
Method Article
Rapid Optical Clearing for Semi-High-Throughput Analysis of Tumor Spheroids
In This Article
Summary
Tumor spheroids are becoming increasingly utilized to assess tumor cell-microenvironment interactions and therapy response. The present protocol describes a robust but simple method for semi-high-throughput imaging of 3D tumor spheroids using rapid optical clearing.
Abstract
Tumor spheroids are fast becoming commonplace in basic cancer research and drug development. Obtaining data regarding protein expression within the spheroid at the cellular level is important for analysis, yet existing techniques are often expensive, laborious, use non-standard equipment, cause significant size distortion, or are limited to relatively small spheroids. This protocol presents a new method of mounting and clearing spheroids that address these issues while allowing for confocal analysis of the inner structure of spheroids. In contrast to existing approaches, this protocol provides for rapid mounting and clearing of a large number of spheroids using standard equipment and laboratory supplies. Mounting spheroids in a pH-neutral agarose-PBS gel solution before introducing a refractive-index-matched clearing solution minimizes size distortion common to other similar techniques. This allows for detailed quantitative and statistical analysis where the accuracy of size measurements is paramount. Furthermore, compared to liquid clearing solutions, the agarose gel technique keeps spheroids fixed in place, allowing for the collection of three-dimensional (3D) confocal images. The present article elaborates how the method yields high-quality two- and 3D images that provide information about inter-cell variability and inner spheroid structure.
Introduction
Three-dimensional (3D) cell cultures, such as spheroids, provide biologically realistic and reproducible models of aggregate cell growth1,2. These models are fast becoming commonplace in both basic research and drug development, where differences in spheroid size and structure are examined between treatments to ascertain drug efficacy3,4. In these contexts, the ability to collect detailed information from a large number of spheroids is highly advantageous, both from a statistical power perspective and to allow rapid assessment of cell behavior across several treatments.
Commonly used techniques for obtaining detailed microscopy images of spheroid structure are either time-consuming, expensive, or produce poor quality images that do not retain key quantitative features such as spheroid size4,5. For example, histological techniques based upon cryosectioning can provide high-quality images but are often time-consuming, require skilled labor, and often create sectioning artefacts6,7, while elegant technologies, such as single plane illumination microscopy (SPIM)8 and multiphoton microscopy9 require specialized microscopes that are not readily available. Modern microscopy technologies have recently enabled the so-called optical sectioning, where spheroids are placed within a refractive index matched clearing solution and images are obtained using confocal microscopy4,5. While these techniques have the potential to produce a high yield, common problems include spheroid movement while imaging, size distortion while clearing, and the high expense of proprietary clearing solutions. Furthermore, many existing protocols apply only to relatively small spheroids of less than 300 µm in diameter or depth up to 100 µm, limiting the technology to the early stages of tumor growth5,10,11.
The present protocol allows semi-high-throughput, high-yield collection of detailed spheroid images using a low-cost refractive index-matched clearing solution derived from whole organ clearing procedures12,13. To prevent spheroid movement during imaging and provide structural support to reduce size distortion, the spheroids are mounted in agarose-PBS gel in a 24-well #1.5 glass bottom plate. Since this technique allows for multiple spheroids to be mounted in each well in a 24-well plate, up to 360 spheroids (15 spheroids/well) can be rapidly mounted and imaged across various experimental conditions. A refractive-index-matched clearing solution constructed from readily available consumables is used to clear mounted spheroids and the surrounding gel optically. After a settling period of 24 h, this protocol provides high-quality 2D and 3D images of spheroid structure, even for relatively large spheroids (approximately 700 µm in diameter), with less than 2% size distortion.
Protocol
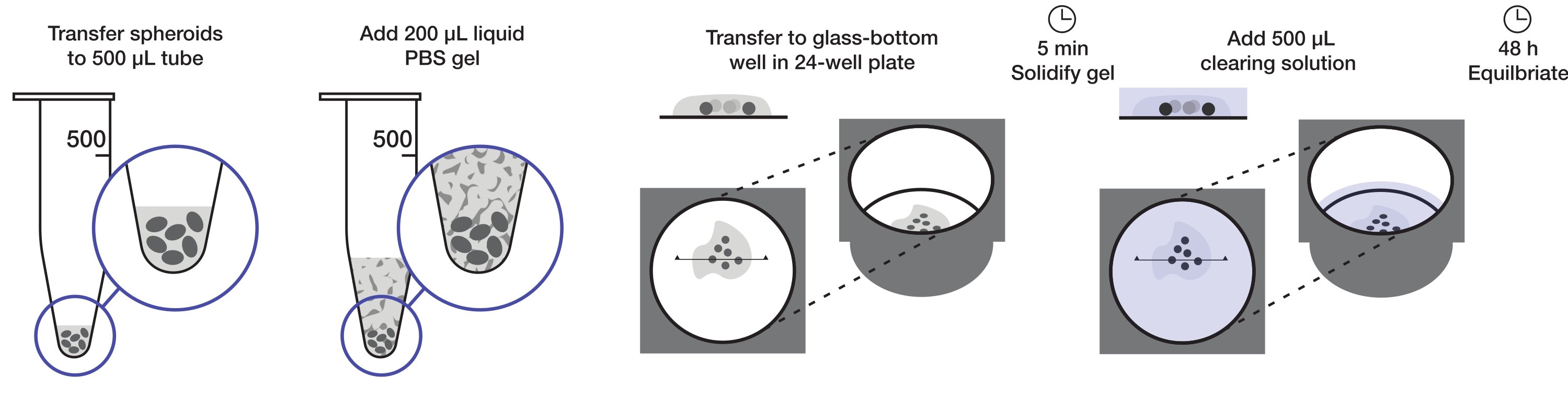
The protocol describes the preparation of a sufficient quantity of tumor spheroids to mount one 24-well plate (approximately 240-360 spheroids or 10-15 spheroids/well) at 200 µL of agarose gel per well and 500 µL of clearing solution per well. The complete procedure is illustrated in Figure 1.
1. 2% agarose-PBS gel preparation
- Weigh 0.5 g of low-melting agarose (see Table of Materials) in a glass bottle and add 25 mL of phosphate-buffered saline (PBS).
- Melt the agarose by boiling the solution in a microwave for 30-60 s with the lid closed but not sealed and with constant swirling. Ensure agarose is fully dissolved and the solution does not boil over.
NOTE: Gel can be stored at room temperature; check the volume before use to account for evaporation. Bring to the liquid state (microwave; 20-60 s with swirling) before using.
2. Preparation of the clearing solution
- Weigh 9 g of N,N,N′,N′-Tetrakis(2-Hydroxypropyl) ethylenediamine, 22 g of urea, 44 g of sucrose, 0.1 g of Triton X-100, and 24.9 g of deionized water (see Table of Materials).
NOTE: It is easier to weigh the approximate amount of N,N,N′,N′-Tetrakis(2-Hydroxypropyl)ethylenediamine and adjust the weight of other components to obtain the desired concentration. - Heat the solution in a 56 °C water bath with constant mixing and continue till all the crystals dissolve.
- Rest the solution at room temperature to allow the bubbles formed during mixing to rise to the surface. The solution is stable at room temperature for up to 2 months.
3. Spheroid preparation
- Generate spheroids using the agarose overlay method as previously described1,3,14.
NOTE: Spheroids generated by other methods are also compatible with this imaging protocol. - Cut the tip of a 1 mL pipette tip using a scalpel to enlarge the orifice.
NOTE: All spheroids were handled with 1 mL or 200 µL pipettes with cut tips. - Using the pipette, aspirate the spheroids from the wells and transfer them to a clear 1.5 mL conical tube. Multiple spheroids from the same conditions can be combined in the same tube.
NOTE: Always allow the spheroids to settle to the bottom of the tube to aspirate the medium. - Wash the spheroids in 1 mL PBS twice, add neutral 4% pre-warmed paraformaldehyde (PFA) solution, and incubate the spheroids at 37 °C for 20 min.
- Remove the PFA solution and wash the spheroids twice with 1 mL of PBS.
4. Spheroid staining
- Using a pipette, transfer up to 15 spheroids into a 200 µL PCR tube.
NOTE: For fragile spheroids, mount the spheroids in gel (steps 5.1-5.4) before proceeding. - Permeabilize the cells using 200 µL of 0.5% Triton X-100 in PBS. Place PCR tubes inside 50 mL screw-cap tubes and incubate the spheroids for 2 h at room temperature with mild agitation.
NOTE: All incubations are done with agitation on a rotor, roller, or shaker (see Table of Materials) unless specified otherwise. This is important to achieve uniform staining. Fixed spheroids in PBS can sometimes stick to the sides of the pipette tip. Rinsing the tip in 0.5% Triton X-100 minimizes the spheroids from sticking to the tips. - Replace the solution (step 4.2) with 200 µL of Antibody dilution buffer (ABDIL)15 and incubate overnight at room temperature.
- Remove ABDIL, add 75 µL of primary antibody in ABDIL, and incubate at 4 °C for 2.5 days.
- Remove the supernatant and wash with 200 µL of PBS with 0.1% Tween and 0.1% Triton X-100 (PBS-TT) twice and incubate with 200 µL of PBS-TT for 4 h at room temperature.
- Remove PBS-TT, add 100 µL of secondary antibody in ABDIL, and incubate at 4 °C for 2.5 days.
NOTE: DAPI or other nuclear dye can be added at this stage. Most dyes require less incubation time than antibodies. - Remove the supernatant and wash with 200 µL of PBS-TT twice and incubate with 200 µL of PBS-TT for 4 h. The spheroids are ready for mounting.
5. Mounting
- Using a 200 µL pipette, transfer fixed and stained spheroids to 500 µL PCR tubes at one tube per condition (approximately 10-15 spheroids).
- Replace the solution with 200 µL of 2% (w/v) liquid agarose gel and centrifuge on the quick spin (at the fixed maximum speed, see Table of Materials) at room temperature for 30 s.
NOTE: Unless mounting immediately, place in a heating block at 50 °C to avoid gel hardening. - Aspirate the spheroids in ~50 µL liquid agarose gel and dispense in well of a 24-well glass-bottom plate.
- Before the gel hardens, separate the spheroids using a pipette tip in the surrounding gel, and ensure the spheroids are covered with gel. Optionally, place the plate on ice to rapidly set the gel.
NOTE: The number of spheroids per well depends on the size of the spheroids. Spheroids are placed far away from each other so that only one spheroid enters the field of view while imaging. This is important for automated image processing. - Add 500 µL of clearing solution (step 2) per well, ensuring the gel is submerged. Incubate at RT for at least 24 h and image the spheroids in the clearing solution. Spheroids are stable in this solution for up to a month at room temperature and when protected from evaporation.
6. Imaging
- Choose an objective with a working distance long enough to encompass the entire spheroid, including the mounting height of the spheroid in the vessel.
NOTE: Objectives with a working distance of 3 mm or longer are required to image the equatorial plane of the spheroids. Higher magnification objectives with higher NA can allow for better resolution, but will limit the maximum depth at which the spheroids can be imaged. - To identify the equatorial plane, adjust the focus till the largest surface area is reached in the XY plane and image with the required laser power percentage, detector voltage, gain, and offset settings.
NOTE: The laser power percentage, detector voltage, gain, and offset will vary greatly depending on the fluorophore, staining intensity, laser intensity, detector sensitivity, etc. - For 3D images, set the start and end of the spheroids, and choose appropriate signal intensity at various z-depth using z intensity correction settings before imaging.
NOTE: For best image resolution, use the Nyquist sampling rate for x, y, and z (for details, see reference16).
Results
To demonstrate the ability of this clearing method to provide high-quality two- and three-dimensional images, spheroids with diameters of 300-600 µm were grown from Fluorescent Ubiquitination-based Cell Cycle Indicator (FUCCI) transduced melanoma cell lines FUCCI-WM164 and FUCCI-WM983b9,17, which express monomeric Kusabira Orange2 (mKO2) and monomeric Azami Green (mAG) protein when in Gap1, and early S-phase/Gap2/mitosis phase of the cell cycle, respectively according to procedures provided in step 31,3,14. The spheroids were then fixed with 4% formaldehyde solution at 37 °C, permeabilized, and stained with anti-p27kip1/anti-rabbit Alexa Fluor 647 anti-pimonidazole/anti-mouse Alexa Fluor 647, DAPI, or DRAQ7 (see Table of Materials) (Figure 2). All microscopy files are uploaded to the GitHub repository (https://github.com/ap-browning/SpheroidMounting). Compared with PBS-mounted spheroids, the clearing solution provides high-clarity images with minimal size distortion (Figure 2A). The protocol allows for high-resolution imaging of cellular-level details deeper into the spheroids without histological sectioning, and the cross-sectional image was obtained from a 20x air objective (0.7 NA) at a resolution of 4096 x 4096 px without stitching (Figure 2B). Using a lower magnification and lower numerical aperture objective with a longer working distance, 3D confocal images that provide cellular-level detail at a depth of at least 200 µm can be obtained (Figure 2C). Spheroids were also cryosectioned and stained as per the protocol by Spoerri et al.4 and compared to whole spheroid staining (Figure 2D,E). Figure 2D shows the hypoxic region of the spheroid stained by pimonidazole, and Figure 2E shows p27kip1 staining that marks cell cycle arrest (yellow) and DAPI nuclear stain (gray). Protein localization and staining pattern are similar between cryosectioning and clearing and hence are unaffected by this clearing method.
Signal intensity correction in z allows the imaging of the whole spheroid. However, light-scattering due to the necrotic core limits the ability to image the far side of the spheroid. Deeper penetration and less scattering of a far-red fluorophore, such as DRAQ7 nuclear stain, allows for even further improved 3D spheroid structure representation (Figure 3). Movie 1 shows the 3D rendering of the FUCCI spheroids stained with DRAQ7. A thinner z-slice may allow for better z-resolution, but this significantly increases the imaging time and photobleaching of the fluorophores.
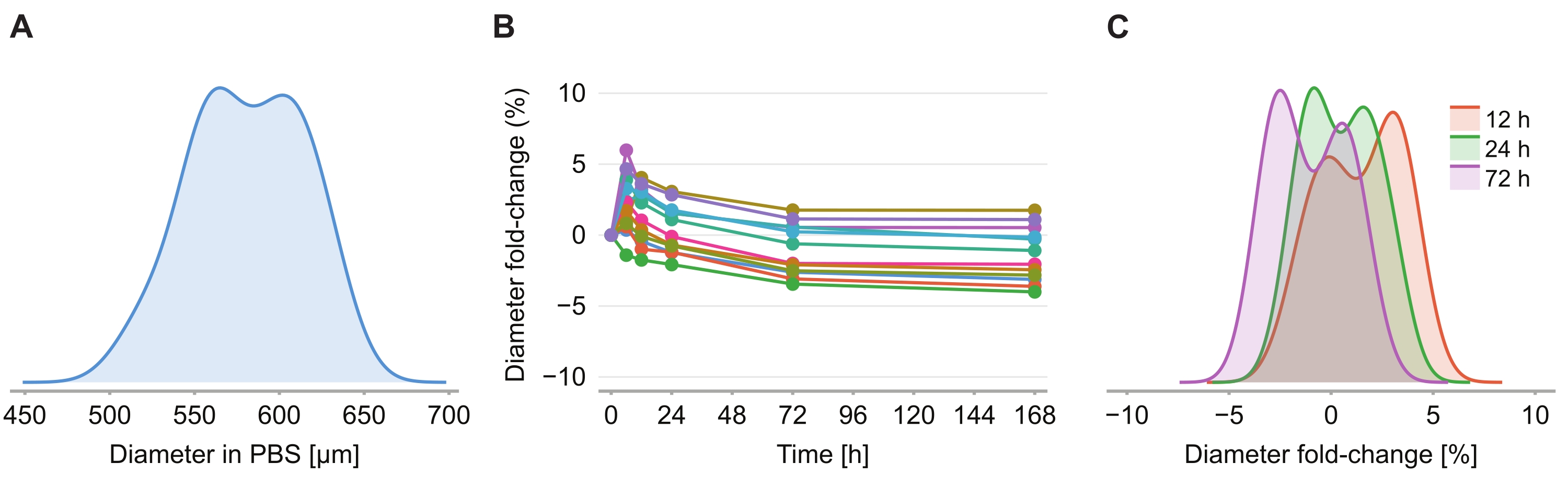
To determine whether the clearing solution causes size distortion, twelve spheroids in 2% agarose-PBS gel were imaged at 6 h, 12 h, 24 h, 72 h, and 168 h following the introduction of the clearing solution. The images were summarized by determining the diameter of the spheroid, defined based on a sphere with the same cross-sectional area as the spheroid (Figure 4A). While the spheroids are observed to slightly increase in size over the first 6 h, indicated by a diameter fold-change of between 2% and 6% (Figure 4B), after 24 h to 72 h, the spheroids return to a size approximately equal to the corresponding size in PBS post PFA fixation (Figure 4C).

Figure 1: Illustration of the spheroid mounting and clearing protocol. Fixed and stained spheroids are transferred to a 500 µL tube; excess fluid is replaced by 200 µL agarose-PBS gel and centrifuged. Spheroids are then transferred to a glass-bottom well in a 24-well plate. After the gel is allowed to solidify, 500 µL clearing solution is added, and spheroids are allowed to equilibrate. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Comparison of cleared and uncleared FUCCI human melanoma spheroids. Coloring indicates cell nuclei positive for mKO2 (red), which indicates cells in gap 1; and cell nuclei positive for mAG (green), which indicates cells in gap 2. (A) Spheroids grown from 5000 FUCCI-WM983b cells, harvested at day 10 and imaged in agarose-PBS gel and 24 h after clearing solution is added. Comparing brightfield and confocal images before and after clearing solution shows minimal size distortion and a large gain in clarity. Images are obtained using a 10x objective. (B) Spheroids grown from FUCCI-WM164 cells permeabilized using Triton X-100 and stained with DRAQ7, staining all cell nuclei. Image is obtained using a 20x objective (0.75 NA), demonstrating the clearing solution allows high-resolution imaging of cell-level details. (C) 3D images (10x, 0.4 NA) were obtained of FUCCI-WM164 spheroids in PBS, and 24 h after the clearing solution was added. Adjusting laser power, voltage, and offset at different z-plane allows imaging deeper inside the spheroid. (D,E) Comparison between cryosection and cleared whole spheroid stained for pimonidazole and p27kip1. (D) Pimonidazole staining in magenta shows the hypoxic region in the spheroids. Red and green indicate FUCCI. (E) Cryosections and cleared spheroid showing DAPI (gray) and p27kip1 (yellow). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Clearing allows for imaging deeper into the spheroid with minimal light loss. Confocal microscopy images of a FUCCI human melanoma spheroid at 10x magnification and lower NA (0.4), allowing imaging at higher z-depth with minimal signal loss. (A) 3.88 µm slices of spheroid nuclei stained with DRAQ7. (B-D) y/z-resolution in the 488 (mAG), 568 (mKO2), and 647 nm (DRAQ7) channels, respectively. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Clearing solution has minimal impact on spheroid size. (A) Distribution of initial spheroid size (equivalent diameter) in PBS gel (n = 12 spheroids). (B) Diameter fold-change over time since addition of clearing solution. At 0 h, spheroids are in PBS gel only. (C) Distribution of diameter fold-changes at 12 h, 24 h, and 72 h. Please click here to view a larger version of this figure.
{kind=link}
Movie 1: 3D rendering of a FUCCI spheroid stained with DRAQ7. Please click here to download this Movie.
Discussion
A protocol for obtaining high-quality two- and three-dimensional images of tumor spheroids is presented here. Existing methods, such as CLARITY, See deep brain (SeeDB), and ScaleS, often cause size distortion by up to 30%, while techniques such as Benzyl Alcohol/ Benzyl Benzoate (BABB) and 3D imaging of solvent-cleared organs (3DISCO) can quench fluorescent protein18. Many of these methods are designed for clearing tissue with structural integrity and distort the size and structure when applied to spheroids18. In contrast to other protocols that use commercially available expensive clearing solutions, this protocol uses readily available consumables while maintaining optical clarity and endogenous fluorescence and minimizing size distortion. Embedding spheroids in agarose-PBS gel provides structural support for spheroids and minimizes osmotic shock when the clearing solution is added. This is crucial when imaging fragile spheroids post drug treatment. It is assumed that this optical clearing method is suitable for spheroids formed by any method since this protocol is adapted from whole tissue clearing. The assumption is based on the similarity in the spheroids obtained by different spheroid formation methods. The choice of fixative can affect the spheroid size as well as endogenous fluorescence. This clearing method is suitable for spheroids fixed with neutral 4% PFA solution. Further testing is required to check its compatibility with other fixatives.
Given that this technique allows multiple spheroids to be mounted simultaneously in a multi-well plate, it is well suited to quantitative analysis pipelines that require spheroid structure information from up to 360 spheroids per 24-well plate. Microscopes with automated stage and plate mapping functions can make imaging less manual. Even though this method is quicker and easier than sectioning, it is currently unsuitable for complete automation. However, images obtained by this method are suitable for automated image processing4,19, and the rate at which spheroids can be mounted using this protocol lends to quantitative analysis of spheroid inner structure20,21,22.
For staining whole spheroids, the antibody concentration, volume, and incubation time need to be optimized for every antibody. As a guide, use 2.5x of the recommended 2D immunofluorescence antibody concentration and 100-200 µL of antibody, depending on the number of spheroids per tube. Ensure that all spheroids are covered in staining solution when on the rotor. The incubation time depends on many factors, including the size and density of spheroids and the antibody, and can range from 16-72 h. Despite the method allowing for signal detection deeper within the spheroid, fluorophores excited by UV cause significant light scattering, leading to a low signal-to-noise ratio. Care must be taken when choosing fluorophores to visualize the target protein. For example, staining less abundant and structural proteins with longer wavelength fluorophores and more abundant protein or nuclear stains with shorter wavelength fluorophores will achieve the best result. Finally, cleared spheroids still show light loss due to scattering in the necrotic core, evident in the y/z-dimension of images obtained of 600 µm spheroids (Figure 2C).
Higher magnification imaging with higher NA objectives is possible with spheroids mounted and cleared using this protocol, but the objective's working distance limits the imaging depth. For an oil immersion lens, it is important to use oil that has an RI of 1.51 for the best result.
To summarize, the agarose-PBS gel embedded clearing method allows for visualization of cells deep inside spheroids using commonly available consumables. Spheroids mounted and cleared using this method undergo minimal size distortion and maintain their structural integrity, allowing the collection of high-quality data relating to the inner spheroid structure and subsequent automated quantification.
Disclosures
Olympus contributed to the costs of publication.
Acknowledgements
This research was carried out at the Translational Research Institute (TRI), Woolloongabba, QLD. TRI is supported by a grant from the Australian Government. We thank the staff in the microscopy core facility at TRI for their outstanding technical support. We thank Prof. Atsushi Miyawaki, RIKEN, Wako-city, Japan, for providing the FUCCI constructs, Prof. Meenhard Herlyn and Ms. Patricia Brafford, The Wistar Institute, Philadelphia, PA, for providing the cell lines. We thank Dr Loredana Spoerri for providing C8161 cryosection images.
This work was supported by project grants to N.K.H.: Australian Research Council (DP200100177) and Meehan Project Grant (021174 2017002565).
Materials
| Name | Company | Catalog Number | Comments |
| #1.5 glass bottom 24-well plate | Celvis | P24-1.5H-N | |
| 500 µL clear PCR tubes | Sigma | HS4422 | |
| Alexa Fluor 647 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno research | 715-605-151 | Dilution used 1:500 |
| Bovine serum albumin | Sigma | A7906 | Final concentration 2% w/v |
| DAPI | Sigma | D9542-10MG | Final concentration 5 µg/mL |
| Deionized water | MILLI Q | ||
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermofisher | A-31573 | Dilution used 1:500 |
| DRAQ7 | Thermofisher | D15106 | Dilution used 1:250 |
| Heating block | Ratek | DBH10 | or similar equipment |
| Hypoxyprobe Kits | Hypoxyprobe | HP1-1000Kit | Antibody dilution used 1:500 |
| Low-melting agarose powder | Sigma | A9414 | Final concentration 2% w/v |
| Microwave | Sharp | ||
| N,N,N′,N′-Tetrakis(2-Hydroxypropyl)ethylenediamine | Sigma | 122262 | Final concentration 9% w/w |
| NaCl | Sigma | S9888 | Final concentration 150 mM |
| NaN3 | Sigma | S2002 | Final concentration 0.10% w/v |
| p27 Kip1 (D69C12) XP | Cell Signalling technology | 3686S | Dilution used 1:500 |
| Paraformaldehyde solution | Proscitech | C004 | Final concentration 4% w/v |
| Phosphate Buffered Saline (PBS) | Thermofisher | 18912014 | Final concentration 1x |
| Pipette | Eppendorf | ||
| Quickspin minifuge | or similar equipment | ||
| Roller | Ratek | BTR10-12V | or similar equipment |
| Rotor | Ratek | RSM7DC | or similar equipment |
| Shaker | Ratek | EOM5 | or similar equipment |
| Sucrose | Sigma | S9378 | Final concentration 44% w/w |
| Tris-HCl pH 7.4 | Sigma | T5941 | Final concentration 20 mM |
| Triton X-100 | Sigma | X100 | Final concentration 0.10% v/v |
| Triton X-100 | Sigma | X100 | Final concentration 0.1% v/v |
| Tween 20 | Sigma | P1379 | Final concentration 0.1% v/v |
| Urea | Sigma | U5379 | Final concentration 22% w/w |
References
- Beaumont, K. A., Anfosso, A., Ahmed, F., Weninger, W., Haass, N. K. Imaging- and flow cytometry-based analysis of cell position and the cell cycle in 3D melanoma spheroids. Journal of Visualized Experiments. (106), e53486 (2015).
- Hirschhaeuser, F., et al. Multicellular tumor spheroids: an underestimated tool is catching up again. Journal of Biotechnology. 148 (1), 3-15 (2010).
- Smalley, K. S., Lioni, M., Noma, K., Haass, N. K., Herlyn, M. In vitro three-dimensional tumor microenvironment models for anticancer drug discovery. Expert Opinion on Drug Discovery. 3 (1), 1-10 (2008).
- Spoerri, L., Gunasingh, G., Haass, N. K. Fluorescence-based quantitative and spatial analysis of tumour spheroids: A proposed tool to predict patient-specific therapy response. Frontiers in Digital Health. 3, 668390 (2021).
- Nürnberg, E., et al. Routine optical clearing of 3D-cell cultures: Simplicity forward. Frontiers in Molecular Biosciences. 7, 20 (2020).
- Kabadi, P. K., et al. Into the depths: Techniques for in vitro three-dimensional microtissue visualization. BioTechniques. 59 (5), 279-286 (2015).
- Ivanov, D. P., Grabowska, A. M. Spheroid arrays for high-throughput single-cell analysis of spatial patterns and biomarker expression in 3D. Scientific Reports. 7, 41160 (2017).
- Spoerri, L., et al. Phenotypic melanoma heterogeneity is regulated through cell-matrix interaction-dependent changes in tumor microarchitecture. bioRxiv. , (2021).
- Haass, N. K., et al. Real-time cell cycle imaging during melanoma growth, invasion, and drug response. Pigment Cell & Melanoma Research. 27 (5), 764-776 (2014).
- Ahmad, A., et al. Clearing spheroids for 3D fluorescent microscopy: combining safe and soft chemicals with deep convolutional neural network. bioRxiv. , 428996 (2021).
- Villani, T., Rossi, A. E., Sherman, H. Image-based characterization of 3-D cell culture models grown in spheroid microplates. American Laboratory. 50 (6), 12 (2018).
- Lloyd-Lewis, B., et al. Imaging the mammary gland and mammary tumours in 3D: optical tissue clearing and immunofluorescence methods. Breast Cancer Research. 18 (1), 127 (2016).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159 (4), 911-924 (2014).
- Spoerri, L., Beaumont, K. A., Anfosso, A., Haass, N. K. Real-time cell cycle imaging in a 3D cell culture model of melanoma. Methods in Molecular Biology. 1612, 401-416 (2017).
- Cold Spring Harbor. Antibody Dilution Buffer (Abdil). Cold Spring Harbor Protocols. , (2018).
- Pawley, J. B., Pawley, J. B. . Handbook Of Biological Confocal Microscopy. , 20-42 (2006).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Tian, T., Yang, Z., Li, X. Tissue clearing technique: Recent progress and biomedical applications. Journal of Anatomy. 238 (2), 489-507 (2021).
- Browning, A. P., Murphy, R. J. . Zenodo. , (2021).
- Browning, A. P., et al. Quantitative analysis of tumour spheroid structure. eLife. 10, 73020 (2021).
- Klowss, J. J., et al. A stochastic mathematical model of 4D tumour spheroids with real-time fluorescent cell cycle labelling. Journal of The Royal Society Interface. 19 (189), 20210903 (2022).
- Murphy, R. J., Browning, A. P., Gunasingh, G., Haass, N. K., Simpson, M. J. Designing and interpreting 4D tumour spheroid experiments. Communications Biology. 5 (1), 91 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved