jensen, S. M. R., et al. Differential inhibitory effects of Cyanovirin-N, Griffithsin, and Scytovirin on entry mediated by envelopes of gammaretroviruses and deltaretroviruses. Journal of Virology. 88 (4), 2327-2332 (2014).
- Lan, J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 581 (7807), 215-220 (2020).
- Lingwood, D., et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 489 (7417), 566-570 (2012).
- Ekiert, D. C., et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 324 (5924), 246-251 (2009).
- Pinto, D., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 583 (7815), 290-295 (2020).
- Keeffe, J. R., et al. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proceedings of the National Academy of Sciences of the United States of America. 108 (34), 14079-14084 (2011).
- Yang, F., et al. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. Journal of Molecular Biology. 288 (3), 403-412 (1999).
- Stemmer, W. P., Crameri, A., Ha, K. D., Brennan, T. M., Heyneker, H. L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 164 (1), 49-53 (1995).
- Fischer, M. J. E., Mol, N., Fischer, M. Amine coupling through EDC/NHS: A practical approach. Surface Plasmon Resonance. Methods in Molecular Biology (Methods and Protocols). 627, (2010).
- Novoradovsky, A., et al. Computational principles of primer design for site directed mutagenesis. Technical Proceedings of 2005 NSTI Nanotechnology Conference and Trade Show. , 532-535 (2005).
- . QuikChange Site-Directed MutagenesisKit, User Manual Available from: https://users.drew.edu/jliu3/Docs/Stratagene%20Quikchange%20mutagenesis.pdf#:~:text=The%20QuikChange%20sitedirected%20mutagenesis%20kit%20is%20used%20to (2005)
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- . Expasy.org Available from: https://web.expasy.org/cgi-bin/protparam/protparam (2022)
- Karlsson, R. Real-time competitive kinetic analysis of interactions between low-molecular-weight ligands in solution and surface-immobilized receptors. Analytical Biochemistry. 221 (1), 142-151 (1994).
- Schuck, P., Zhao, H. The role of mass transport limitation and surface heterogeneity in the biophysical characterization of macromolecular binding processes by SPR biosensing. Methods Molecular Biology. 627, 15-54 (2020).
- Barnes, O., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 588 (7839), 682-687 (2020).
- . Using reichert Surface Plasmon Resonance (SPR) for Antiviral Development, Application Note 28 Available from: https://www.reichertspr.com/clientuploads/directory/application_notes/Application_Note_28__Using_Reichert_Surface_Plasmon_Resonance_for_Antiviral_Testing.pdf (2022)
- Sundberg, E. J., Andersen, P. S., Gorshkova, I. I., Schuck, P., Schuck, P. Surface plasmon resonance biosensing in the study of ternary systems of interacting proteins. Protein Interactions: Biophysical Approaches for the Study of Complex Reversible Systems. 5, 97-141 (2007).
- . Method Development Notes Available from: https://www.reichertspr.com/applications/method-development-notes/ (2022)
- Angulo, J., Enríquez-Navas, P. M., Nieto, P. M. Ligand-receptor binding affinities from saturation transfer difference (STD)-NMR spectroscopy: the binding Isotherm of STD initial growth rates. Chemistry. 16 (26), 7803-7812 (2010).
- Goldflam, M., Tarragó, T., Gairí, M., Giralt, E. NMR studies of protein-ligand interactions. Methods in Molecular Biology. 831, 233-259 (2012).
- Kumar, S., Maurya, V. K., Prasad, A. K., Bhatt, M. L. B., Saxena, S. K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease. 31 (1), 13-21 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved