
Method Article
Robust and Highly Reproducible Generation of Cortical Brain Organoids for Modelling Brain Neuronal Senescence In Vitro
* These authors contributed equally
In This Article
Summary
In this study, we provide a detailed technique for a simple yet robust cortical organoid culture system using standard feeder-free hPSC cultures. This is a rapid, efficient, and reproducible protocol for generating organoids that model aspects of brain senescence in vitro.
Abstract
Brain organoids are three-dimensional models of the developing human brain and provide a compelling, cutting-edge platform for disease modeling and large-scale genomic and drug screening. Due to the self-organizing nature of cells in brain organoids and the growing range of available protocols for their generation, issues with heterogeneity and variability between organoids have been identified. In this protocol paper, we describe a robust and replicable protocol that largely overcomes these issues and generates cortical organoids from neuroectodermal progenitors within 1 month, and that can be maintained for more than 1 year. This highly reproducible protocol can be easily carried out in a standard tissue culture room and results in organoids with a rich diversity of cell types typically found in the developing human cortex. Despite their early developmental make-up, neurons and other human brain cell types will start to exhibit the typical signs of senescence in neuronal cells after prolonged in vitro culture, making them a valuable and useful platform for studying aging-related neuronal processes. This protocol also outlines a method for detecting such senescent cells in cortical brain organoids using senescence-associated beta-galactosidase staining.
Introduction
Our current knowledge of the human brain has largely been based on animal models and post-mortem brain specimens. Stem cell biology is a rapidly advancing field that provides new insights into the basic biology of human brain development and the pathological drivers of human brain disorders. Human pluripotent stem cells (hPSCs) are an invaluable tool for modeling the human brain via the generation of organoids, organ-like three-dimensional (3D) tissue that typically recapitulates the developmental trajectories, cellular make-up, and architecture of the developing human brain. Brain organoids are self-assembled and composed of neural stem cells, specified neural progenitors, mature neurons, and glial cell types. Organoids, therefore, provide a unique opportunity to study the early human brain, which is often inaccessible for direct experimentation but also has intrinsic limitations such as the absence of vasculature and an immune system.
Methodologies to generate brain organoids have been pursued in two different ways: unguided and guided differentiation. Unguided brain organoid methods rely on the spontaneous intrinsic differentiation capacities of the stem cells that drive tissue morphogenesis1,2 and allow for the emergence of a variety of cell lineage identities ranging from forebrain, midbrain, and hindbrain, to choroid plexus, retina, and mesoderm. In contrast, guided brain organoid methods require substantial use of external factors to drive hPSCs toward the desired patterning of neuronal lineages representing one brain region type, such as medial ganglionic eminence3, forebrain4, midbrain5, hypothalamus6, cerebellum7, and choroid plexus8. This ability to generate different brain regions with different cell lineages, and the potential to fuse these at will, makes brain organoids an excellent model for investigating human brain development and deciphering the underlying mechanisms of brain-related diseases. Although these methods for generating brain organoids offer a breakthrough in modeling human brain regions, the variability and heterogeneity between organoids remain a significant limitation for systematic and quantitative studies, such as drug screening.
The current protocol is based on a method developed in our recent paper9 and involves the selective differentiation of hPSC colonies toward neuroectoderm (NEct) identity with dual SMAD inhibitors (SB-431542 and LDN 193189), which then have the ability to self-organize within 4 days into 3D neuroepithelium spheroids under the influence of FGF2 signaling. These neuroepithelium spheroids reliably generate homogenous cortical organoids with an in vivo-like cellular composition within 4 weeks of differentiation. The protocol described here is built on our previous findings showing that inhibition of dual SMAD (Suppressor of Mothers Against Decapentaplegic) signaling promotes the differentiation of hPSCs toward rostral neural stem cells derived from neuroectodermal progenitors10 by, among others, inhibiting endodermal, mesodermal, and trophectoderm cell fate choice11. Furthermore, the embedding of the neuroepithelium spheroids in the hESC-qualified basement membrane matrix triggers significant budding of the neuroepithelia, forming ventricles with apicobasal polarity. Large-scale culture showed reproducibility and homogeneity of cortical organoids independent of cell lines, clones, or batches, and thus represents a reliable and stable stem cell system to mimic early human cortical development in health and disease in vitro. We further outline a protocol for detecting senescent neuronal cell markers in hPSCs-derived cortical brain organoids that have been cultured for prolonged periods of time.
After plating hPSCs at a seeding density of 20%-30%, the cells are treated with dual SMAD inhibitors for 3 days to differentiate hPSC colonies toward neuroectodermal colonies. These colonies are then gently lifted with dispase and seeded into ultra-low attachment 6-well plates supplemented with FGF2. The floating 2D colonies self-organize into 3D neuroectodermal spheroids overnight and are maintained for 4 days in N2 medium supplemented daily with FGF2. Once the spheroids have established the neuroepithelial layer, they can be embedded in the basement membrane matrix. By routinely adding fresh terminal differentiation medium, the researchers will observe progressive expansion and budding of neuroepithelia in cortical organoids. Researchers may wish to dissociate these organoids to conduct transcriptional and proteomic profiling. Additionally, brightfield imaging is recommended for monitoring the quality of the cortical organoids. Analysis can be performed by fixation, cryosection, and immunostaining. Descriptions and methods for these techniques have been previously described12. Ultimately, this protocol allows researchers to rapidly and robustly generate homogeneous cortical brain organoids for modeling the developing human cortical brain, with low cost and limited equipment, and for studying aspects of cellular neuronal senescence, as outlined in this paper.
Protocol
1. Cortical brain organoids generation
NOTE: All the steps in this section of the protocol will occur in a Class 2 biosafety hood, unless stated otherwise.
- Induction of 2D neuroectodermal colonies from hPSC 2D culture (Days -1 to 3)
- Before induction, plate the hPSC colonies on a hESC qualified basement membrane matrix in a 6-well plate at 20%-30% density. Achieve this density by passaging hPSC colonies from one well of a 6-well plate at 60% confluency into three wells of a 6-well plate.
- For basement membrane matrix coating, dilute the basement membrane matrix at a ratio of 1:50 in a plain basal medium. Evenly deposit 1 mL/well of a 6-well plate, incubate for 1 h at room temperature (RT), and then aspirate.
- Maintain hPSC colonies for 1 day in 2 mL of a serum-free cell culture medium prior to differentiation.
- On the day of hPSC differentiation, inspect the hPSC colonies using brightfield microscopy at 4x to 10x magnification to ensure healthy colonies with no detectable differentiation.
NOTE: Healthy hPSCs will form tight edge colonies with cells that have a large nucleus, very small cytoplasm, and prominent nucleoli. Differentiated iPSC colonies will exhibit clear morphological differences to that of the described hPSC colonies above, particularly around the outer edges of the colonies or at the center. - Add reagents listed in Table 1 to make up the N2 medium required for differentiation. Bring this medium to RT before use.
- Once at RT, aspirate the serum-free cell culture medium from each well of the 6-well plate and replace with 2 mL of N2 medium gently added with a 5 mL serological pipette.
- Add the dual SMAD inhibitors, SB-431542 (10 µM) and LDN 193189 (100 nM).
NOTE: The SMAD inhibitors can be added to the N2 medium after the medium has been placed in each well, or to the required amount of N2 medium prior to replacing the serum-free cell culture medium. Inhibitors can be evenly incorporated into the medium by gently swirling the plate or inverting the tube containing the medium and inhibitors 3-4 times. - Add fresh N2 media supplemented with SB-431542 (10 µM) and LDN 193189 (100 nM) daily to each well for the next 2 days.
NOTE: Fresh N2 medium is added to reduce the prolonged exposure of cells to the DMSO that is used to dissolve SB-431542 and LDN 193189 compounds and prevent cytotoxicity.
- Generation of 3D neuroectodermal spheroids from induced 2D neuroectodermal colonies (Days 3 to 7)
- Lift the induced neuroectodermal colonies using dispase following steps 1.2.2-1.2.8.
- First, remove 2 mL of N2 medium from the 6-well plate, and wash 1x with HBSS to ensure that all of the N2 medium is removed.
NOTE: N2 medium can interfere with the enzyme activity of dispase, preventing adequate detachment of neuroectodermal colonies from the well. - Add 1 mL of 2.4 unit/mL dispase to each colony-containing well.
- Incubate the well for 20-25 min (maximum 30 min) at 37 °C. Check for colony detachment regularly.
NOTE: Small colonies may detach within 20 min. Any colonies that remain stuck down after 30 min should be ignored. - After incubation, add 1 mL of N2 medium to the well to stop the activity of the dispase enzyme and transfer the colonies into a 15 mL tube using a wide-bore P1000 pipette tip or a modified P1000 pipette tip cut with sterile scissors (making it a wide-bore P1000 tip).
- Allow the colony clumps to sink to the bottom of the tube with gravity.
NOTE: This process will take approximately 1 min. - Once the clumps have sunk, carefully remove the supernatant with a standard P1000 pipette tip and replace it with 1 mL of fresh N2 medium. Repeat this washing step three times to ensure complete removal of dispase.
NOTE: Any remaining dispase will prevent a uniform formation of neuroectodermal spheroids and induce cell death. - After washing, resuspend the cell clumps in 3 mL of N2 medium and transfer to one well of a 6-well plate and add 40 ng/mL of bFGF.
NOTE: If a high number of neuroectodermal colonies were detached, these colonies could be plated across two or more wells of a 6-well plate to prevent spheroids fusion. 24 h after dispase detachment of the colonies, check whether the spheroids have formed. - Maintain the spheroids in the same media for the next 3-4 days but add fresh bFGF (40 ng/mL) to each well daily to promote neuroectodermal cell proliferation, self-organizing, and induce and expand neuroepithelia.
NOTE: If spheroids have been plated at a higher density, the media is likely to turn yellow and require replacing every 2 days with fresh bFGF (40 ng/mL). However, this is not recommended, and instead, a lower number of spheroids should be maintained in each well to avoid this issue. Spheroids can be embedded in the basement membrane matrix after 3 days if neuroepithelia are evident. If neuroepithelia are not evident or do not look strong, then maintain the spheroids for another day and check again.
- Cortical brain organoid differentiation and maintenance (Day 8)
- Prepare the terminal differentiation media (DM) using the reagents listed in Table 2. Bring this media to RT.
- Thaw 100% hESC-qualified basement membrane matrix on ice.
- Prepare a sheet of parafilm with dimples sterilized with 70% ethanol in a 10 cm Petri dish and place it on a stereomicroscope under a hood.
NOTE: An empty tray of 200 µL pipette tips can be used to generate a grid of dimples. - Cut the end off a 100 µL pipette tip (to make it wide-bore). This will be used for embedding neural spheroids in the basement matrix without breaking them.
- Using a stereomicroscope, select similar-sized neural spheroids (500 µm) from the 6-well plate and transfer them to the parafilm-dimples using the wide-bore 100 µL pipette tip, placing a single neural spheroid per dimple.
- Gently remove any excess media, leaving just enough to cover the spheroids.
NOTE: This is to maintain the quality of the spheroids before adding the basement matrix and to ensure they do not dry out. - Gently add 18 µL of basement matrix over the spheroid, positioning the spheroid within the center of the matrix drop. Use the end of a 10 µL pipette tip to center the spheroids in the matrix.
NOTE: Try to add the basement matrix as quickly as possible to avoid the excess media around the spheroids from drying out. - Cover the 10 cm Petri dish and transfer into the incubator and incubate the parafilm dish with basement matrix embedded spheroids at 37 °C for 25 min.
- After incubation, rinse off the embedded spheroids into a low attachment 24-well plate using a P1000 tip with 0.5 mL of the DM media, making sure individual embedded spheroids are placed in one well each.
NOTE: If more than one embedded spheroid falls into one well, use a wide-bore P1000 tip to transfer the other spheroid into a new well. - Maintain the differentiated cortical brain organoids in the DM media for prolonged periods of time, with media changes occurring every 2 days when the organoids become larger and older.
NOTE: During the first week of differentiation, the medium can be changed every 3 days.
2. Characterization of neuronal aging in Cortical organoids
- Process cortical organoids for cryosections:
NOTE: The steps were performed in a Class 2 biosafety hood.- Prepare 2 mL tubes, each filled with 1.5 mL of 4% paraformaldehyde (PFA).
- Cut the end of a P1000 pipette tip (to make it a wide-bore) and gently transfer each organoid to one of the 2 mL tubes prepared above (one organoid per tube).
NOTE: To prevent excess DM media mixing with the PFA, allow the organoid to sink toward the opening of the P1000 pipette tip and rest the tip just above the top of the PFA in the tube before pipetting the organoid out. This will enable the researcher to transfer just the organoid and very minimal media. - Allow the fixation process to take place at 4°C for 1 h.
- Using an uncut P1000 pipette tip, carefully aspirate excess PFA and add 1.5 mL of cold 1x PBS.
- Transfer the tubes to an orbital shaker set at 70 rpm for 10 min at RT.
- Repeat the washing process with cold 1x PBS three times to ensure all PFA has been thoroughly removed.
NOTE: Do not dispose of PFA in regular waste containers; instead, prepare a specific chemical waste disposal container for this, as PFA is a hazard. - Immerse the organoids in 1x PBS containing 30% sucrose and incubate at 4 °C until all organoids have sunk to the bottom of the tube.
NOTE: The time required to allow the organoids to sink depends on the size/age of the organoids. 3-month-old organoids may take up to 5 h. - Using a cut, wide-bore P1000 pipette tip, gently transfer three to five organoids into a mounting mold containing a mounting solution made of 30% sucrose and 100% optimal cutting temperature (OCT) medium, at a ratio of 3:2.
- Use a 10 µL pipette tip with the aid of a stereomicroscope to orientate and position organoids in a grid-like pattern.
- Place the mold on dry ice to solidify the sucrose OCT solution before proceeding with cryo-sectioning (16-20 µm) using a cryostat.
NOTE: For senescence-associated beta-galactosidase, all tissues must be processed for sectioning once they have sunk. For immunofluorescence, tissues can be processed for sectioning the next day. All the slides containing sections must be stored at -20 °C before subsequent immunofluorescence or beta-galactosidase, if not stained immediately.
- Process for analysis of senescence in the cortical brain organoids:
NOTE: The following steps can be performed on a regular lab bench.- Transfer the slides into a microscope slide staining container with a lid and wash the sectioned organoid tissue three times with 1x PBS for 10 min at RT to remove any excess mounting solution.
- Following this, incubate the washed tissue with freshly made beta-galactosidase staining solution overnight at 37 °C.
NOTE: The beta-galactosidase staining solution is made of phosphate buffer (for 10 mL of phosphate buffer: 8.15 mL of 1 M NaH2PO4, 1.85 mL of 1M Na2HPO4) adjusted pH = 6, 100 mM of potassium hexacyanoferrate (III), 100 mM of potassium hexacyanoferrate (II) trihydrate, 5 M of NaCl, 1 M of MgCl2, 20 mg/mL of X-Gal. Avoid using a standard cell culture incubator containing CO2 as the CO2 will alter the pH of the beta-galactosidase staining solution. - Wash the stained tissues with 1x PBS three times for 10 min each at RT to remove the beta-galactosidase solution.
- Mount the washed tissues with a glass antifade mountant and allow the mounting solution to solidify for 30 min at RT before viewing under the microscope.
Results
Here we have described a robust protocol that allows researchers to generate homogenous hPSC-derived cortical brain organoids that mimic the in vivo human cortical brain region within 1-3 months of culture. hPSC colonies are first cultured in differentiation media to generate neuroectodermal colonies, which can then be used to form neural spheroids. These spheroids are subsequently embedded in a basement membrane matrix and maintained for prolonged periods of time to produce organoids that can be used to model neuronal aging (see Figure 1 for an outline of the protocol). It is worth noting that culturing these organoids in ultra-uncoated 24-well plates causes cellular stress and promotes senescence-associated phenotypes over 13 weeks of in vitro culture. Organoids derived from this protocol can also be maintained in stirred bioreactors for optimum growth and differentiation of cortical plate neural cells or at the air-liquid interface.
To begin, hPSC colonies are cultured for 1 day prior to neuroectodermal differentiation. It is critical that these hPSC colonies are cultured to only 20%-30% confluency and are of the highest possible quality: a tight flat monolayer with no differentiated cells contaminating the colonies (Figure 2A,B). The pluripotency of the hPSC colonies should be confirmed by the expression of markers such as NANOG (Figure 2C). The validated hPSC colonies are then exposed to the N2 neuroectodermal differentiation media with SB-431542 and LDN 193189. After 3 days of maintenance in this media, the hPSC colonies should have differentiated into neuroectodermal colonies and no longer show the same tight flat monolayer morphology of the hPSCs (Figure 2B), but rather, they will become longer columnal-shaped cells (Figure 2B). These cells will also be negative for pluripotency markers such as NANOG (Figure 2C).
It is at this stage that the neuroectodermal colonies are enzymatically detached with dispase, and each healthy and successfully detached colony is allowed to self-organize and form a young neural spheroid (Figure 2D, Supplementary Video 1). Only healthy, clean neuroectodermal colonies will detach in the timeframe specified for dispase activity; all other colonies should be ignored as they will result in a poorer quality of spheroid. With daily exposure to FGF2 in the N2 media, the neural stem cells (SOX2+) in these spheroids (Figure 2D, day 1) will proliferate and form a significant number of neural rosettes (Figure 2D, day 4). These rosettes will express the tight junction and epithelial marker ZO1 in cells located within the center of the rosettes and along the outer edge of the spheroid, demonstrating the apical-basal polarity of the spheroid (Figure 2D, day 4). The method for wholemount 3D imaging of spheroids has been described before13. Daily inspection of the spheroids should elucidate the formation of a tight, dark outer edge and bright periphery of the spheroids, this being the neuroepithelial layer. This layer should be sufficiently formed after 3-4 days with an approximate diameter of 500 µm, at which time the spheroids can be embedded in the basement matrix. If this layer is not present or is only weakly formed, the spheroids are not sufficiently developed enough to take forward. It is recommended to wait another day to observe any change, but if this is not observed, disregard these spheroids.
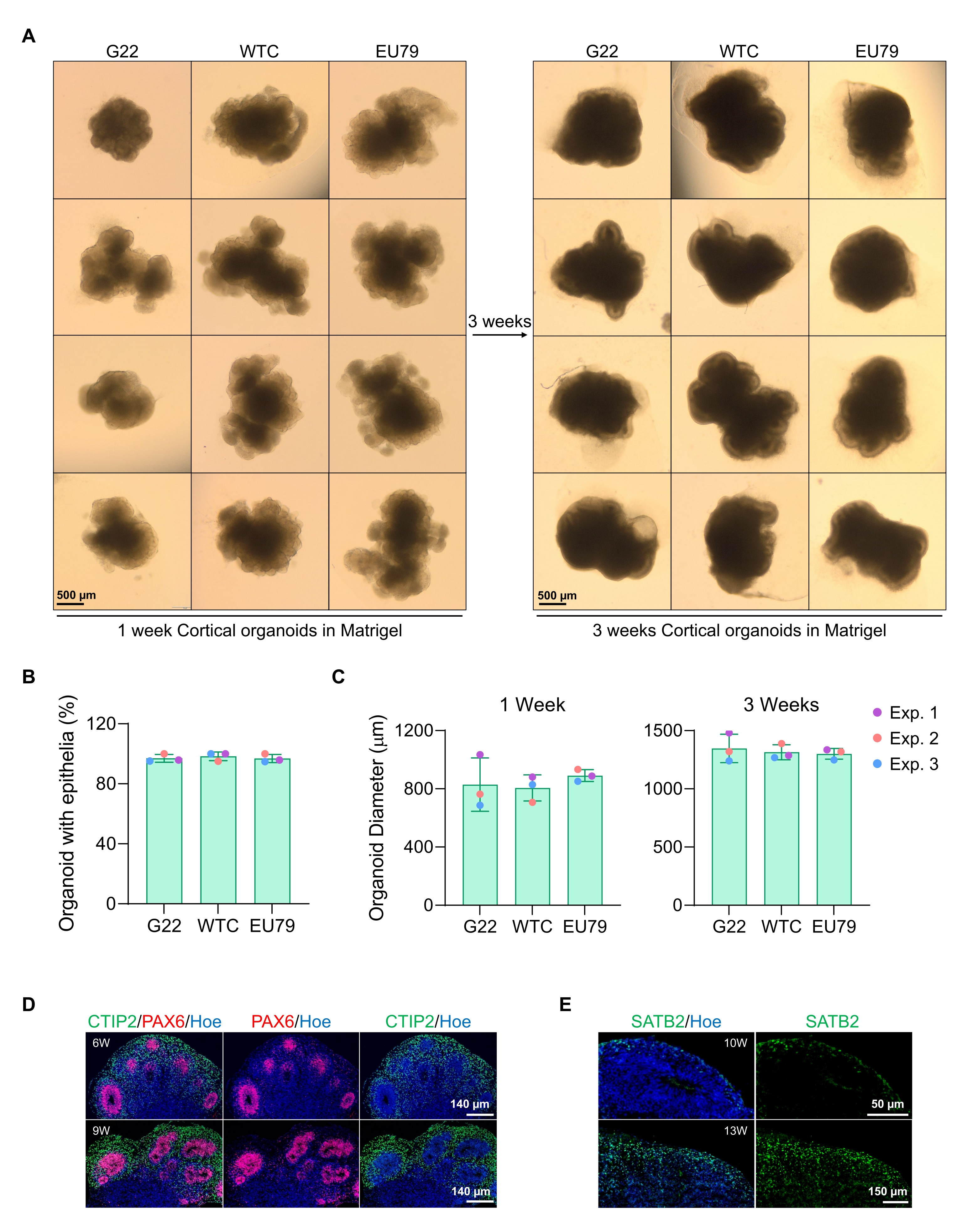
A representative brightfield image of the spheroids after 3 days of culture can be seen in Figure 2D. Spheroids with a tight neuroepithelial layer that have not fused with other neighboring spheroids, have semi-transparent tissue, and demonstrate neural rosette formation, are chosen to be embedded in the basement matrix. Once embedded, the spheroid will proliferate rapidly and start budding: nodes of compact tissue will appear, expanding outward from the main body of the spheroid. This is evident between 1-3 weeks in the basement matrix and can be observed across multiple cell lines (Figure 3A). Quantitative analysis of the embedded spheroids confirms the presence of epithelial cells in up to 100% of spheroids across three different cell lines, affirming the homogeneity and reproducibility expected from this protocol (Figure 3B). The quantification of organoids diameter during in vitro differentiation further confirms the reproducibility across different lines of hPSC (Figure 3C). If budding does not occur, the spheroids are not developing appropriately and should be discarded. Once the spheroids have been embedded in a matrix, their development progresses, and the spheroids are now referred to as organoids. Immunofluorescence staining also confirms the presence of neural progenitor cells (PAX6) as well as cortical layer markers stained with CTIP2 and SATB2 in the organoids with clear layering (Figure 3D,E). This layering is observable across different time points of organoid maintenance (Figure 3D,E). The method of immunohistochemistry of tissues has been described before14.
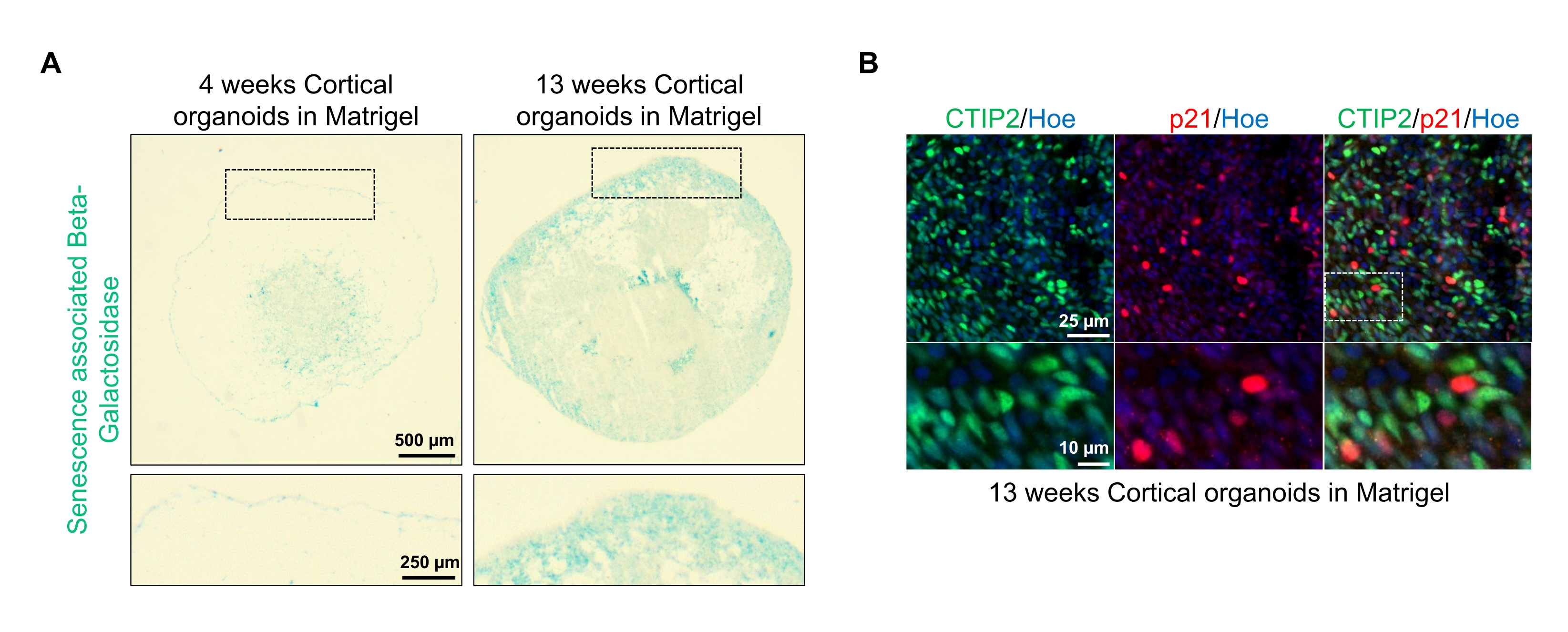
One possible application of these organoids is to study how neuronal aging-related processes affect the brain. To investigate this, successfully generated organoids are harvested from multiple different time points for sectioning and staining for standard molecular biomarkers of senescence such as senescence-associated beta-galactosidase and p21. Figure 4A shows a representative image of senescence-associated beta-galactosidase staining of organoids 4 and 13 weeks after embedding in the basement membrane matrix. Between weeks 4 and 13, there is a marked increase in the presence of senescence-associated beta-galactosidase, suggesting that cellular senescence, a recognized driver of organismal aging, has occurred over this time in culture. Immunofluorescence staining of organoids at week 13 confirmed the presence of another senescence marker, p21, co-labeled with the mature cortical neuronal marker (CTIP2) and can be seen in Figure 4B. It should, however, be noted that the presence of p21 is a marker of cell cycle arrest and by itself is not a definitive marker of senescence, and detection of other markers of senescence such as p16 and SASP (senescence-associated secretory phenotype) factors are recommended to definitively identify cells as senescent.

Figure 1: Schematic diagram for generating reproducible cortical brain organoids. Schematic workflow of the experimental procedure for the generation of cortical brain organoids from hPSCs maintained in the feeder-free medium. The workflow provides an overview of six steps involved to differentiate the 2D hPSCs into 3D patterned cortical plate human tissues in organoids. Please click here to view a larger version of this figure.
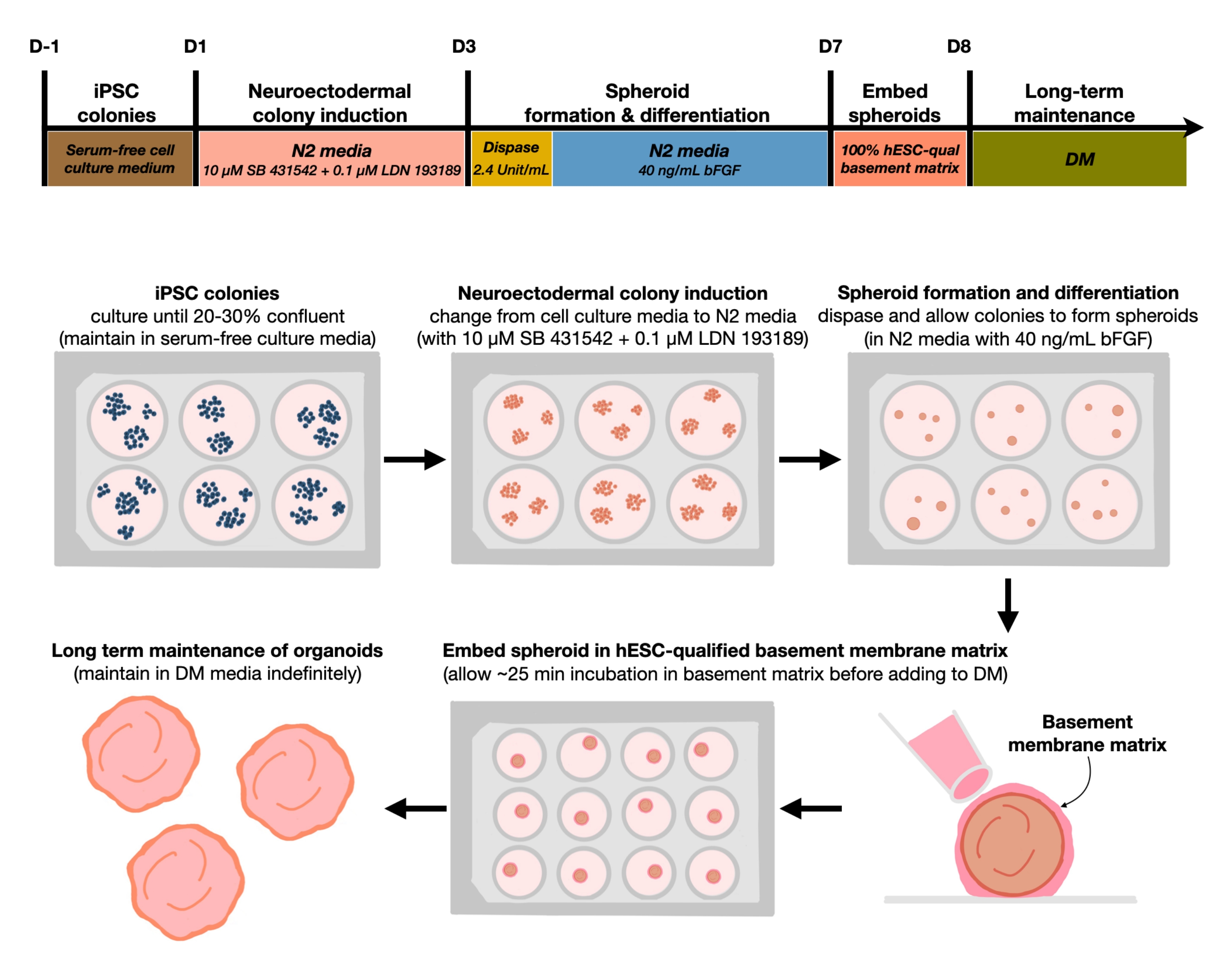{kind=link}

Figure 2: Generation of neural spheroids derived from neuroectodermal colonies-hPSCs. (A) Representative images of human PSC exhibiting optimum (white tick) and differentiated colonies (white cross). Scale bar: 200 µm, 4x magnification. (B) Representative image of neuroectodermal colony derived from hPSCs after 3 days of dual SMAD inhibitor treatments. Scale bar: 200 µm, 4x magnification. (C) Human PSC colonies were differentiated toward neuroectodermal colonies. Images represent staining of PSC (at day 1) and neuroectodermal (at day 3) colonies with SOX2 (Red), NANOG (Green), all nuclei were counterstained with Hoechst 33342 (blue). Scale bar: 40 µm, 100x magnification. (D) Images showing the developmental stages of cortical brain spheroids over time in culture in vitro under brightfield, and wholemount immunostained with SOX2 (Red) at day 1, and double immunostained with SOX2 (red) and ZO1 (Green) at day 4. The scale bar of the brightfield image is 500 µm, 4x magnification, scale bars of the bottom images are 40 µm, 20x magnification. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Characterization of cortical brain organoids derived from different hPSC lines. (A) Representative images of cortical brain organoids derived from G22, WTC, and EU79 human iPSC lines cultured over 3 weeks in vitro. The scale bar of all images is 500 µm, 2x magnification. (B) Percentages of the successful generation of cortical brain organoids at 3 weeks of in vitro differentiation in different hPSC lines (G22, WTC, and EU79). N = 3. Data are presented as mean ± standard deviation. (C) Bar graphs showing the growth of cortical brain organoids (based on average diameter) at weeks 1 and 3 of in vitro differentiation in different lines of human pluripotent stem cell lines (G22, WTC, and EU79). N = 3. Data are presented as mean ± standard deviation. (D) Representative images of sections of 6-week and 9-week-old cortical brain organoids derived from G22 hPSCs, immunostained for ventricle zone PAX6 (red) and cortical plate CTIP2 (green). All sections were counterstained with Hoechst 33342 (blue). Scale bar = 140 µm, 20x magnification. W is week. (E) Representative images of sections of 10-week and 13-week-old cortical brain organoids derived from WTC hPSCs, immunostained for cortical layer IV SATB2 (green). All sections were counterstained with Hoechst 33342 (blue). 10 weeks image Scale bar = 50 µm, 40x magnification. 13 weeks image Scale bar = 150 µm, 40x magnification. W is week. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Characterization of senescence in cortical brain organoids derived from hPSCs. (A) Representative images of sections of human cortical brain organoids derived from WTC hPSCs cultured for 4 and 13 weeks in vitro and stained with SA-β-gal. Scale bar = 500 µm, scale bar of zoomed images = 250 µm, 4x magnification. The dotted box indicates a magnified image. (B) Representative images of sections of 13 week-old cortical brain organoids derived from human EU79 hPSCs, immunostained for cortical neurons CTIP2 (green) and p21 (red). All the sections were counterstained with Hoechst 33342 (blue). Scale bar = 25 µm, scale bar of zoomed images = 10 µm, 40x magnification. Please click here to view a larger version of this figure.
{kind=link}
| Media components | Concentration |
| DMEM Nutrient mix F12 10x 500 mL (DMEM/F-12) | |
| N2 Supplement 5 mL (100x) | Supplmented at 1% |
| B 27 Supplement 10 mL | Supplemented at 2% |
| MEM Non-Essential Amino Acids Solution (100x) | Supplemented at 1% |
| Penicillin-Streptomycin (10,000 U/mL) | Supplemented at 1% |
| 2-Mercaptoethanol 50 mL(1000x ) | Supplemented at 0.1% |
Table 1: N2 Medium. The table lists the reagents required to prepare the N2 medium.
| Media components | Concentration |
| DMEM Nutrient mix F12 10x 500 mL (DMEM/F-12) | DM media is made with 1:1 ratio of DMEM/F12 and Neurobasal media |
| Neurobasal Medium | |
| N2 Supplement 5 mL (100x) | Supplmented at 0.5% |
| B 27 Supplement 10 mL | Supplemented at 1% |
| MEM Non-Essential Amino Acids Solution (100x) | Supplemented at 1% |
| GlutaMAX Supplement 100x | Supplemented at 1% |
| Penicillin-Streptomycin (10,000 U/mL) | Supplemented at 1% |
| Insulin Solution Human Recombinant | 12.5 µL for 50 mL of media |
| 2-Mercaptoethanol 50 mL (1000x ) | 17.5 µL for 50 mL of media |
Table 2: Differentiation medium (DM). The table lists the reagents required to prepare the differentiation medium.
Supplementary Video 1. Live imaging of induced hNEct 2D sheet/colony conversion to 3D under the treatment of bFGF. Induced colonies of hNEct were gently detached from the dish with dispase as outlined above and transferred to a low attachment 6-well culture plate. 2D hNEct colonies were converted to 3D hNEct spheroids within 12 h. Serial images were captured every 5 min. Scale bar = 100 µm. Please click here to download this Video.
Discussion
To enable the use of hPSC-derived brain organoids in drug screening and disease modeling, it is crucial to make organoids following a replicable and reliable protocol15. Brain organoids are commonly generated from embryoid bodies derived from hPSCs, which are then embedded in an extracellular matrix that promotes tissue expansion and neural differentiation. When compared to such protocols as Lancaster's1,16,17 and Velasco18, which begin from embryoid bodies and allow for a default differentiation pathway to be followed by the developing organoids, we have found that commencing cortical brain organoid creation with human NEct cells rather than with embryoid bodies improves the consistency of cortical brain organoid formation. This consequently also allows for the scaling required for drug and phenotypic screening. Since human NEct cells can not only be expanded into considerable quantities but can also be readily cryopreserved, this approach also improves replicability between experiments. It should also be noted that, compared to other protocols that have adopted the use of Bioreactors and similar technologies, no specialized equipment is required for this protocol, making it suitable for any lab6. Finally, the time required to generate mature organoids that are positive for cortical layer markers such as SATB2 is reduced compared to both Lancaster1 and bioreactor protocols6,19 making it more suitable for studying the developmental trajectory of human cortical development in health and diseases1,6,16.
Furthermore, given the continuously growing global health care impact of aging-related diseases such as dementia, which are associated with an increase in senescent cell types in the brain that contributes to pathogenesis, the ability to identify and test compounds that can ameliorate brain aging are of enormous interest. Despite hPSCs being known to be epigenetically rejuvenated during the reprogramming process20, we find robust increases in senescent cells in cortical brain organoids cultured for prolonged periods of time. This is a promising development that now enables the screening of drugs that eliminate such senescent cells from the brain (senolytics) or that slow this process down (senostatics)21. Since human NEct-derived cortical brain organoids are of human origin, this approach will likely shorten the traditional path to market such novel therapeutics.
There are two critical steps in this protocol. The first is the correct level of confluency of the hPSC colonies at the time of differentiation. hPSC colonies must be at most 30% confluent to ensure that generated NEct colonies do not fuse with neighboring colonies and that individual organoids are clonally driven. The second critical step involves the correct use of dispase to lift the NEct colonies and produce the neural spheroids. The timing of incubation with dispase is critical to the eventual quality of the neural spheroids generated. This is because over-exposure of colonies with dispase is toxic to the cells22 and eventually affects the quality of generated organoids. The limitation of this protocol is that it is difficult to control the size of the neural spheroids because it is dependent on the size of the initial colonies that are lifted with dispase. However, this issue can be overcome by selecting neural spheroids that are of a similar size when proceeding to the embedding stage.
Finally, future applications could extend to the use of these reproducible cortical organoids in robotic analysis and biopharmaceutical screening approaches typically used in that industry. This is supported by preliminary data from our laboratory indicating that the generation of cortical brain organoids from human NEct cells can be readily automated, making it compatible with these approaches.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work is supported by the Medical Research Future Fund-Accelerated Research, Leukodystrophy flagship Massimo's Mission (EPCD000034), Medical Research Future Fund-Stem Cell Mission (APP2007653). Authors would like to thank Dr. Ju-Hyun Lee (Korea University) for generating data in Supplementary Video 1.
Materials
| Name | Company | Catalog Number | Comments |
| 16% Formaldehyde (W/V) Methanol-free | Thermo Fisher Scientific | 28908 | 4% of PFA are diluted in 1x PBS |
| 2-Mercaptoethanol 50 mL(1000x) | Life Technologies Australia (TFS) | 21985023 | Used in NM and DM media |
| B 27 Supplement 10 mL | Life Technologies Australia (TFS) | 17504044 | Used in NM and DM media |
| CKX53 microscope with SC50 camera | Olympus | ||
| Corning Costar 6 well cell culture plates | Sigma Aldrich Pty Ltd | CLS3516-50EA | |
| Dispase II powder | Thermo Fisher Scientific | 17105041 | Powder is dissolve in HBSS, filtered through 0.22 µm filter, aliquote at 10 mL and store at -20 °C |
| DMEM Nutrient Mix F12 10x 500 mL (DMEM/F-12) | Thermofisher | 11320082 | Used in NM and DM media |
| DMSO Dimethyl Sulfoxide | Sigma Aldrich Pty Ltd | D2650-100ML | |
| Dulbecco's Phosphate Buffered Saline | Sigma Aldrich Pty Ltd | D1408-500ML | |
| Falcon Matrigel hESC-qualified Matrix | In Vitro Technologies Pty Ltd | FAL354277 | Make aliquotes of 100 µL and stored at -20 °C |
| GlutaMAX Supplement 100x | Thermo Fisher Scientific | 35050061 | Used in NM and DM media |
| Hanks Balanced Salt Solution | Sigma Aldrich Pty Ltd | H8264 | |
| Human induced pluripotent stem cells (EU79) | In-house reporogrammed from skin fibroblast | ||
| Human induced pluripotent stem cells (G22) | Genea Biocells | Obtained from Genea Biocells (San Diego, United States) | |
| Human induced pluripotent stem cells (WTC) | Gift from Professor Bruce Conklin | ||
| InSolution TGF-Β RI Kinase Inhibitor VI, SB431542 | Merck | US1616464-5MG | |
| Insulin Solution Human Recombinant | Sigma Aldrich Pty Ltd | I9278 | Used in NM and DM media |
| LDN193189 Dihydrochloride | Sigma Aldrich Pty Ltd | SML0559-5MG | Used during differentiation |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | 11140050 | Used in NM and DM media |
| mTeSR Plus | STEMCELL TECHNOLOGIES | 100-0276 | Used to maintain hiPSC colonies prior to differentiation with NM media |
| N2 Supplement 5 mL (100x) | Life Technologies Australia Pty Ltd | 17502048 | Used in NM and DM media |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 | Used in DM media |
| OCT Embedding Compound Sakura Clear (118 mL/Bottle) | Tissue Tek | 4583 | |
| Parafilm M Roll Size 4 in. x 125 Ft | Sigma Aldrich Pty Ltd | P7793 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | Used in NM and DM media |
| Potassium Hexacyanoferrate (II) Trihydrate | Sigma Aldrich Pty Ltd | CP1087 | |
| Potassium hexacyanoferrate(III) | Sigma Aldrich Pty Ltd | 455946 | |
| Prolong Glass Antifade Mountant | Life Technologies Australia (TFS) | P36980 | |
| Recombinant Human FGF basic | R&D Systems | 233-FB-01M | Aliquotes are made at 20 µg/mL and stored at -20 °C |
| SB431542 | Tocris | 1614 | Used during differentiation |
| Sucrose | Sigma Aldrich Pty Ltd | PHR1001-1G | 30% of sucrose are diluted in 1x PBS |
| Ultra-Low attachment multiwell plates , 24 well plate, polystyrene | Sigma Aldrich Pty Ltd | CLS3473-24EA | |
| X-GAL EA | Life Technologies Australia (TFS) | R0404 | Make aliquotes of 20 mg/mL and storde at -80 °C |
References
- Lancaster, M. A., Knoblich, J. A. Generation of cerebral organoids from human pluripotent stem cells. Nature Protocols. 9 (10), 2329-2340 (2014).
- Mansour, A. A., et al. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology. 36 (5), 432-441 (2018).
- Xiang, Y., et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 21 (3), 383-398 (2017).
- Bagley, J. A., Reumann, D., Bian, S., Lévi-Strauss, J., Knoblich, J. A. Fused cerebral organoids model interactions between brain regions. Nature Methods. 14 (7), 743 (2017).
- Kwak, T. H., et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. 38 (6), 727-740 (2020).
- Qian, X., et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nature Protocols. 13 (3), 565-580 (2018).
- Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K., Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports. 10 (4), 537-550 (2015).
- Shaker, M. R., Cooper-White, J., Wolvetang, E. J. Self-organizing 3D human choroid plexus-ventricle-cortical organoids. BioRxiv. , (2020).
- Shaker, M. R., Aguado, J., Chaggar, H. K., Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging and Mechanisms of Disease. 7 (1), 1-12 (2021).
- Shaker, M. R., et al. Anteroposterior Wnt-RA gradient defines adhesion and migration properties of neural progenitors in developing spinal cord. Stem Cell Reports. 15 (4), 898-911 (2020).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shaker, M. R., et al. Rapid and efficient generation of myelinating human oligodendrocytes in organoids. Frontiers in Cellular Neuroscience. 15, 631548 (2021).
- Lee, J. -. H., Shaker, M. R., Lee, E., Lee, B., Sun, W. NeuroCore formation during differentiation of neurospheres of mouse embryonic neural stem cells. Stem Cell Research. 34, 101691 (2020).
- Shaker, M. R., et al. Spatiotemporal contribution of neuromesodermal progenitor-derived neural cells in the elongation of developing mouse spinal cord. Life Sciences. 282, 119393 (2021).
- Shaker, M. R., et al. Neural epidermal growth factor-like like protein 2 Is expressed in human oligodendroglial cell types. Frontiers in Cell and Developmental Biology. 10, 803061 (2022).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Giandomenico, S. L., Sutcliffe, M., Lancaster, M. A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nature Protocols. 16 (2), 579-602 (2021).
- Velasco, S., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 570 (7762), 523-527 (2019).
- Qian, X., et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 165 (5), 1238-1254 (2016).
- Hunter, Z. L., Leeson, H. C., Shaker, M. R., Wolvetang, E. J., Vadlamudi, L. Human induced pluripotent stem cells generated from epilepsy patients for use as in vitro models for drug screening. Stem Cell Research. 60, 102673 (2022).
- Kaur, A., Macip, S., Stover, C. M. An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Frontiers in Cell and Developmental Biology. 8, 218 (2020).
- Wang, F., et al. Safety and efficacy of dispase and plasmin in pharmacologic vitreolysis. Investigative Ophthalmology & Visual Science. 45 (9), 3286-3290 (2004).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved